

Abstract
Purpose
Metastasis and drug resistance are the major limitations in the survival and management of cancer patients. This study aimed to identify the mechanisms underlying HT29 colon cancer cell chemoresistance acquired after sequential exposure to 5-fluorouracil (5FU), a classical anticancer drug for treatment of epithelial solid tumors. We examined its clinical relevance in a cohort of colon cancer patients with liver metastases after 5FU-based neoadjuvant chemotherapy and surgery.
Results
We show that a clonal 5F31 cell population, resistant to 1μM 5FU, express a typical cancer stem cell-like phenotype and enter into a reversible quiescent G0-state upon re-exposure to higher 5FU concentrations. These quiescent cells overexpressed the tyrosine kinase c-Yes that became activated and membrane-associated upon 5FU exposure. This enhanced signaling pathway induced the dissociation of the Yes/YAP (Yes-associated protein) molecular complex and depleted nuclear YAP levels. Consistently, c-Yes silencing decreased nuclear YAP accumulation and induced cellular quiescence in 5F31 cells cultured in 5FU-free medium. Importantly, c-Yes and YAP transcript levels were higher in liver metastases of colon cancer patients after 5FU-based neoadjuvant chemotherapy. Moreover, the c-Yes and YAP levels positively correlated with colon cancer relapse and shorter patient survival (p<0.05 and p<0.025, respectively).
Conclusions
We identified c-Yes and YAP as potential molecular targets to eradicate quiescent cancer cells and dormant micrometastases during 5FU chemotherapy and resistance and as predictive survival markers for colon cancer.
Keywords: Chemoresistance, Colon cancer stem cells, Colon liver metastases, Adjuvant chemotherapy, Predictive survival markers
INTRODUCTION
Treatment of colon cancer involves the surgical resection of the primary tumor. For patients with stage III disease, a survival advantage is obtained with 5FU/leucovorin-based adjuvant chemotherapy used in combination with oxaliplatin or irinotecan (1–3). In the last 8 years, cetuximab and panitumumab, two monoclonal antibodies that target the epidermal growth factor receptor, were shown to be effective in combination with chemotherapy or as single agents in patients with wild-type KRAS tumors (4–6). In addition, anti-angiogenic therapy targeting vascular endothelial growth factor (bevacizumab) confers a benefit when used in combination with chemotherapy (7). However, the development of drug resistance remains a major limitation in the efficacy of the clinical response to chemotherapeutic and targeted therapy regimens.
A growing body of evidence suggests that the majority of tumors comprise a population of tumor-initiating or cancer stem cells (CSCs) that are responsible for the development and maintenance of tumors and resistance to cytotoxic drugs (8). In breast cancer, studies using clinical tumor samples support the hypothesis that the residual disease after neo-adjuvant chemotherapy is enriched with CSCs. Cell suspensions derived from chemotherapy-treated patients showed an increase in mammosphere formation, self-renewal and enrichment in CD44+CD24−/low stem-like cells (9). In murine models of colon cancer cell xenografts, the treatment of mice with chemotherapeutic agents enriched the tumor xenografts in ESA+CD44+ and ESA+CD44+CD166+ CSCs (10). In other studies, CSCs were isolated from various types of tumors and analyzed for chemoresistance ex vivo. In human colon cancer, CD133-positive CSCs were highly resistant to 5FU and oxaliplatin (11). When the colon cancer cell line HT29 was treated continuously with 5FU or oxaliplatin, the emergent resistant subpopulations were highly enriched in cells expressing stem cell markers, including CD133 (16- to 30-fold) and CD44 (2-fold) (12). These findings suggest that drug-resistant subpopulations may be enriched in CSC.
The intrinsic resistance of CSCs to chemotherapeutic agents may be explained by high expression of ABC multidrug transporters, anti-apoptotic proteins and by the resistance to DNA damage. Accumulating evidence has indicated that CSC quiescence may also account for a possible mechanism of resistance because the activity of several cytotoxic agents is dependent on cell cycle progression (13–16). Cellular quiescence is a basic mechanism of clinical tumor dormancy, although angiogenic dormancy and escape from immune-system control also play important roles (17).
We previously reported that chronic treatment of the HT29 colon cancer cell line with chemotherapeutic agents resulted in the emergence of drug-resistant HT29 subpopulations overexpressing the chemokine (C-X-C) motif receptor 4 (CXCR4). In addition, overexpression and autocrine activation of CXCR4 played a role in the metastatic spreading to the lungs in immunodeficient mice (18, 19). Here, we report the acquisition of a complex mechanism of chemoresistance to 5FU involving selection for colon cancer stem cells and their quiescence linked to the activation of the c-Yes tyrosine kinase. We show that c-Yes controls the balance between quiescence and cycling of 5FU chemoresistant cells as well as the cytoplasmic/nuclear ratio of the Yes-associated protein (YAP) transcription co-activator. Finally, we discovered a direct relationship between c-Yes/YAP expression levels and overall and disease-free survival of patients with colorectal liver metastases after 5FU-based neoadjuvant therapy.
MATERIALS AND METHODS
Human colorectal carcinoma cell lines and patients tissue samples
HT29 parental cell line, 5FU (10−6M and 10−6M)-resistant HT29 subpopulations (HT29FU) and 5F7 and 5F31 clonal derivatives as well as 5FU-resistant (8×10−6M) RKO cells (RKO-FU) and oxaliplatin-resistant (2×10−6M) RKO cells (RKO-OXA) and HT29OXA cells subpopulations were cultured as previously described (20–22). Liver metastases of colon adenocarcinoma were resected from 49 patients and were processed and stored by the Tumor Cell and Tissue Bank of the Regional Reference Cancer Center of Lille. After hepatic resection, fragments were taken from macroscopic metastases, snap-frozen in liquid nitrogen and stored at −80°C. The whole remaining tissue was fixed in 10% formalin and several other fragments were taken from the fixed metastases and embedded in paraffin. Hematoxilin, eosin, saffron and Astra blue-stained sections were examined by an experienced pathologist to establish the histological diagnosis. Fragments used in this study contained at least 60% malignant cells. Informed consent was obtained from all patients. The curative microscopically-complete R0 liver resections in all patients were performed between February 2002 and May 2009. Neoadjuvant chemotherapy was administered for 28 patients before surgery according to the recommendations of the French Thesaurus of Digestive Cancerology: Folfox in 17 patients, Folfiri in 1 patient, Folfiri-bevacizumab in 10 patients. Information about sites of tumor recurrence or metastasis development after curative hepatectomy and delay of recurrence were collected for each patient.
c-Yes silencing
c-Yes silencing was performed by retroviral infection of 5F31 cells using pRETRO-Super vector as previously described (23). Sh RNAs targeting c-Yes sequences were also previously described (24). Selection of the c-Yes silenced populations was performed with puromycin (1μg/ml).
Statistical analyses
All data were expressed as mean (standard deviation) and categorical variables by percentage (frequency). Overall survival (OS) and disease-free survival (DFS) were calculated by the Kaplan-Meier method, and the differences between groups were compared using the log-rank test. To identify predictive variables of OS or DFS, continuous variables were analyzed by a Cox proportional hazards model and qualitative variables using the log-rank test. Expectation maximization algorithm was used to determine the cut-off values of Yes and Yap. All analyses were performed using SAS software version 9.2 (SAS Institute Inc., Cary, NC 25513). A p-value <0.05 was considered significant.
RESULTS
5FU-resistant HT29 clones exhibited heterogeneity in their chemoresistance
Previous studies have reported the emergence of stable HT29 cell subpopulations resistant to 5FU that were capable to differentiate into goblet-cell like or enterocyte-like phenotypes (20). Cloning of the HT29 subpopulation resistant to 10−6M 5FU (HT29FU) by limiting-dilution gave rise to several HT29 cell clonal subpopulations (HT29 5F clones) (21). The analysis of IC50 values in HT29 5F clones towards newly added 5FU revealed differential intrinsic levels of chemoresistance to this drug. As shown in Fig. 1, the 5F31 clone was highly 5FU-resistant (IC50=19.6×10−6M 5FU) whereas 5F7 clone was much less resistant (IC50=1.9×10−6M) but still more resistant than the parental HT29 cell line (IC50=1.1×10−6M). The highly 5FU-resistant 5F31 cells were able to recover proliferative capacity after drug withdrawal.
Figure 1.
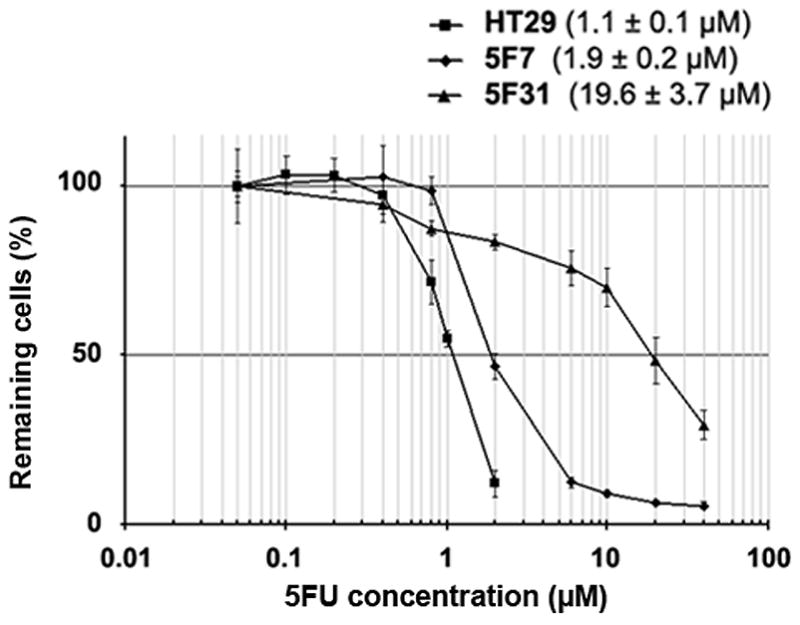
5FU-resistance of 5F7 and 5F31 cells. Survival curves of 5FU-resistant 5F7 and 5F31 clones after subsequent exposure to 5FU concentrations for 5 days. The IC50 was defined as the concentration of 5FU producing a 50% decrease in the number of cells compared with untreated controls. Error bars represent the mean±SD for six replicates.
Drug-resistance is associated with a diverse expression pattern of stem cell markers
5FU-resistant clones highly expressed several colon cancer stem cell surface markers as shown by flow cytometry using a quadruple labelling of CD24, CD44, CD133 and CXCR4 (Supplementary Fig. S1A). In contrast, parental HT29 cell population contained a low percentage of cells expressing stem cell markers, ranging from 4% (CXCR4) to 32% (CD24) (Supplementary Table S1). The two 5FU-resistant clones contained more than 90% of cells expressing at least one marker. The 5F31 cell population mainly contained CD24+/CD44+ cells (55%), CD24+/CD44+/CD133+ cells (14%) and CD24+ cells (14%). The 5F7 cell population was composed of CD24+/CD44+/CD133+/CXCR4+ cells (50%) and of CD24+/CD44+/CXCR4+ cells (31%). The 5FU-resistant (HT29FU) and oxaliplatin-resistant (HT29OXA) subpopulations (22) also contained an important proportion of CD24, CD44, CD133 and CXCR4-positive cells (Supplementary Table S1). Moreover the percentages of CD133− and CXCR4-positive cells varied substantially in 5F7 and 5F31. This heterogeneity was further confirmed using the cancer stem cell marker ALDH1A3, an isoform of ALDH (25) (Supplementary Fig. S1B). ALDH1A3 was strongly downregulated (74-fold) in 5F7 cells and conversely upregulated in 5F31 cells (3-fold), as compared to the parental HT29 cells. Total ALDH activity was also higher in 5F31 vs 5F7 cells. Altogether, our data show that resistant clones were enriched in stem cell-like cancer cells that differ in the expression pattern of stem cell markers.
Drug-resistant HT29 clones differed in their proliferation and metastatic potentials
Stem cells are defined by their combined ability to self-renew and to differentiate. We investigated the self-renewal potential of 5F7, 5F31 and HT29 cells and their 5FU-treated counterparts using anchorage-independent growth (Fig. 2A). 5F31 cells formed well-delimitated regular spheres whereas 5F7 and parental cells produced irregular spheres with a strong tendency to form aggregates. The self-renewal potential of spheres was evaluated by an increase ratio in the number of spheres formed over three consecutive generations (Fig. 2B). The ratio between third and first generation of spheres was significantly enhanced in 5FU-treated 5F7 and 5F31 cells (1.9- and 5.8-fold, respectively) as compared to their untreated counterparts (1.3- and 2.5-fold, respectively). Also, the HT29FU and HT29OXA subpopulations were enriched in the sphere-forming, self-renewing cells when compared with their parental cell line.
Figure 2.
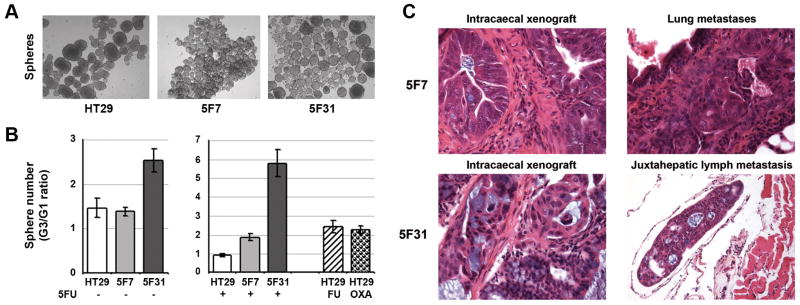
5FU-resistant 5F7 and 5F31 clones differ in their self-renewal and metastatatic potentials. A- Sphere formation in anchorage-independent culture conditions, using parental HT29 cells and the drug-resistant clones. B- Self-renewal capacity in spheres arising from untreated or treated cells. The spheres obtained from cells cultured in the absence of 5FU (left panel) or presence of 5FU (4 days, ½IC50, right panel) were dissociated and subsequently reseeded over three passages. The HT29FU and HT29OXA subpopulations were analyzed in comparison (right panel). Results are the ratio of the number of counted spheres between the third and first generations of spheres. Histograms are the means from at least two independent experiments. HT29 parental population (white), 5F7 (bright grey), 5F31 (dark grey), HT29FU (hatched), HT29OXA (grid). C- Tumorigenic and metastatic potential of 5F7 and 5F31 clones in immunodeficient mice using orthotopic intracaecal xenografts. Intracaecal tumors and metastases were analysed by conventional histology (hematoxylin, eosin, saffron and Astra blue, HESAB), magnification, ×200. Intracaecal xenografts appear as adenocarcinoma with cell rows and mucus-secreting glands. Both 5F7 and 5F31 tumors produce lung metastases but only 5F31 tumors generate extrahepatic lymph metastases.
The 5F7 and 5F31 clones were then assessed for their tumorigenic and metastatic potential using orthotopic xenografts. Both clones produced fairly well-differentiated adenocarcinomas with cell layers and glands containing mucus in their lumen (Fig. 2C). The presence of lung micrometastases was observed in both 5F7- and 5F31-xenografted mice, however, the number of lung metastases was higher in 5F7 xenografts in comparison to 5F31 xenografts (9 vs 2 per mouse). In addition, only 5F31 cells generated metastases in lymph vessels around the liver, demonstrating that these two clones have similar tumorigenic but differential metastatic abilities.
Variations in drug-resistance of HT29 clones are connected to different phases of the cell cycle
Cellular responses to 5FU treatments using ½IC50 and 2×IC50 5FU concentrations for 5F7 cells (1 and 4μM) and 5F31 cells (10 and 40μM) were examined by the flow cytometry cell cycle analysis (Supplementary Table S2). Treatment of HT29 parental cells by high 5FU concentrations induced typical cell accumulation in the S-phase as previously shown (26). Similarly, 5F7 cells accumulated at the S-phase in response to graded high 5FU concentrations, as well as at G2/M-phase. In contrast, subsequent exposure of 5F31 cells to 5FU induced a selective and concentration-dependent accumulation of cells at the G0-phase of the cell cycle, from 2.9% in control cells to 12.3 and 50.4% in 5F31 cells exposed to 10 and 40μM 5FU, respectively. In addition, the percentage of quiescent cells in 5FU-treated 5F31 cell line gradually increased over time, from 8.0% (1h) to 14.7% (4h), 32.5% (72h) and 50.4% (96h). Since non-dividing cells are not targeted by chemotherapeutic drugs targeting proliferating cells, our data suggest that 5F31 cells evade 5FU treatment through entry in a quiescent G0-state. Importantly, a high percentage of quiescence was observed in other drug-resistant colon cancer cell subpopulations, i.e. RKO-OXA (8.5%), -FU (9.8%) vs control RKO cells, (3.1%) and HCT-116-OXA (11.1%), -FU (21.8%) vs control HCT-116 cells (4.7%). As these 5FU- and OXA-resistant cells are enriched in stem cell markers (our data and 12), we concluded that the entry into quiescence is a more general mechanism regulating colon cancer stem survival and chemoresistance.
5FU-resistance of 5F7 and 5F31 HT-29 clones was connected to differential activation of Chk-2 and c-Yes kinases
Control and subsequently 5FU-treated 5F7 and 5F31 resistant cells were analyzed by phospho-kinase array and western blotting (Fig. 3A and 3B). Treatment of 5F7 cells with 5FU primarily induced Chk-2 (checkpoint kinase 2) phosphorylation at the threonine 68 residue. As shown previously, Chk-2 is activated by the ataxia telangiectasia mutated (ATM)-mediated responses to DNA damage (27). Activation of this DNA damage-sensing pathway prevents progression through the cell cycle and recruits the DNA repair machinery. Our result showing accumulation of 5F7 cells in the S- and G2/M-phase upon 5FU treatment are consistent with these observations. In contrast, the level of phosphorylated Chk-2 remained unchanged in 5FU-treated 5F31 cells (Fig. 3A and B). Instead, they had higher levels of phosphorylated c-Yes. This finding was confirmed by western blotting of c-Yes in anti-phosphotyrosine precipitates (Fig. 3C). The c-Yes phosphorylation level was increased by 3.8-fold upon 5FU exposure. c-Yes phosphorylation was further confirmed by ELISA assay (Fig. 3D). Control and 5FU-treated 5F31 cells showed higher c-Yes protein levels than the parental HT29 or 5F7 cells (4.5-fold and 4.9-fold, respectively, p<0.01, Fig. 3B). Also, they expressed 7.7-fold higher levels of c-Yes transcripts while 5F7 cells only 3-fold higher than parental HT29 cells (Fig. 3E).
Figure 3.

5FU-resistant 5F7 and 5F31 clones selectively activate different signaling pathways following subsequent exposure to 5FU. A- Total cell lysates from control and 5FU-treated 5F7 and 5F31 cells (4 days) were analyzed using phospho-kinase arrays. Chk-2 serine/threonine kinase, c-Yes tyrosine kinase (arrows). Other kinases explored in these phospho-antibody arrays are involved in cancer cell proliferation and cell cycle controls, survival, adhesion and transcription (see the phospho-array coordinates in the methods section). F17, F18, G5, G6: negative controls; A1, A2, A17, A18, G1, G2, positive controls. B- Western blots of the total cell lysates using specific antibodies against P-Chk-2 (T68), Chk-2 and c-Yes. Actin was used as loading control. The data are representative of two independent experiments. C- Analysis of c-Yes phosphorylation by immunoprecipitation using the anti-phosphotyrosine antibody followed by western blotting with the anti-c-Yes antibody. D- Analysis of c-Yes phosphorylation using the phospho-c-Yes (Y426) ELISA assay. The error bars represent the mean ± SD for three replicates. E- Analysis of c-Yes mRNA levels in 5F7 and 5F31 cells in comparison to HT29 cells.
c-Yes-silencing in 5F31 cells induced quiescence and restricted YAP nuclear accumulation
To investigate whether c-Yes is involved in the 5FU-induced cellular quiescence of 5F31, we silenced c-Yes in these cells using small hairpin RNAs (shRNAs). Two silenced sh1 and sh2 5F31 populations showing differential levels of c-Yes silencing were obtained, as compared to control transfected cells with the scrambled shRNA sequence (Scr) (Fig. 4A). Consistent with recent data published on c-Yes-silenced HT29 cells (24), c-Yes-silencing in 5F31 cells was associated with increased β-catenin levels (Fig. 4A). Most interestingly, c-Yes silencing levels were directly correlated with the emergence of G0 quiescent 5F31 cells (Fig. 4B), as observed in sh2 (50% silencing, 10% in G0 state) and sh1 cells (86% silencing, 29% cells in G0 state) as compared to Scr cells (4.6% in G0 state). Next, we compared the proliferation capacity of 5FU-treated sh1 cells vs control Scr cells after 5FU re-treatment (40μM, 5 days) and subsequent drug withdrawal. The population doubling time of silenced sh1 cells was 57h while the control Scr cells divided every 36h. These data demonstrate that depletion of c-Yes arrests 5F31 cells in the G0 phase of the cell cycle and suggest that c-Yes is required for the reentry of 5F31 quiescent cells into the proliferative state. As Yes-associated protein (YAP) is a growth-promoting transcriptional coactivator that promotes stem cell self-renewal and progenitors expansion (28), we analyzed the consequences of c-Yes silencing on its fate. As shown in Fig. 4A, total YAP levels remained unchanged upon c-Yes silencing, however the YAP nuclear/cytoplasmic ratio decreased in sh1 cells as compared to control Scr cells (39%–61% vs 48%–52% Fig. 4C), implying that c-Yes controls levels of nuclear YAP.
Figure 4.
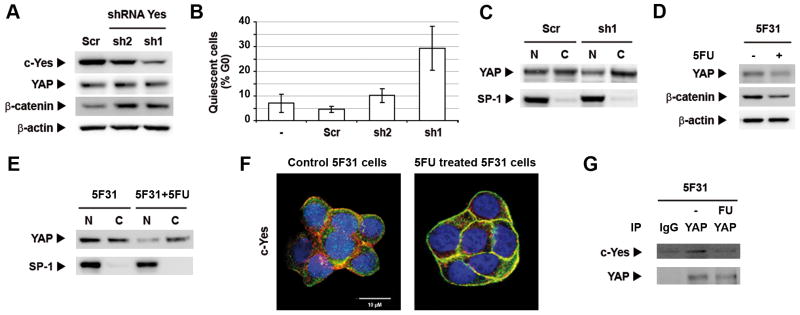
c-Yes-silencing and 5FU inhibits YAP nuclear translocation. A- Western blot analysis of c-Yes, YAP and β-catenin in c-Yes silenced Sh1 and Sh2 cells as compared to control Scr cells. B- Percentage of cells arrested at the quiescent G0 state in c-Yes silenced Sh1 and Sh2 cells. Results are expressed in percentage of quiescent cells in G0 state. C- Immunoblot of YAP and transcription factor Sp1 (nuclear marker) in nuclear and cytoplasmic fractions from c-Yes silenced Sh1 cells and control Scr cells. D- Immunoblots of YAP and β-catenin in control and 5FU-treated (2×IC50, 4 days) 5F31 cells. E- Impact of subsequent 5FU treatement (2×IC50, 4 days) on the nuclear distribution of YAP in 5F31 cells. Western blots are representative of at least two independent experiments. F- Subcellular localization of c-Yes by confocal microscopy analysis in control and 5FU-treated 5F31 cells. G- Coimmunoprecipitation (IP) of c-Yes with the anti-YAP antibody followed by immunoblotting using the c-Yes antibody and the YAP antibody in control and 5FU-treated 5F31 cells. Non-specific c-Yes immunoprecipitation was monitored using a non-relevant IgG.
The nuclear accumulation of YAP was restricted in 5FU-treated quiescent 5F31 cells
Since the majority of 5F31 cells submitted to high 5FU concentration enter quiescence, we examined the effect of 5FU treatment upon the nuclear distribution of YAP. As shown in Fig. 4D and E, the nuclear pool of YAP decreased markedly upon 5FU exposure in 5F31 cells (27% vs 55%) whereas total YAP level was not significantly altered. Our data support a possible link between nuclear YAP depletion and cellular quiescence. As stated above, c-Yes becomes phosphorylated in 5F31 cells re-exposed to 5FU. Confocal microscopy analysis showed that 5FU-exposure induced the recruitment of cytoplasmic c-Yes to the cell membrane (Fig. 4F). This shift in c-Yes subcellular distribution is consistent with its activation since the c-Yes kinase inhibitor SU6656 prevents c-Yes interaction with integral membrane proteins (29, 30). Thus, we hypothesized that 5FU treatment induces c-Yes phosphorylation and dissociates the c-Yes/YAP complex in 5F31 cells and that the released c-Yes is targeted to the plasma membrane. In order to test this hypothesis, YAP coimmunoprecipitation experiments were performed using the anti-YAP antibody. Interestingly, c-Yes was found in YAP immunoprecipitates prepared from control 5F31 cells but not from 5FU-treated 5F31 cells, showing that the c-Yes/YAP complex was lost upon 5FU-exposure (Fig. 4G).
c-Yes and YAP expression levels were increased by chemotherapy in human colon liver metastases and are correlated with colon cancer relapse
To examine the clinical significance of our data, we analyzed the level of c-Yes and YAP transcripts in a cohort of 49 colon cancer patients who underwent surgical resection of liver metastases. Twenty-eight patients received the chemotherapeutic regimens prior surgery. As shown in Fig. 5A, quantification of c-Yes and YAP expressions by qRT-PCR showed enhanced c-Yes (4.2-fold) and YAP (1.7-fold) transcript levels in patients treated by chemotherapy (p=0.035 and 0.026, respectively, Fig. 5A). Consistently, increased c-Yes protein levels were observed in 5FU- and OXA-resistant HT29 and RKO colon cancer cells, respectively. Of note, YAP protein levels were strongly elevated in OXA-resistant HT29 and RKO cells (Fig. S2). This indicates that chemotherapy may preselect for quiescent colon cancer stem cells harboring a deregulation of the c-Yes/YAP axis. Most importantly, c-Yes expression negatively correlated with the overall survival (OS, threshold of 2.25, p=0.0231) and disease-free survival (DFS, threshold of 1.35, p=0.0433) (Fig. 5B). Interestingly, also YAP expression correlated with shorter overall (threshold of 2.62, p=0.025) and disease-free survival (threshold of 2.75, p=0.0088). The neo-adjuvant chemotherapy treatment is administered when liver metastases are initially unresectable or marginally resectable (≥ 5 bilateral nodules). Thus, the treated patients have generally a more severe disease. As expected, in the studied population there was no difference in OS and DFS between the groups of treated and non-treated patients because of the advantages provided by neo-adjuvant chemotherapy. In the non-treated group, there was no correlation between c-Yes and YAP levels and the clinical outcomes (DFS and OS). In the group of treated patients, c-Yes overexpression was negatively correlated with OS, with statistical significance (p=0.022) and with near significance with DFS (p=0.071). Regarding YAP, a trend was observed for both OS and DFS (p=0.092 and p=0.1, respectively). Thus, the subgroup of treated patients with liver metastases expressing high c-Yes levels is associated with poorer outcomes. This clinical data is consistent with our experimental study showing a direct correlation between c-Yes expression and the quiescence of chemoresistant HT29 colon cancer cells treated with 5FU. Altogether, these results show that a poorer clinical outcome segregates with the increased c-Yes and YAP transcript levels and concerns patients who received 5FU-based neoadjuvant chemotherapy.
Figure 5.

Correlations between c-Yes and YAP expression and colon cancer patient survival. A- Comparison of c-Yes and YAP transcript levels in colon liver metastases resected from patients who have received or not neoadjuvant chemotherapy. B- Kaplan-Meier survival analysis of colon cancer patients according to the expression of c-Yes and YAP transcripts.
DISCUSSION
Currently, treatment of synchronous or metachronous colorectal liver metastases requires neoadjuvant chemotherapy before hepatic surgery. Chemotherapeutic regimens consisting of 5FU and folinic acid combined with oxaliplatin (Folfox), irinotecan (Folfiri), or irinotecan and bevacizumab (Folfiri-bevacizumab) are scheduled every 15 days during 3 months (1–3). Drugs are withdrawn one month before surgery. Although temporarily efficient, this treatment rarely cures cancer and disease relapses from the drug-resistant cells. To investigate chemoresistance mechanisms occurring upon sequential chemotherapy regimens, 5FU-resistant clones propagated in drug-free medium were re-exposed to 5FU. We show that 5FU-resistant cells could use different strategies to survive re-challenge with 5FU. On one hand, 5F7 cells which show a weak chemoresistant potential respond to the drug by activating a typical ATM/Chk-2 pathway that prevents cell cycle progression until DNA repair is completed (27). On another hand, 5F31 cells although derived from the same parental chemoresistant subpopulation resist high 5FU concentrations by entering a protective quiescent state, reversible upon drug-withdrawal. The escape into quiescence may recapitulate clinical observations linked to tumor dormancy and cancer relapse following chemotherapy. This is an important observation that should be investigated further to determine therapeutic regimens targeting also quiescent cancer stem cells. Interestingly, both 5FU- and OXA-resistant cells expressed colon cancer stem cell markers, implying that 5FU-based chemotherapy fails to eradicate quiescent cancer stem cells. The variability in the expression pattern of several stem cell markers identified in 5FU-resistant 5F7 and 5F31 cells suggests that the resistant clones emerge from different cancer stem cell subtypes. This notion reflects again clinical observations that a tumor may contain heterogeneous populations of cancer stem cells (31). Almost an exclusive expression of cancer stem cell markers CXCR4 by 5F7 cells and ALDH1A3 by 5F31 cells might be predictive of the metastatic potential (32, 33). This is consistent with our previous finding that CXCR4 overexpression stimulates metastatic spreading of 5F7 subcutaneous xenografts to the lungs (19). Using orthotopic murine xenografts, we found here that 5F31 cells expressing high ALDH1A3 levels disseminated to more metastatic sites than 5F7 cells over-expressing CXCR4, emphasizing a strong link between the tumorigenic and metastatic potential and the cancer stem cell phenotype. We argue that one mechanism responsible for such a link may involve c-Yes/YAP signaling pathways.
c-yes gene is amplified in 5FU resistant cancer cell lines due to its chromosomal localization close to the thymidylate synthase gene, a 5FU target (19, 34, 35). 5F31 cells, characterized by the high chemoresistance potential and the ability to enter quiescence upon 5FU exposure, displayed a particularly elevated level of c-Yes. We have therefore explored the possible implication of c-Yes and YAP in driving 5FU resistance and quiescence of 5F31 cells. YAP stimulates the proliferation of mouse intestinal stem cells and maintains their pluripotency (28, 36, 37). YAP is kept under an inactive state in differentiated cells through the Hippo pathway that phosphorylates YAP at the 127 serine residue and induces its cytoplasmic sequestration and degradation (38). Here, we found that the entry of 5FU-resistant 5F31 cells in quiescence upon a 5FU re-challenge induced the repression of nuclear YAP levels, suggesting that this repression contributes to cancer cell dormancy. In agreement, we observed that the 5FU-induced c-Yes phosphorylation and its translocation to the plasma membrane are associated with a loss of the c-Yes/YAP complex and depletion of nuclear YAP. In addition, we have shown that c-Yes silencing induced the accumulation of 5F31 cells at the G0 phase of the cell cycle as well as depletion of nuclear YAP levels. Consequently, the c-Yes-dependent regulation of YAP activity in 5FU-resistant 5F31 cells can be considered as potential mechanism involved in the control of the quiescence/proliferation balance of these cells. Taken together, our data support the notion that the c-Yes/YAP signaling pathway contributes to 5FU resistance through the combined acquisition of both cellular quiescence and stem cell-like phenotype. In support of this conclusion, recent data indicate that the Yes/YAP-TEAD2 signaling cascade downstream of the leukemia inhibiting factor (LIF) is necessary for the YAP nuclear translocation and self-renewal of mouse embryonic stem cells (39). YAP growth-promoting activity is mediated by the interaction between its N-terminal domain and the TEA domain (TEAD) family of DNA-binding proteins (40). Oncogenic YAP is overexpressed in most colon tumors and promotes the proliferation of colon cancer cell lines (37, 41, 42). Consistently, YAP was recently incriminated in the metastatic potential of breast cancer and melanoma cells through its TEAD-interaction domain (43). In cancer-associated fibroblasts, YAP promotes matrix stiffening, cancer cell invasion and angiogenesis (44). YAP and its paralog TAZ, are two downstream targets of the Hippo pathway regulated by Lats 1/2 kinases. Upstream Hippo signals are initiated by extracellular diffusible signals and receptor/non-receptor tyrosine kinases such as c-Yes and c-Abl (45). Accordingly, several survival effectors are known to act through G-protein receptors and tyrosine phosphorylations, including the src family kinases. Further studies are needed to determine the possible connections of the c-Yes/YAP axis with the core components of the Hippo pathway. In our study, it is also plausible that the oxidative stress response induced by 5FU is implicated in c-Yes activation (46) Finally, the c-Yes/cdc42 pathway was shown to repress the transcription factor NFAT1 by inducing the formation of a cytoplasmic complex with casein kinase 1α (CK1α), and that Lats-induced YAP phosphorylation primes YAP for CK1-induced phosphorylation (47, 48).
We have shown that 5FU-based neoadjuvant chemotherapy of metastatic colon cancer patients increased levels of both c-Yes and YAP transcripts in liver metastases, thus providing one explanation for the higher aggressiveness and metastatic potential of relapsed drug-resistant tumors. Furthermore, we found that c-Yes and YAP transcript levels were correlated with the reduced disease-free and overall survival, supporting the potential implication of the c-Yes/YAP signaling cascade in tumor relapse. Our data demonstrate that 5FU chemotherapy in the HT29 colon cancer cell line preselects two distinct drug-resistant clonal populations each enriched in cells expressing their specific set of stem cell markers and different self-renewing potential, suggesting that in clinic, 5FU chemoresistant cancer cells may require clone-selective and adapted therapeutic strategies to be eradicated. Our data points to the c-Yes/YAP axis as signaling elements involved in the acquisition and maintenance of cellular quiescence linked to 5FU resistance in colon cancer. Taken together with our clinical study, we demonstrated here that the c-Yes/YAP-signaling pathway should be considered as a potential therapeutic target to kill drug-resistant quiescent cancer cells. Alternatively, identification of new compounds targeting cancer stem cells in quiescent state might prove to be beneficial for cancer patients with colon liver metastases and detectable alterations of the c-Yes/YAP signaling axis.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Metastasis, chemoresistance and dormancy are the major limitations in the management of cancer patients. Here, we show that 5FU chemoresistance in human colon cancer cells preselects two distinct drug-resistant clonal populations each enriched in cells expressing specific stem cell markers and differential self-renewing and metastatic potentials. The 5FU-induced quiescence is linked to activation of the c-Yes tyrosine kinase and nuclear depletion of the c-Yes-associated YAP oncogene. Consistently, c-Yes silencing decreased nuclear YAP accumulation and induced cellular quiescence in 5FU-free conditions. We further demonstrate the prognostic relevance of our data in colon cancer liver metastases treated by neoadjuvant chemotherapy regimens based on 5FU. High c-Yes and YAP transcript levels measured in residual liver metastases after adjuvant chemotherapy are observed in patients at risk for colon cancer relapse and shorter survival. Therefore, these two markers identify candidates for adapted treatment targeting the c-Yes/YAP axis in chemoresistant quiescent colon carcinomas and liver metastases.
Acknowledgments
Financial support:
ARC, Ligue contre le Cancer (Comité du Nord), INSERM, SIRIC ONCOLille Grant INCa-DGOS-inserm 6041 and IRCL
We are grateful to Nathalie Jouy (Flow Cytometry Core Facility IFR114/IMPRT), M.H. Gevaert and R. Siminsky (Department of Histology, Faculty of Medicine, University of Lille 2), M. Samyn and V. Dumetz (Laboratory of Immunohistochemistry, Centre de Biologie-Pathologie, CHRU-Lille), and Laurence Wicquart (“Tumorothèque du Centre Regional de Référence en Cancérologie”).
Footnotes
Disclosures:
No potential conflicts of interest were disclosed.
References
- 1.Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041–7. doi: 10.1016/s0140-6736(00)02034-1. [DOI] [PubMed] [Google Scholar]
- 2.Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. N Engl J Med. 2000;343:905–14. doi: 10.1056/NEJM200009283431302. [DOI] [PubMed] [Google Scholar]
- 3.de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938–47. doi: 10.1200/JCO.2000.18.16.2938. [DOI] [PubMed] [Google Scholar]
- 4.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 5.Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23:1803–10. doi: 10.1200/JCO.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 7.Ellis LM. Mechanisms of action of bevacizumab as a component of therapy for metastatic colorectal cancer. Semin Oncol. 2006;33:S1–7. doi: 10.1053/j.seminoncol.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 9.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 10.Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G, 2nd, et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–7. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lacerda L, Pusztai L, Woodward WA. The role of tumor initiating cells in drug resistance of breast cancer: Implications for future therapeutic approaches. Drug Resist Updat. 2010;13:99–108. doi: 10.1016/j.drup.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Rasheed ZA, Kowalski J, Smith BD, Matsui W. Concise review: Emerging concepts in clinical targeting of cancer stem cells. Stem Cells. 2011;29:883–7. doi: 10.1002/stem.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–9. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 16.Wells A, Griffith L, Wells JZ, Taylor DP. The Dormancy Dilemma: Quiescence versus Balanced Proliferation. Cancer Res. 2013;73:3811–3816. doi: 10.1158/0008-5472.CAN-13-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sosa MS, Avivar-Valderas A, Bragado P, Wen HC, Aguirre-Ghiso JA. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clin Cancer Res. 2011;17:5850–7. doi: 10.1158/1078-0432.CCR-10-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gouyer V, Fontaine D, Dumont P, de Wever O, Fontayne-Devaud H, Leteurtre E, et al. Autocrine induction of invasion and metastasis by tumor-associated trypsin inhibitor in human colon cancer cells. Oncogene. 2008;27:4024–33. doi: 10.1038/onc.2008.42. [DOI] [PubMed] [Google Scholar]
- 19.Dessein AF, Stechly L, Jonckheere N, Dumont P, Monte D, Leteurtre E, et al. Autocrine induction of invasive and metastatic phenotypes by the MIF-CXCR4 axis in drug-resistant human colon cancer cells. Cancer Res. 2010;70:4644–54. doi: 10.1158/0008-5472.CAN-09-3828. [DOI] [PubMed] [Google Scholar]
- 20.Lesuffleur T, Kornowski A, Luccioni C, Muleris M, Barbat A, Beaumatin J, et al. Adaptation to 5-fluorouracil of the heterogeneous human colon tumor cell line HT-29 results in the selection of cells committed to differentiation. Int J Cancer. 1991;49:721–30. doi: 10.1002/ijc.2910490516. [DOI] [PubMed] [Google Scholar]
- 21.Leteurtre E, Gouyer V, Rousseau K, Moreau O, Barbat A, Swallow D, et al. Differential mucin expression in colon carcinoma HT-29 clones with variable resistance to 5-fluorouracil and methotrexate. Biol Cell. 2004;96:145–51. doi: 10.1016/j.biolcel.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12:4147–53. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- 23.Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2:243–7. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 24.Sancier F, Dumont A, Sirvent A, Paquay de Plater L, Edmonds T, David G, et al. Specific oncogenic activity of the Src-family tyrosine kinase c-Yes in colon carcinoma cells. PLoS One. 2011;6:e17237. doi: 10.1371/journal.pone.0017237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. 2011;10:1378–84. doi: 10.4161/cc.10.9.15486. [DOI] [PubMed] [Google Scholar]
- 26.Pizzorno G, Sun Z, Handschumacher RE. Aberrant cell cycle inhibition pattern in human colon carcinoma cell lines after exposure to 5-fluorouracil. Biochem Pharmacol. 1995;49:553–7. doi: 10.1016/0006-2952(94)00444-q. [DOI] [PubMed] [Google Scholar]
- 27.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–55. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–60. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 29.Chen YH, Lu Q, Goodenough DA, Jeansonne B. Nonreceptor tyrosine kinase c-Yes interacts with occludin during tight junction formation in canine kidney epithelial cells. Mol Biol Cell. 2002;13:1227–37. doi: 10.1091/mbc.01-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao X, Mruk DD, Lee WM, Cheng CY. c-Yes regulates cell adhesion at the blood-testis barrier and the apical ectoplasmic specialization in the seminiferous epithelium of rat testes. Int J Biochem Cell Biol. 2011;43:651–65. doi: 10.1016/j.biocel.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baccelli I, Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol. 2012;198:281–93. doi: 10.1083/jcb.201202014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Letters. 2008;267:226–244. doi: 10.1016/j.canlet.2008.04.050. [DOI] [PubMed] [Google Scholar]
- 33.Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29:32–45. doi: 10.1002/stem.563. [DOI] [PubMed] [Google Scholar]
- 34.Silverman GA, Kuo WL, Taillon-Miller P, Gray JW. Chromosomal reassignment: YACs containing both YES1 and thymidylate synthase map to the short arm of chromosome 18. Genomics. 1993;15:442–5. doi: 10.1006/geno.1993.1086. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Marsh S, Cassidy J, McLeod HL. Pharmacogenomic dissection of resistance to thymidylate synthase inhibitors. Cancer Res. 2001;61:5505–10. [PubMed] [Google Scholar]
- 36.Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24:1106–18. doi: 10.1101/gad.1903310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci U S A. 2011;108:E1312–20. doi: 10.1073/pnas.1110428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2008;24:862–74. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamm C, Bower N, Anneren C. Regulation of mouse embryonic stem cell self-renewal by a Yes-YAP-TEAD2 signaling pathway downstream of LIF. J Cell Sci. 2010;124:1136–44. doi: 10.1242/jcs.075796. [DOI] [PubMed] [Google Scholar]
- 40.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001;15:1229–41. doi: 10.1101/gad.888601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103:12405–10. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, et al. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39:1582–9. doi: 10.1016/j.humpath.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109:E2441–50. doi: 10.1073/pnas.1212021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–46. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piccolo S, Cordenonsi M, Dupont S. Molecular Pathways: YAP and TAZ Take Center Stage in Organ Growth and Tumorigenesis. Clin Cancer Res. 2013;19:4925–30. doi: 10.1158/1078-0432.CCR-12-3172. [DOI] [PubMed] [Google Scholar]
- 46.Reinehr R, Becker S, Wettstein M, Haussinger D. Involvement of the Src family kinase yes in bile salt-induced apoptosis. Gastroenterology. 2004;127:1540–57. doi: 10.1053/j.gastro.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 47.Dejmek J, Safholm A, Kamp Nielsen C, Andersson T, Leandersson K. Wnt-5a/Ca2+-induced NFAT activity is counteracted by Wnt-5a/Yes-Cdc42-casein kinase 1alpha signaling in human mammary epithelial cells. Mol Cell Biol. 2006;26:6024–36. doi: 10.1128/MCB.02354-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2012;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.