

Abstract
Aim:
Atazanavir (ATV) is a HIV-1 protease inhibitor for the treatment of AIDS patients, which is recently reported to provoke excessive prolongation of the QT interval and torsades de pointes (TdP). In order to elucidate its arrhythmogenic mechanisms, we investigated the effects of ATV on the hERG K+ channels expressed in HEK293 cells.
Methods:
hERG K+ currents were detected using whole-cell patch clamp recording in HEK293 cells transfected with EGFP-hERG plasmids. The expression of hERG protein was measured with Western blotting. Two mutants (Y652A and F656C) were constructed in the S6 domain within the inner helices of hERG K+ channels that were responsible for binding of various drugs. The trafficking of hERG protein was studied with confocal microscopy.
Results:
Application of ATV (0.01–30 μmol/L) concentration-dependently decreased hERG K+ currents with an IC50 of 5.7±1.8 μmol/L. ATV (10 μmol/L) did not affect the activation and steady-state inactivation of hERG K+ currents. Compared with the wild type hERG K+ channels, both Y652A and F656C mutants significantly reduced the inhibition of ATV on hERG K+ currents. Overnight treatment with ATV (0.1–30 μmol/L) concentration-dependently reduced the amount of fully glycosylated 155 kDa hERG protein without significantly affecting the core-glycosylated 135 kDa hERG protein in the cells expressing the WT-hERG protein. Confocal microscopy studies confirmed that overnight treatment with ATV obstructed the trafficking of hERG protein to the cell membrane.
Conclusion:
ATV directly blocks hERG K+ channels via binding to the residues Y652 and F656 in the S6 domain, and indirectly obstructs the transport of the hERG protein to the cell membrane.
Keywords: ether-a-go-go potassium channels, HIV protease inhibitors, long QT syndrome, atazanavir, torsades de pointes, patch-clamp techniques, protein trafficking, HEK293 cells
Introduction
Atazanavir (ATV) is one of the newer protease inhibitors (PIs) approved for the treatment of human immunodeficiency virus (HIV)-1 infected patients1. The benefits encouraging the use of ATV include a favorable effect on lipid profiles, convenient once-daily dosing, low capsule burden, and a relatively favorable resistance profile compared with other PIs1. Despite the known benefits of ATV, there are concerns about its potential adverse effects of inducing cardiac arrhythmia2,3. Similar to other PIs4,5,6,7, ATV has previously been reported to be specifically associated with acquired long-QT syndrome, drug-induced QT prolongation, and torsades de pointes (TdP) in HIV-infected patients8,9,10. However, these results contradict other recently published studies11,12.
Most drugs that affect the QT interval inhibit the rapid delayed rectifier (IKr) potassium (K+) channels in cardiac myocytes encoded by the human ether-a-go-go-related gene (hERG)13. Recently, it has been reported that four PIs (lopinavir, nelfinavir, ritonavir, and saquinavir) can inhibit the hERG K+ current in vitro, and monitoring of the QT interval in patients treated with PIs was recommended14.
However, information regarding the effects and mechanisms of ATV on recombinant hERG channels is lacking. Therefore, the goal of this study was to investigate the effects of ATV on cloned hERG K+ channels expressed heterologously in human embryonic kidney (HEK293) cells. This study is geared at ultimately providing a theoretical basis for guidance in the individually tailored administration of this drug in a clinical setting to reduce the incidence of adverse cardiac reactions.
Materials and methods
Cell culture and molecular biology
The pcgi-enhanced green fluorescent protein (EGFP)-hERG plasmid was provided by Dr Zhao ZHANG (College of Life Sciences, Nanjing Normal University, China) for electrophysiological recordings and Western blot analysis, and the pcDNA3-hERG plasmid was provided by Dr Gail A ROBERTSON (University of Wisconsin, Madison, WI, USA) for confocal microscopy.
HEK293 cells were cultured and transiently transfected as previously described15. Briefly, HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, in a humidified 5% CO2 incubator at 37 °C. The transfection was performed using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA).
Y652A and F656C mutations were constructed using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the manufacturer's instructions and were verified by sequencing and subsequently subcloned into the full-length pcgi-EGFP vector.
Electrophysiological recordings
EGFP-positive cells were selected using an epifluorescence system, and the whole-cell patch-clamp technique was used to measure the hERG current at room temperature, as previously described15. The cells were superfused with bath solution containing 140 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L glucose, and 10 mmol/L HEPES (pH 7.4 with NaOH). For the measurement of the current through the F656C mutant16, which showed a comparatively low level of expression, a bath solution containing 94 mmol/L KCl was used, and the NaCl concentration was correspondingly reduced. A pipette with a tip resistance of 2–5 mega-ohm (MΩ) filled with an internal solution containing 130 mmol/L KCl, 1 mmol/L MgCl2, 5 EGTA, 5 mmol/L Mg-ATP, and 10 mmol/L HEPES (pH 7.2 with KOH) was used.
A giga-ohm (GΩ) seal resistance was achieved in all experiments. After cell membrane rupture, at least 3–5 min was allowed to elapse to ensure cell dialysis before recording the current. For potency tests, a baseline control of 3–5 min of stable recording was obtained prior to the application of ATV. In the presence of ATV, a steady-state response was achieved before applying each subsequent concentration.
The current and kinetic characteristics of the hERG channel were recorded as previously described15,17, using an EPC-10 (HEKA electronic, Lambrecht-Pfalz, Germany) patch-clamp amplifier with Pulse 8.67 software (HEKA electronic, Lambrecht-Pfalz, Germany). Voltage commands and data acquisition were controlled by Pulsefit (v10.0) software (HEKA electronic, Lambrecht-Pfalz, Germany).
Western blot analysis
Western blot analysis of hERG protein was performed as described previously15,17. Briefly, ATV was diluted in serum- and antibiotic-free DMEM and added to transfected HEK293 cells for 36–48 h. The cells were then scraped into ice-cold phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mmol/L Tris-HCl at pH 7.5, 150 mmol/L NaCl, 5 mmol/L EDTA, 1% Nonidet P-40 buffer, and 10% glycerol) containing a protease inhibitor cocktail (Roche, Mannheim, Germany). For each sample, 10 μg of protein was electrophoresed on 9% Tris-acetate gels and transferred onto polyvinylidene difluoride membranes (PVDF). The membranes were blocked with 5% non-fat dry milk before incubation with rabbit anti-hERG antibody (1:400, APC-109; Alomone Labs, Jerusalem, Israel) overnight. To normalize the hERG expression levels to the reference protein, the membranes were also probed with a rabbit anti-β-actin antibody (1:800, Santa Cruz Biotechnology, CA, USA). The membranes were then washed and incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:10 000, Santa Cruz Biotechnology, CA, USA) for 1 h. After being washed, the membranes were developed using enhanced chemiluminescence (ECL). Blots were analyzed and quantified using Quantity One software (Bio-Rad, Hercules, CA, USA).
Confocal microscopy
A pcDNA3-hERG plasmid was used and transfected with HEK293 cells for the immunostaining test to avoid EGFP fluorescent interference. Cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min after 24–28 h incubation in normal or ATV-containing medium. Fixed cells were permeabilized with 0.1% Triton X-100 for 10 min and preblocked with 2% bovine serum albumin (BSA) at room temperature. The cells were then incubated with rabbit polyclonal anti-hERG antibodies (1:50, Alomone Labs, Jerusalem, Israel) or chicken polyclonal anti-calreticulin antibodies (1:50, Abcam, Cambridge, MA, USA) overnight at 4 °C, followed by incubation with Alexa Fluor 488-conjugated donkey anti-rabbit IgG secondary antibodies (1:100, Invitrogen, Carlsbad, CA, USA) and Alexa Fluor 594-conjugated goat anti-chicken IgG secondary antibodies (1:100, Jackson ImmunoResearch Laboratories Inc, West Grove, PA, USA) at 37 °C for 2 h. Stained cells were viewed under a confocal laser scanning microscope (FV10, Olympus, Tokyo, Japan).
Chemicals
ATV (Sigma, Saint Louis, MO, USA) was dissolved in methanol to generate a stock solution of 10 mmol/L, which was stored at room temperature. The final concentration of methanol used in each experiment was less than 0.1%, which would have no significant effect on the hERG current recording18.
Statistical analysis
All data are presented as the mean±SEM. The data were analyzed, and illustrations were drawn using the statistical package for the social sciences (SPSS) v12.0 (SPSS Inc, Chicago, IL, USA) and Origin 6.0 software (OriginLab Corp, Northampton, MA, USA). The significant differences between paired and unpaired samples were evaluated by Student's t-test or one-way repeated measures analysis of variance (ANOVA). P<0.05 was considered statistically significant.
Results
ATV inhibits hERG K+ current in a concentration-dependent manner
The hERG currents were elicited by a 3-s step to +20 mV from a holding potential of -80 mV, followed by repolarization to -40 mV for 2 s to produce tail currents. As shown in Figure 1A, ATV (10 μmol/L) reduced the steady-state current amplitude and tail current peak amplitude, and the effect was partially reversible after a 15 min wash-off. Figure 1B shows the time-course of the blockade of the hERG current for the same cell with a maximum block occurring after 5 min of ATV exposure. Figure 1C shows the concentration-dependent inhibition of the hERG current at six different concentrations of ATV, including 0.01, 0.1, 1, 3, 10, and 30 μmol/L, and their inhibition ratios for the tail current were 11.9%±2.6%, 22.6%±5.3%, 34.2%±8.5%, 41.3%±9.8%, 55.8%±4.7%, and 73.2%±7.0%, respectively. The IC50 for the inhibition of hERG tail currents by ATV was 5.7±1.8 μmol/L, with a Hill coefficient of 0.3±0.1.
Figure 1.

Concentration-dependent inhibition of human ether-a-go-go-related gene (hERG) potassium (K+) currents by atazanavir (ATV). (A) Representative currents in the absence, presence (5 min), and following wash-off (15 min) of 10 μmol/L ATV, elicited by the voltage protocol shown in the inset. (B) Representative currents from the same cell in the absence of ATV and during ATV exposure. (C) Concentration-response curve of ATV-induced inhibition of hERG K+ currents in HEK293 cells. The concentration-response relationship shows that the IC50 was 5.7±1.8 μmol/L, and the Hill coefficient was 0.3±0.1. Each drug concentration was evaluated on at least five cells.
Effects of ATV on the activation and inactivation of hERG channel gating
Representative hERG currents recorded under control conditions and 5 min after the application of ATV in the same cell are shown in Figure 2A. As shown in Figure 2B and 2C, ATV (10 μmol/L) strongly reduced the steady-state current amplitude and tail current peak amplitude.
Figure 2.

Effects of atazanavir (ATV) on human ether-a-go-go-related gene (hERG) potassium (K+) currents expressed in HEK293 cells. (A) Original traces of hERG K+ currents elicited by the pulse protocol shown in the inset under control conditions (left) and 5 min after incubation with 10 μmol/L ATV (right). (B and C) I–V relationships for (B) currents measured at the end of depolarizing steps and (C) tail currents in control cells and in cells exposed to 10 μmol/L ATV (bP<0.05, cP<0.01 vs control, n=6, mean±SD). (D) Peak tail currents were normalized to their respective maximum current amplitude (control and drugs) to illustrate changes in half-maximal activation voltages. (E) Representative current traces showing steady-state inactivation using the pulse protocol (upper panel).
We evaluated the voltage dependence of activation by plotting the normalized tail current as a function of voltage (Figure 2D). The curve was fitted to a Boltzmann equation:
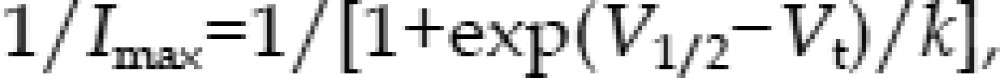 |
where V1/2 is the voltage at which the current is activated to half the maximum, and k is the slope factor15. The V1/2 was 15.5±0.4 mV in the control and 17.8±0.8 mV in the ATV-treated cells (n=6, P>0.05 compared with the control). The corresponding k values were 13.6±0.6 and 15.0±0.7 for the control and ATV-treated cells, respectively (P>0.05 compared with the control). Furthermore, the voltage dependence of the activation relations in control and ATV overlapped closely with each another.
To analyze the steady-state inactivation, test potentials between -130 and +20 mV in 10-mV increments per 20 ms were applied after a depolarizing pulse of up to +20 mV for 4 s. This step was followed by a test pulse back to +20 mV for 500 ms before a final return of the voltage to the -80 mV holding potential (Figure 2E, inset). Figure 2E shows the steady-state inactivation curves of the hERG current in the absence and presence of 10 μmol/L ATV. This curve was fitted with a Boltzmann equation to yield the inactivation V1/2 and k values. In the control and ATV-treated cells, the values of V1/2 were -61.1±0.9 mV and -56.3±1.1 mV (n=6, P>0.05 compared with the control), with corresponding k values of 24.1±0.8 mV and 23.7±1.0 mV (P>0.05 compared with the control), respectively. Although the half-inactivation voltage was slightly shifted in the positive direction, the shift was not statistically significant.
Attenuation of ATV blockade by Y652A and F656C mutations
It has been reported that two aromatic residues, Y652 and F656, which are located in the S6 domain within the inner helices of the hERG K+ channel, are important components of the binding site for various drugs19. Therefore, the effects of the Y652A and F656C mutants on the inhibitory action of ATV were examined. Drug concentrations of 3, 10, and 30 μmol/L were used to attain a profound blockade of the wild type (WT)-hERG. ATV blocked the Y652A-hERG channel by 6.3%±5.4%, 20.4%±9.7%, and 30.5%±9.1% at 3, 10, and 30 μmol/L, respectively (n=5, P<0.05 compared with WT-hERG). Figure 3A and 3C show that Y652A reduced the inhibitory effect of ATV on the current flow through the hERG channel.
Figure 3.

Effects of atazanavir (ATV) on Y652A and F656C human ether-a-go-go-related gene (hERG) potassium (K+) channels. (A) Representative WT-hERG and Y652A-hERG currents in the absence and presence of 30 μmol/L ATV. (B) Representative WT-hERG and F656C-hERG K+ currents in the absence and presence of 30 μmol/L ATV. (C) Mean fractional block produced by ATV for WT-hERG and Y652A-hERG. (D) Mean fractional block produced by ATV for WT-hERG and Y652A-hERG. bP<0.05 vs wild type. n=5 cells per group; WT, wild type.
The F656C mutant was further investigated by measuring the inward tail currents elicited at -120 mV in the presence of high level of extracellular K+(94 mmol/L). At concentrations of 3, 10, and 30 μmol/L, ATV blocked current flow through the WT-hERG channel by 16.5%±4.9%, 28.9%±8.0%, and 40.7%±10.1%, respectively, and through the F656C-hERG channel by 10.5%±4.5%, 19.5%±8.3%, and 21.9%±8.2%, respectively (n=5, P<0.05 compared with WT-hERG, Figure 3B and 3D). These results indicated that Y652A- and F656C-hERG channels exhibit a reduced blockade compared with the WT-hERG, suggesting that ATV binds to the pore cavity on the hERG channel gate.
Disruption of hERG protein trafficking
Previous studies reported that some drugs such as fluconazole15, cardiac glycosides20, and pentamidine21 are able to reduce hERG surface expression by interrupting protein trafficking to the cell membrane. Therefore, we studied the effect of ATV on hERG channel protein trafficking. Cells expressing the WT-hERG channel protein were incubated with ATV (0.1, 1, 10, and 30 μmol/L) for 36–48 h, and the lysates were subjected to Western blot analysis. Figure 4A shows representative cell lysates probed for hERG. The WT channel protein bands appeared at 135 kDa (immature, core-glycosylated protein in the endoplasmic reticulum, ER) and 155 kDa (mature, complexly glycosylated channel protein in the cell membrane). Incubation with ATV produced a concentration-dependent decrease in the amount of mature, fully glycosylated hERG protein. At the doses of ATV tested, the amount of mature hERG protein was significantly reduced to 88.9%±3.9% (0.1 μmol/L), 72.6%±6.0% (1 μmol/L), 39.8%±4.3% (10 μmol/L), and 23.6%2.3% (30 μmol/L) compared with the untreated conditions (n=5, P<0.05, ANOVA, Figure 4B). Immature, core-glycosylated hERG protein levels were not significantly affected by ATV treatment. The protein ratio of 155/135 kDa was 90.7%±4.4%, 71.6%±5.2%, 39.7%±4.4%, and 25.1%±1.9% at 0.1, 1, 10, and 30 μmol/L ATV, respectively. The results suggested that the mature hERG proteins (155 kDa) in the membrane gradually decreased with increasing concentrations of ATV.
Figure 4.

Atazanavir (ATV) reduced maturation and surface expression of human ether-a-go-go-related gene (hERG) channels. (A) Western blot showing effects of overnight treatment with increasing concentrations of ATV (0.1, 1.0, 10, and 30 μmol/L) on hERG protein transiently expressed in HEK293 cells. (B) Image densities of fully glycosylated 155 kDa and core-glycosylated 135 kDa hERG proteins were quantified as a function of ATV concentrations using a PhosphorImager. All image densities were normalized to the β-actin protein form measured under control conditions. bP<0.05, cP<0.01 vs control. n=5. (C) Representative current traces under control conditions and after long-term application of ATV at 1 and 30 μmol/L (see Figure 2A for pulse protocol). (D) Peak tail current densities at each concentration normalized to the value under control conditions. bP<0.05, cP<0.01 compared with control. n=8.
Next, we recorded hERG currents from the transfected cells incubated with ATV for 24–48 h to examine the functional consequences of inhibiting hERG protein trafficking in HEK293 cells. Figure 4C shows representative traces of currents elicited by the same voltage protocol described in Figure 2A. The tail current densities normalized to the value under control conditions were also decreased in a concentration-dependent manner (n=8, P<0.05, ANOVA, Figure 4D). Chronic disruption of hERG expression on the cell surface led to reduced hERG current amplitudes.
To further confirm these results, double immunofluorescence staining with hERG and calreticulin (an ER marker) proteins was used to evaluate the subcellular localization of hERG proteins in HEK293 cells. As shown in Figure 5A, hERG proteins were mainly expressed on the cell membrane and within the cytoplasm in cells. Figure 5B demonstrated that WT-hERG channels were mainly expressed in the cytoplasm when the cells were incubated with ATV. These results further suggest that ATV inhibited the trafficking of the mature hERG channels, reducing the number of such channels expressed on the membrane.
Figure 5.

Double immunofluorescence staining of human ether-a-go-go-related gene (hERG) protein and the endoplasmic reticulum (ER) marker protein calreticulin in HEK293 cells incubated in normal (A) or 30 μmol/L ATV-containing medium (B) for 36–48 h.
Finally, we tested whether Y652A and F656C mutations altered the ATV-induced disruption of hERG protein trafficking. Under control conditions, the two mutations showed both the 135 and 155 kDa protein bands, similar to the WT protein. Cells expressing the Y652A and F656C mutant channel proteins were also treated with ATV for 36–48 h, and the lysates were compared with the control (untreated) cells using Western blots. We observed that the normalized image density of the 155 kDa band was significantly decreased in a concentration-dependent manner for both Y652A (Figure 6A and 6C) and F656C (Figure 6B and 6D) mutant channel-expressing cells. However, there was no significant difference compared with the WT-hERG-expressing cells. Therefore, common drug-binding sites (Y652 and F656) are involved in the direct blockade of the hERG channel but not in the hERG with the ATV-induced trafficking defect.
Figure 6.

Western blot analysis of cells expressing human ether-a-go-go-related gene (hERG) Y652A and F656C mutant channels. Cells expressing hERG Y652A and F656C mutant channels were analyzed by Western blot under control conditions and after 36–48 h incubation with ATV (1.0, 10, and 30 μmol/L). bP<0.05 compared with control. n=4 per group.
Discussion
This report is the first to describe the effects of the protease inhibitor drug ATV on cloned hERG K+ channels expressed in HEK293 cells. ATV is known to produce QT prolongation and TdP in clinical use8,9,10. In this study, we demonstrated that ATV directly inhibits both the cellular hERG K+ channel current and hERG K+ channel protein trafficking at similar concentrations.
In this study, ATV directly inhibited hERG currents in HEK cells at an IC50 of 5.7±1.8 μmol/L. The IC50 value was similar to other PIs such as lopinavir (8.6 μmol/L), nelfinavir (11.5 μmol/L), ritonavir (8.2 μmol/L), and saquinavir (15.3 μmol/L) reported previously in studies using HEK293 cells14. In this study, ATV showed no significant effects on the voltage dependence of activation or inactivation.
The structural requirements for the drug-binding site in hERG K+ channels have been previously studied in detail19,22. The Y652 and F656 residues are located in the S6 domain and within the inner helices of the hERG channel. Here, these residues have been implicated as critical molecular determinants of channel blockade by drugs that gain access to the inner cavity of the channel upon gating of the channel23. However, not all hERG blocking drugs interact with these residues. For example, the blockade of hERG by propafenone24 is substantially reduced by F656 but not by Y652. Our study showed that Y652A and F656C mutations markedly weakened the blockade of the hERG current by ATV (3-fold increase in the IC50 for Y652A and F656C) compared with the WT-hERG. It is possible that in addition to Y652 and F656, other amino-acid residues may be molecular determinants of drug binding.
Several drugs, including pentamidine21, fluoxetine25, celastrol26, escitalopram27, citalopram27, and desipramine28, are known to inhibit hERG indirectly. This inhibition, which occurs by the disruption of hERG channel protein trafficking to the plasma membrane, reduces the cell surface hERG channel density. Drugs may cause acute channel blockade or inhibition of forward trafficking or both29. To date, however, no information has been published on the disruption of protein trafficking by PIs, including ATV. In this study, Western blot analysis revealed that the reduction in current density caused by ATV was associated with a decrease in the fully glycosylated cell surface form (155 kDa) of the protein. The propensity to disrupt protein trafficking exhibited by ATV was similar to its blockade of the direct current. The precise mechanism by which ATV reduces the cell surface expression of the channel protein remains to be determined.
According to our study, the Y652 and F656 mutations did not significantly alter the ATV-induced disruption of channel protein trafficking. This result indicated that the binding domain in the pore-S6 region mediates direct ATV-induced current blockade but does not affect protein trafficking. The trafficking function was virtually intact in the two mutants, suggesting that different interaction sites on the protein or different proteins in the secretory pathway may regulate processing. Importantly, our study reveals that the PIs can inhibit channels in in vitro experiments. Therefore, we propose that this protease inhibitor class has the potential to induce a risk of QT interval prolongation.
PIs are among the most frequently prescribed drugs for the treatment of HIV-infected patients. However, PIs have been associated with a rare but potentially fatal and specific prolonged QT interval, a cardiotoxicity that leads to TdP30,31. Over the past four years, the Food and Drug Administration (FDA) has issued warnings that ritonavir boosted with lopinavir or saquinavir may cause prolongation of the QTc and PR intervals.
The cardiotoxicity of PIs as a class of drugs remains controversial partly because of the lack of supporting data. In 2007, Ly et al presented the first reported case of TdP in association with ATV use8. In 2013, Santimaleeworagun et al reported that ATV induced first-degree atrioventricular block and ventricular tachycardia10. In addition, there has also been a report of QT interval prolongation or the risk of ventricular tachycardia (VT) with combined treatment using ATV and other drugs such as methadone9. Recently, it was reported that ATV did not prolong the QTc interval and did not increase QTc dispersion11. Charbit et al12 studied the relationship between PIs and QT interval prolongation in patients with AIDS and showed that PIs inhibit the hERG current in vitro but are not determinant or independent factors of QT prolongation in patients with AIDS. QT prolongation in AIDS patients is largely dependent on the presence of other risk factors. These factors include older age, female sex, bradycardia, electrolyte abnormalities, congestive heart failure, a baseline prolonged QT interval, and possible drug-drug interactions12,32. The results of the study by Soliman et al33 also supported this finding.
In HIV-infected patients, the mean maximal total plasma concentration of ATV following the once-daily oral administration of 400 mg is approximately 1.3–7.6 μmol/L. ATV is 86% bound to human serum proteins, namely alpha-1-acid glycoprotein and albumin, to a similar extent, leading to an unbound fraction of <20%1. ATV has a peak free concentration of 0.18–1.06 μmol/L, which is less than the IC50 value of 5.7±1.8 μmol/L obtained in our results. This result could explain the lack of a significant influence of PI on QTc prolongation, which is consistent with the recent data showing that ATV has no effect on ventricular repolarization with repeated administration11.
Considering the foregoing, clinicians should bear in mind that the use of PIs should be closely monitored, particularly in patients with a high risk for QTc prolongation. In addition, periodic electrocardiogram monitoring is advisable in patients treated with ATV. Such monitoring is particularly important if the activity of ATV is enhanced in the presence of other drugs that can potentially induce QT interval prolongation.
In conclusion, our findings indicate that ATV inhibits hERG K+ channels in HEK293 cells via directly inhibiting the flow of current through the hERG channel and indirectly disrupting hERG protein trafficking. In addition, the Y652 and F656 residues are critical molecular determinants of the blockade of hERG channels by ATV.
Author contribution
Sheng-na HAN and Xiao-yan SUN performed research and analyzed data; Zhao ZHANG contributed new reagents; Sheng-na HAN and Li-rong ZHANG designed research and wrote the paper.
Acknowledgments
We thank Dr Gail A ROBERTSON for providing us the plasmids of pcDNA3-hERG. This work was supported by the National Natural Science Foundation of China (No 81200138) and the Key Technologies R&D Program of Henan Province (No 92102310225).
References
- Rivas P, Morello J, Garrido C, Rodriguez-Novoa S, Soriano V. Role of atazanavir in the treatment of HIV infection. Ther Clin Risk Manag. 2009;5:99–116. [PMC free article] [PubMed] [Google Scholar]
- Reyataz product information [homepage on the Internet]. Bristol-Myers Squibb Company; Princeton, NJ 08543, USA: [cited 8 March 2014]. Available from: : http://packageinserts.bms.com/pi/pi_reyataz.pdf .
- Bristol-Myers Squibb Company. BMS-232632, atazanavir briefing document, May 2003. [homepage on the Internet]. [cited 8 March 2014]. Available from: : http://www.fda.gov/ohrms/dockets/ac/03/briefing/3950B1_01_BristolMyersSquibb-Atazanavir.pdf
- Girgis I, Gualberti J, Langan L, Malek S, Mustaciuolo V, Costantino T, et al. A prospective study of the effect of IV pentamidine therapy on ventricular arrhythmias and QTc prolongation in HIV-infected patients. Chest. 1997;112:646–53. doi: 10.1378/chest.112.3.646. [DOI] [PubMed] [Google Scholar]
- Castillo R, Pedalino RP, El-Sherif N, Turitto G. Efavirenz-associated QT prolongation and Torsade de Pointes arrhythmia. Ann Pharmacother. 2002;36:1006–8. doi: 10.1345/aph.1A454. [DOI] [PubMed] [Google Scholar]
- Saidi AS, Moodie DS, Garson A, Jr, Lipshultz SE, Kaplan S, Lai WW, et al. Electrocardiography and 24-h electrocardiographic ambulatory recording (Holter monitor) studies in children infected with human immunodeficiency virus type 1. The Pediatric Pulmonary and Cardiac Complications of Vertically Transmitted HIV-1 Infection Study Group. Pediatr Cardiol. 2000;21:189–96. doi: 10.1007/s002460010038. [DOI] [PubMed] [Google Scholar]
- Vallejo Camazon N, Rodriguez Pardo D, Sanchez Hidalgo A, Tornos Mas MP, Ribera E, Soler Soler J. Ventricular tachycardia and long QT associated with clarithromycin administration in a patient with HIV infection. Rev Esp Cardiol. 2002;55:878–81. doi: 10.1016/s0300-8932(02)76720-1. [DOI] [PubMed] [Google Scholar]
- Ly T, Ruiz ME. Prolonged QT interval and torsades de pointes associated with atazanavir therapy. Clin Infect Dis. 2007;44:e67–8. doi: 10.1086/511875. [DOI] [PubMed] [Google Scholar]
- Gallagher DP, Kieran J, Sheehan G, Lambert J, Mahon N, Mallon PW. Ritonavir-boosted atazanavir, methadone, and ventricular tachycardia: 2 case reports. Clin Infect Dis. 2008;47:e36–8. doi: 10.1086/589869. [DOI] [PubMed] [Google Scholar]
- Santimaleeworagun W, Pattharachayakul S, Chusri S, Chayagul P. Atazanavir induced first degree atrioventricular block and ventricular tachycardia: a case report. J Med Assoc Thai. 2013;96:501–3. [PubMed] [Google Scholar]
- Busti AJ, Tsikouris JP, Peeters MJ, Das SR, Canham RM, Abdullah SM, et al. A prospective evaluation of the effect of atazanavir on the QTc interval and QTc dispersion in HIV-positive patients. HIV Med. 2006;7:317–22. doi: 10.1111/j.1468-1293.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- Charbit B, Rosier A, Bollens D, Boccara F, Boelle PY, Koubaa A, et al. Relationship between HIV protease inhibitors and QTc interval duration in HIV-infected patients: a cross-sectional study. Br J Clin Pharmacol. 2009;67:76–82. doi: 10.1111/j.1365-2125.2008.03332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Anson BD, Weaver JG, Ackerman MJ, Akinsete O, Henry K, January CT, et al. Blockade of HERG channels by HIV protease inhibitors. Lancet. 2005;365:682–6. doi: 10.1016/S0140-6736(05)17950-1. [DOI] [PubMed] [Google Scholar]
- Han S, Zhang Y, Chen Q, Duan Y, Zheng T, Hu X, et al. Fluconazole inhibits hERG K(+) channel by direct block and disruption of protein trafficking. Eur J Pharmacol. 2011;650:138–44. doi: 10.1016/j.ejphar.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Park SJ, Kim KS, Kim EJ. Blockade of HERG K+ channel by an antihistamine drug brompheniramine requires the channel binding within the S6 residue Y652 and F656. J Appl Toxicol. 2008;28:104–11. doi: 10.1002/jat.1252. [DOI] [PubMed] [Google Scholar]
- Guo J, Han SN, Liu JX, Zhang XM, Hu ZS, Shi J, et al. The action of a novel fluoroquinolone antibiotic agent antofloxacin hydrochloride on human-ether-a-go-go-related gene potassium channel. Basic Clin Pharmacol Toxicol. 2010;107:643–9. doi: 10.1111/j.1742-7843.2010.00550.x. [DOI] [PubMed] [Google Scholar]
- Hu HN, Zhou PZ, Chen F, Li M, Nan FJ, Gao ZB. Discovery of a retigabine derivative that inhibits KCNQ2 potassium channels. Acta Pharmacol Sin. 2013;34:1359–66. doi: 10.1038/aps.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA. 2000;97:12329–33. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wible BA, Wan X, Ficker E. Cardiac glycosides as novel inhibitors of human ether-a-go-go-related gene channel trafficking. J Pharmacol Exp Ther. 2007;320:525–34. doi: 10.1124/jpet.106.113043. [DOI] [PubMed] [Google Scholar]
- Kuryshev YA, Ficker E, Wang L, Hawryluk P, Dennis AT, Wible BA, et al. Pentamidine-induced long QT syndrome and block of hERG trafficking. J Pharmacol Exp Ther. 2005;312:316–23. doi: 10.1124/jpet.104.073692. [DOI] [PubMed] [Google Scholar]
- Perry M, de Groot MJ, Helliwell R, Leishman D, Tristani-Firouzi M, Sanguinetti MC, et al. Structural determinants of HERG channel block by clofilium and ibutilide. Mol Pharmacol. 2004;66:240–9. doi: 10.1124/mol.104.000117. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Menchaca A, Ferrer-Villada T, Lara J, Fernandez D, Navarro-Polanco RA, Sanchez-Chapula JA. Block of HERG channels by berberine: mechanisms of voltage- and state-dependence probed with site-directed mutant channels. J Cardiovasc Pharmacol. 2006;47:21–9. doi: 10.1097/01.fjc.0000191564.52242.00. [DOI] [PubMed] [Google Scholar]
- Witchel HJ, Dempsey CE, Sessions RB, Perry M, Milnes JT, Hancox JC, et al. The low-potency, voltage-dependent HERG blocker propafenone-molecular determinants and drug trapping. Mol Pharmacol. 2004;66:1201–12. doi: 10.1124/mol.104.001743. [DOI] [PubMed] [Google Scholar]
- Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, et al. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol. 2006;149:481–9. doi: 10.1038/sj.bjp.0706892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Liu X, Xiong Q, Shikano S, Li M. Chronic inhibition of cardiac Kir2.1 and HERG potassium channels by celastrol with dual effects on both ion conductivity and protein trafficking. J Biol Chem. 2006;281:5877–84. doi: 10.1074/jbc.M600072200. [DOI] [PubMed] [Google Scholar]
- Chae YJ, Jeon JH, Lee HJ, Kim IB, Choi JS, Sung KW, et al. Escitalopram block of hERG potassium channels. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:23–32. doi: 10.1007/s00210-013-0911-y. [DOI] [PubMed] [Google Scholar]
- Staudacher I, Wang L, Wan X, Obers S, Wenzel W, Tristram F, et al. hERG K+ channel-associated cardiac effects of the antidepressant drug desipramine. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:119–39. doi: 10.1007/s00210-010-0583-9. [DOI] [PubMed] [Google Scholar]
- Zhang KP, Yang BF, Li BX. Translational toxicology and rescue strategies of the hERG channel dysfunction: biochemical and molecular mechanistic aspects. Acta Pharmacol Sin. 2014;35:1473–84. doi: 10.1038/aps.2014.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg SD, Moorman AC, Williamson JM, Tong TC, Ward DJ, Wood KC, et al. Protease inhibitors and cardiovascular outcomes in patients with HIV-1. Lancet. 2002;360:1747–8. doi: 10.1016/S0140-6736(02)11672-2. [DOI] [PubMed] [Google Scholar]
- Friis-Moller N, Sabin CA, Weber R, d'Arminio Monforte A, El-Sadr WM, Reiss P, et al. Combination antiretroviral therapy and the risk of myocardial infarction. N Engl J Med. 2003;349:1993–2003. doi: 10.1056/NEJMoa030218. [DOI] [PubMed] [Google Scholar]
- Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–22. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- Soliman EZ, Lundgren JD, Roediger MP, Duprez DA, Temesgen Z, Bickel M, et al. Boosted protease inhibitors and the electrocardiographic measures of QT and PR durations. AIDS. 2011;25:367–77. doi: 10.1097/QAD.0b013e328341dcc0. [DOI] [PMC free article] [PubMed] [Google Scholar]