

Abstract
This work details the design and testing of affinity membrane adsorbers for lectin purifications that incorporate glucose-containing glycopolymers. It is the selective interaction between the sugar residues of the glycopolymer and the complementary carbohydrate-binding domain of the lectin that provides the basis for the isolation and purification of lectins from complex biological media. The design approach used in these studies was to graft glycopolymer ‘tentacles’ from macroporous regenerated cellulose membranes by atom transfer radical polymerization. As shown in earlier studies, this design approach can be used to prepare high-productivity membrane adsorbers. The model lectin, concanavalin A (conA), was used to evaluate membrane performance in bind-and-elute purification, using a low molecular weight sugar for elution. The membrane capacity for binding conA was measured at equilibrium and under dynamic conditions using flow rates of 0.1 and 1.0 mL/min. The first Damkohler number was estimated to relate the adsorption rate to the convective mass transport rate through the membrane bed. It was used to assess whether adsorption kinetics or mass transport contributed the primary limitation to conA binding. Analyses indicate that this system is not limited by the accessibility of the binding sites, but by the inherent rate of adsorption of conA onto the glycopolymer.
1. Introduction
Lectins are proteins that associate with sugars through specific, affinity type interactions. In the past three decades, the importance of these plant-derived proteins has evolved from being the focus of nutritional studies to becoming the tool for investigating cell surface sugars.1 Because of their carbohydrate-specific affinity, lectins are used to assess the role of cell surface sugars in cell growth and differentiation. Additionally, lectins can be used to distinguish between the same protein with differing number of sugar residues, i.e., different degrees of sialylation. When tethered to stationary chromatography media, lectins have been shown to separate antibodies with differing anti-inflammatory activity.2
As lectins find more uses in biological applications, the demand for affordable, high-purity lectins will continue to increase. Carbohydrate affinity chromatography is a high selectivity approach for the purification of lectins, and was introduced for the purification of Concanavalin A (conA) by Agrawal and Goldstein in 1967.3 ConA has since become the basis for many applications, including immobilized lectin affinity chromatography, which incorporates conA onto the stationary phase for the purification of glycoproteins and glycopeptides.4
Many research studies have employed conA for lectin affinity chromatography. Of special interest to the authors, Clemmitt and Chase5 investigated the roles of operating variables and established optimal parameters for designing expanded bed columns for high capacity and high efficiency capture of specific glycosylated cells. Reichelt et al.6 describe efforts to improve the throughput of lectin affinity chromatographic media, including the development of monolithic spin-type columns that employ conA for enriching and purifying glycan mixtures. Babac et al.7 attached conA to the stationary phase of a monolithic cryogel and demonstrated the capture of large proteins with specific sugar moieties from complex feeds, such as IgG and other blood proteins from blood plasma.
The traditional isolation and purification of conA was developed in the late 1960s and consists of a series of initial separation steps including centrifugation and dialysis, after which the conA-containing solution is loaded onto a column of Sephadex G-50, which contains dextran residues that bind conA. An eluent solution containing a competing sugar displaces the lectin, yielding a solution with higher purity and concentration.3
While processes implementing dextran gels (Sephadex) for the selective adsorption of conA achieve a reasonable yield from the crude jack bean meal and high recovery from the chromatography elution step, there are features that make it undesirable. The process has low throughput due to batch operation or because of the low flow rates used for bind-and-elute operations required by these traditional chromatography media. Advances needed in lectin purification include methods with potential for faster processing speeds.
In addition to slow processing speeds using dextran gels, there are issues associated with microbial degradation of the dextran that could be avoided with a different substrate material. Khan et al.8 provide one example of current research to develop and test new materials for conA purification in which calcium alginate-cellulose beads containing transition metal Ni(II) as a substrate for purifying the lectin. In this immobilized metal ion chromatography (IMAC) process, the Ni(II) is believed to associate with the hystidine and tryptophan amino acid residues of con A.8 Despite the widespread lab-scale use of IMAC, there are issues that have limited its use at commercial scale. Two significant disadvantages of IMAC are metal ion transfer and metal ion leakage into the purified protein solution. Metal ion transfer leads to protein loss and low protein yields9, and can disrupt protein bioactivity. Since commonly used metal ions such as Ni(II) and Co(II) are known carcinogens10, protein products may be hazardous for use. Even though additional columns could be added to a manufacturing process to capture metal ion contaminants that leak from the IMAC column, there are other factors that make industrial scale IMAC processes difficult to implement.11 As a result, IMAC separations have low predictability, frustrating optimizing and scale-up.
Thus remains the need for new materials and methods to more effectively recover and purify lectins from crude media. While focus in this paper is given to conA, the need is general, as the cost associated with the isolation and purification of other natural lectins is prohibitively high. In fact, to overcome the costly purification of natural lectins, researchers are looking to create synthetic lectins. For example, Mahalingam et al.12 have studied the anti-HIV activity of synthetic lectins that bind to the surface sugars on the virus, inhibiting the entry of HIV into cells.
New methods and materials for lectin purifications include magnetic separation, aqueous two-phase extraction, affinity precipitation, monoliths, and membranes.13 In 2001, Guo and Ruckenstein14 introduced cellulose membranes as a matrix for purifying conA by immobilizing maltose as the affinity ligand. The binding capacity for conA ranged from 0.48 to 1.05 mg/mL membrane. Khan et al.8 used calcium alginate-cellulose beads as a low-cost matrix for purification of conA from jack bean extract that resulted in higher protein yields compared to the traditional Sephadex process. However, both of these materials were used in a batch process. Percin et al.15 studied the effect of flow rate, temperature, ionic strength, and conA concentration for a mannose affinity ligand attached to a poly(hydroxyethyl methacrylate) cryogel in a bind-and-elute operation and found the highest dynamic binding capacities at the lowest flow rates. However, it is not clear if the lower capacities at high flow rate were due to mass-transfer constraints of conA diffusion through the cryogel or short residence times relative to the timescale for adsorption to occur. One aspect of the work described in this manuscript is to relate the adsorption rate to the convective mass transport rate through the membrane bed and use this relationship to assess whether adsorption kinetics or mass transport contribute the primary limitation to conA binding.
Recent developments in conA affinity membranes include glycosylated microporous polypropylene membranes16, SPG (Shirasu-porous-glass) membranes containing brush-like glycopolymers17, and glycosylated polypropylene non-woven meshes.18 Ulbricht and coworkers19 have shown that polymeric glycoligands selectively bind specific lectins, mimicking the gluco-receptors found on cell surfaces that recognize and bind bacteria. The literature also reports the use of model substrates to elucidate optimal polymerization conditions, and kinetic studies relating polymerization time to the degree of polymerization.20 The affinity binding of a homotetrameric conA molecule to its complementary polysaccharide results from multivalent hydrogen bonds and hydrophobic interactions. Because the number of carbohydrates needed to create a binding site for this lectin molecule is greater than one, the adsorbing material should contain a large number of carbohydrate groups. The location and structure of these carbohydrates influence the capacity of the material to capture lectins, as demonstrated in the glycoside cluster effect observed for the comparison of comb-like versus linear glycopolymer layers grafted from poly(ethylene terephthalate) track-etched membrane surfaces to bind peanut agglutinin.20,21 Thus, methods to prepare membrane adsorbers should yield binding-site architectures with a high density of glycoligand groups.
Surface-initiated atom transfer radical polymerization (ATRP) has been used extensively in the formulation of membrane adsorbers for protein isolation and purification.22–35 ATRP is suitable for incorporating glycol-ligand affinity groups onto membranes for the same reasons it has been a successful method for constructing membrane adsorbers for other chromatographic processes, which have been discussed in detail previously.22,36 Namely, surface-initiated ATRP methods provide the ability to optimize and control the grafting density and chain length of low-polydispersity polymers without significantly altering the substrate structure, and without initiating solution-phase polymerization that can lead to pore blocking. ATRP, among other methods, has been researched as a means of incorporating glycoligands into polymers for interaction with specific lectins. Ting et al.37 provide a review that summarizes synthesis methods of glycopolymers for lectin recognition.
This work contributes a new approach to creating membrane adsorbers for carbohydrate chromatography. The heart of the approach is a graft polymerization step that provides a high number of binding sites per basis of adsorbing material. The objectives of this study were (1) to produce affinity membrane adsorbers with a high-binding capacity of conA and (2) to perform a comparison of kinetic rate of conA adsorption versus convective mass transport of conA to the binding sites on the membrane surface. Adsorptive membranes were prepared under different reaction conditions to incorporate d-glucosamine-HCl into PGMA that was grafted by surface-initiated ATRP from regenerated cellulose membranes. The reaction condition with the highest binding capacity was selected as the focus of the second objective.
2. Experimental section
2.1 Materials
Regenerated cellulose membranes (RC60) with reported nominal pore size of 1.0 μm, thickness of 70 μm and diameter of 47 mm were purchased from Whatman, Inc. (Dassel, Germany).
The following chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, MO) with purities reported in weight percent: α-Bromoisobutyryl bromide, 98% (BIB); concanavalin A from Canavalia ensiformis (Jack bean), type IV (conA); glycidyl methacrylate, 97%, contains 100 ppm monomethyl ether hydroquinone as inhibitor (GMA); inhibitor removers, for removing hydroquinone and monomethyl ether hydroquinone; copper(I) chloride, ≥99.995% (Cu(I)Cl); D-(+)-glucosamine hydrochloride, ≥99%, (dGluc-HCl); dichloromethane, ≥99.5%, contains 50 ppm amylene (DCM); dimethyl sulfoxide, ≥99.9% (DMSO); 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, ≥99.5% (HEPES); methyl α-D-mannopyranoside for microbiology, ≥99.0% (Me-Manp); methyl α-D-glucopyranoside for microbiology, ≥99.0% (Me-Glcp); manganese (II) chloride solution, 1.00 M, tetrahydrofuran anhydrous, ≥ 99.9%; inhibitor-free (THF); triethylamine, ≥99.5% (TEA). 1,1,4,7,7-pentamethyldiethylenetriamine, 98+% (PMDETA) was purchased from Acros Organics (Geel, Belgium). The following chemicals were purchased from Fisher Scientific (Fairlawn, NJ): calcium chloride diydrate, Lab Grade; tetrahydrofuran, certified, contains about 0.025% butylated hydroxytoluene as a preservative (THF). Sodium hydroxide, 97.0% (NaOH) was purchased from Alfa Aesar (Ward Hill, MA). The following were purchased from VWR (Radnor, PA): cellulose acetate syringe filter, 0.45 μm, sodium chloride (NaCl).
Buffers used in this study include binding buffer, B: 10 mM HEPES, 0.1 mM Mn2+, 0.1 mM Ca2+, 0.15 M NaCl adjusted with 5 M NaOH to pH 7.0 and elution buffer, E: 200 mM methyl α-D-mannopyranoside (Me-Manp), 200 mM Methyl α-D-glucopyranoside (Me-Glcp)) in binding buffer, pH 7.0. The lectin solution used was 1 mg/mL conA in B, which was filtered with a 0.45 μm cellulose acetate syringe filter.
2.2 Membrane surface modification
Up to ten membranes were modified at one time; however, reaction amounts are reported on the basis of one 47 mm membrane.
2.2.1 Membrane activation
Membranes were activated by reacting with BIB (18 μM, 0.11 mL) in THF (50 mL) for 2 h under constant stirring at 35 °C in a glovebox (MBRAUN UNIlab) with a nitrogen environment (O2 < 10 ppm, H2O < 1 ppm), as reported earlier.31 Membranes were further modified by surface-initiated atom transfer radical polymerization (ATRP) of glycidyl methacrylate (GMA) (3.6 M, 17.06 g) in THF (16.35 mL) with a catalyst complex of Cu(I)Cl (9 mM, 29.7 mg) and PMDETA (9 mM, 0.063 mL) for 21 h. Details about this procedure were reported earlier by Wang et al.34
2.2.2 Incorporation of lectin-specific glycoligand
Activated membranes were soaked in 12 mL THF twice, followed by 12 mL of DMSO twice, then introduced to a solution of d-glucosamine-HCl (0.080 M, 0.863 g) and TEA (0.120 M, 0.838 mL) in DMSO (49 mL) and heated to 70 °C, as represented in Scheme 1. The reaction solution was stirred at 150 rpm for 8.5 h and then cooled to room temperature. Modified membranes were soaked in DMSO (12 mL, twice) followed by THF (12 mL, twice) and stored in THF. They were dried with nitrogen gas prior to characterization or performance testing.
Scheme 1.

Reaction of D-Gluc to ATRP-grafted PGMA.
2.3 Surface characterization by ATR-FTIR
Attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR) measurements were taken using a Thermo Scientific Nicolet 550 Magna-IR Spectrometer with a diamond crystal equipped with Omnic ESP version 6.1a software. An ATR correction was applied to each spectrum and the baseline was corrected manually. A background spectrum was collected prior to each measurement of a dried membrane sample, taken with 16 scans and a resolution of 4.0 cm−1.
2.4 Membrane performance testing
2.4.1 Static binding capacity measurements
Each membrane was placed in binding buffer B before being introduced to 5 mL of a 1 mg/mL conA solution in buffer B. The volume of solution absorbed in the membrane pores during this equilibration step was accounted for in the mass balance used to calculate conA binding capacity. Membranes were incubated for 21 h, which was sufficient according to other static measurements reported in literature14,15 on a shaker table at 80 rpm at 22 ºC. Initial and final lectin concentrations were measured using a calibration curve relating the conA concentration to absorbance at 280 nm with a Carey UV-vis spectrophotometer. Static binding capacity was calculated as the mass of conA adsorbed per membrane volume (mg/mL) by performing a mass balance.
2.4.2 Dynamic binding capacity measurements
Membranes were cut to the appropriate module size before preparation and soaked in binding buffer B prior to use. Eight membranes of the same degree of modification were loaded into a Pall Mustang Coin® module (ID = 18 mm), sandwiched between two Whatman 5 membranes. The module was connected to an AKTA Purifier (GE Healthcare). The system applied an equilibration step of at least 10 column volumes (CV) until the absorbance baseline (280 nm) was stable. Following column equilibration, the lectin solution was injected (10 mL, 1.0 mg/mL in binding buffer B, pre-filtered through a 0.45 μm syringe filter). Following lectin loading, the system applied a wash step with buffer B (10 mL) to wash unbound lectin, followed by an elution step with buffer E (10 mL). The flow rate during the protein loading step was varied, while the flow rates for column equilibration, washing and elution were 1.0 mL/min. Fractions were collected throughout the load, wash, and elution step for analysis. Protein binding capacities were obtained for various loading flow rates. Data were recorded and processed by Unicorn 5.11 software (GE Healthcare, Bio-sciences). All buffers and protein solutions were used at room temperature. Chromatograms were analyzed by calibrating area under the A280 curves (mAU × mL) to concentration (mg/mL) to estimate protein adsorption.
3. Results and Discussion
To develop a working formulation for reacting the dGluc-HCl with the epoxide side groups of the PGMA chains grown by ATRP from the surface of regenerated cellulose membranes, the glycoligand was tested in multiple solvents to find the solvent condition with the best dissolution. Challenges arose with initial reaction attempts, as heat seemed to be necessary to facilitate ring-opening and incorporate the glycoligand. The dGluc appeared to undergo the Maillard reaction. This reaction occurs between carbonyl groups of the sugar and amines in the presence of heat and is responsible for the browning of foods. Martins et al.38 discuss more information on the Maillard reaction including the effects of temperature, pH, and initial concentration. To identify solutions to the challenges that arose with reaction conditions at elevated temperature, reactions using different concentrations, temperatures, and time-scales were performed and the resulting membranes were tested using static binding of conA. The results of this substantial body of preliminary formulation and characterization work are discussed in Supplementary Materials. Table S1 lists the concentrations of dGluc-HCl in different solvents, and Table S2 summarizes the reaction conditions and observations of the formulation work. Figures S1 and S2 present ATR-FTIR spectra of membrane samples prepared using different formulations. Figure S3 reports the corresponding static binding capacities. The formulation that yielded the best results was used to prepare membranes for the work described in this manuscript. An alternative approach was considered for low temperature coupling using an epoxide ring-opening catalyst (Zn(BF4)2·xH2O), but dGluc had low solubility in the solvents needed for this reaction.
3.1 Surface Characterization
Figure 1 presents the FTIR spectra used to evaluate the difference in membrane surface chemistry after modification with glycopolymer chains. After surface-initiated ATRP was performed to graft PGMA chains from the membrane (spectrum b) a peak change was observed near 1730 cm−1 that is assigned to the stretching of carbonyl groups in PGMA and was anticipated based on previous work.34 Thus, PGMA has been grafted from the membrane surface. Additionally, there is a reduction in the intensity of the strong broad peak centered at 3400 cm−1, (attributed to –OH stretching) and of the peaks in the 1000 – 1200 cm−1 region (C-O stretching and C-H wagging). Because the range of penetration depth of ATR-FTIR is 0.5 to 2.0 μm, and the PGMA dry layer thickness is on the order of 70 nm34, the cellulose bond vibrations are expected to be reduced only slightly. After introduction of the glycoligand along the PGMA backbone according to Scheme 1 (spectrum c), these same regions show a stronger absorbance. The bonds that contribute to these vibrations are common to cellulose and d-Gluc. The peak near 1730 cm−1 is more pronounced after the glycoligand addition, perhaps due to C=O stretching that would be present if some of the attached glycoligand exists in the open-chain form. Peaks unique to the incorporated glycoligand would be N-H stretching and wagging. However, because very little of this compound is present relative to the cellulose substrate, in the case of N-H stretching, the peak in these region is small (700–730 cm−1). In the case of N-H wagging, the peak is undetectable because it appears in the same region as the broad hydroxyl group stretching.
Figure 1.
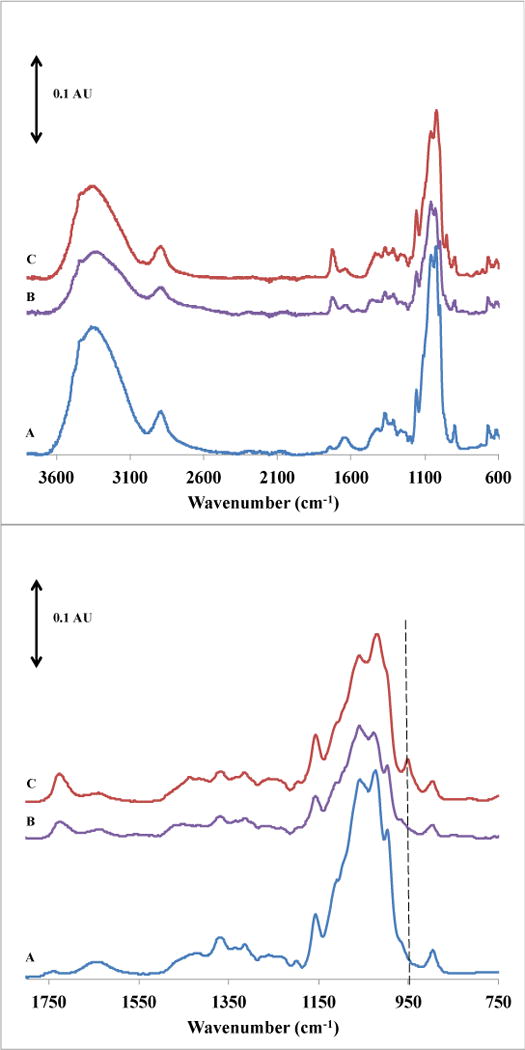
ATR-FTIR spectrum of (A) unmodified RC60 membrane, (B) grafted PGMA layer, (C) glycoligand incorporated via reaction condition E.
3.2 Membrane Performance
3.2.1 Static Binding
The result of the mass balance indicated the unmodified RC60 membrane adsorbed no conA after 21 hours. The membrane modified with PGMA and d-Gluc adsorbed 6.0 ± 0.6 mg conA/mL of membrane, reported as an average and standard deviation of three measurements. This binding capacity is comparable with other materials described in the literature, as summarized in Table 1. For the polypropylene macroporous membrane (PPMM) substrate, a dramatic increase in adsorbed conA occurred when the grafting density was above 90 μg/cm2. For the sake of comparison, the reported values were converted to common units using the approximate surface area. Approximate values were found to be 10 and 45 mg/mL. The hypothesis is that below a certain grafting density, conA capacity is low because of the sparsely distributed sugar molecules. However, above a certain chain density, the proximity of sugar molecules is sufficient to create the necessary multivalent binding sites for conA. Although grafting density was not varied in the current study, it is possible that the current modification technique has not incorporated enough glucosamine to meet the threshold grafting density to observe this glycoside cluster effect. The binding capacities of the PHEMA cryogel and Sephadex G 50 resin are comparable to our membrane platform; however, the membrane-based substrate has the key advantage of easier scale-up to large volumes, which has been demonstrated in the case of ion-exchange membrane chromatography.39 It also has been shown that, under high loading conditions, membranes can offer higher resolution over some resin chromatography columns.40 This is an aspect of membrane chromatography worth exploring for this system.
Table 1.
Summary of conA adsorbing materials.
| Substrate | Chemistry | Grafting Density (μg/cm2) | Adsorbed Species | Adsorbed | Flow Rate | |
|---|---|---|---|---|---|---|
| 1 | PPMM | UV polymerization alda-d-allyl glucoside (AG) | 90 150 |
Con A | 100 μg/cm2 450 μg/cm2 |
Static binding |
| 2 | PHEMA cryogel | covalent attachment of D-mannose | 191.9 [mg/g] | Con A | 11.2 mg/g | 1 mL/min, 2h recirculation |
| 3 | Sephadex G 50 | Cross-linked dextran | – | Con A | 17 mg/g | – |
| 4 | RC | ATRP of GMA / glucosamine addition | – | Con A | 6 mg/cm3 (13 mg/g) | Static binding |
The reported values are from the following studies: (1) Yang et al., 2006, (2) Percin et al., 2012, (3) Akhtar et al., (4) this study.
3.2.2 Effect of flow rate
Dynamic capacities were measured using a bind-and-elute process. In these measurements, one stack of membrane adsorbers was used for all flow rates tested (more than four runs). There was no measureable irreversible binding. The elution peak areas were the same for duplicate runs at the same flow rate. Figures 2 and 3 show representative chromatograms for loading flow rates of 1.0 mL/min and 0.1 mL/min, respectively. For a flow rate of 1.0 mL/min, no lectin was observed when the elution buffer was applied. However, at a flow rate of 0.1 mL/min, a small elution peak was observed. This peak corresponded to a capacity of 0.6 mg/mL. This result is counter to what was expected; that is, dynamic binding capacities often are independent of flow rate using membrane adsorbers. Multiple examples of this phenomenon are presented in a review of affinity membrane materials, in which membranes with porosities suitable for high flow rates provide enough surface area for binding, and access to those binding sites is not restricted by diffusion.41 Additionally, for the same substrate used in this study (Whatman RC60 1 μm regenerated cellulose membrane), this effect has been observed by our group.24,31 In fact, in Bhut and Husson24, flow rates under 1 mL/min presented flow distribution challenges, resulting in lower binding capacity. It appears, therefore, that lectin binding kinetics may be the rate limiting step in this case.
Figure 2.
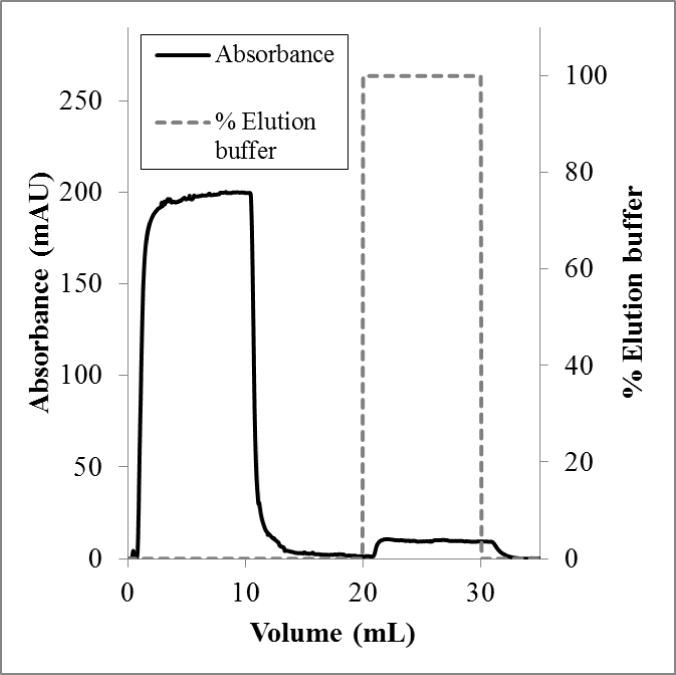
Flow rate 1.0 mL/min. Conductivity remained at 15 ± 1 mS/cm and the pH at 7.4 ± 0.1.
Figure 3.
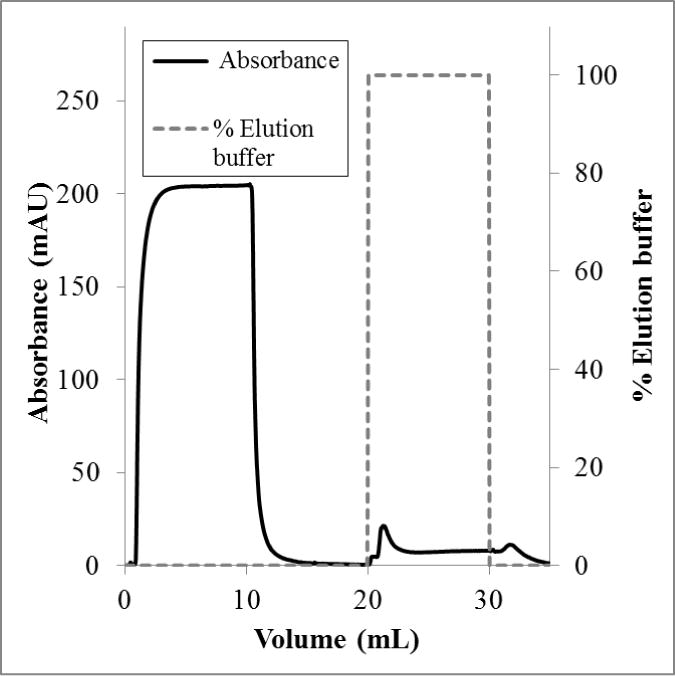
Flow rate 0.1 mL/min. Conductivity remained at 15 ± 1 mS/cm and the pH at 7.3 ± 0.1.
3.3 Reaction rate analysis
It is recognized that the efficiency of many affinity-type chromatography processes may be limited by binding kinetics. To assess whether the rate of adsorption or the convective mass transport of the lectin through the membrane bed was limiting the degree of lectin adsorption, the first Damkohler number was estimated using the following analysis.
The first Damkohler number (Da) is a dimensionless quantity representing the ratio of the reaction rate to the rate of convective mass transport through a reactor and is defined by Equation 1:
| (1) |
where k is the reaction rate constant, CP is the feed protein (i.e., lectin) concentration and τ is the residence time, or space time. The system is modeled as a plug flow reactor with a second order reaction P + L → PL, where P is the protein and L is the binding site comprising one or more sugar groups. For this case, Da > 10 indicates that the rate of adsorption is sufficiently fast that 90% of the available protein will adsorb to the column (X = Da/(1+Da) for conversion, X). As Da decreases, less protein will adsorb due to a slow rate of adsorption and insufficient space time for the adsorption to occur, with mass transport of the protein to the binding site having relatively little impact on degree of binding.
Clegg et al.42 measured the association and dissociation rate constants for various saccharides to conA using a temperature-jump relaxation technique and found the association rate constant to be 5 × 104 M−1s−1 and 4 × 104 M−1s−1 for methyl Me-Glcp and Me-Manp, respectively, at 25 °C and pH 7.2. A value of 4 × 104 M−1s−1 was used to approximate the rate constant for the adsorption of conA to the glucose site on the grafted glycopolymer, recognizing that true value may be much lower for the heterogeneous binding reaction.
The feed concentration in our study CP, was 1 mg/mL and the molar mass of tetrameric conA is estimated at 108 kDa. In the chromatography column used (CV = 0.143 mL), a volumetric flow rate of 1.0 mL/min equates to a residence time of 8.6 s. Together these parameters result in a Da of 3.2. Under these conditions, 69% of the available protein would be expected to adsorb to a binding site as it passes through the membrane bed. Several factors may contribute to the lack of binding seen experimentally. Firstly, if the glucose functional groups are not in a favorable position to form a multivalent binding site, adsorption will be restricted. Secondly, membranes have been shown to exhibit uneven flow distribution especially at low flow rates24 and in columns with short bed height.43 A non-uniform pore size distribution is another factor that can result in less than theoretical performance. In this scenario, proteins follow preferential flow paths through larger pores, reducing the local protein concentration at binding sites in smaller pores. As a result, larger pore size distribution results in broader breakthrough curves, a well-known concept that has been modeled and discussed in detail44 and continues to be the focus of membrane materials research. Finally, and perhaps most importantly, this model uses a literature value for the rate constant. Boi et al.45 studied the adsorption kinetics of galactose-specific Momordica charantia lectin on different membrane supports and found that the rate of adsorption differed. Furthermore, the literature value is for reaction in solution. The true value for this heterogeneous reaction may be significantly lower. Performing a detailed study that measured the kinetic constant for this system would enhance the accuracy of the model.
For the theoretical system to adsorb a high percentage (>90%) of the available protein, such that the rate of adsorption is not hindering the binding of conA to the glycopolymer, the bulk concentration of conA or the residence time would need to be increased by at least 3-fold. For a flow rate of 0.1 mL/min, the residence time is 86 seconds, and the calculated Da is 32, which corresponds to theoretically 97% protein adsorption. In this case, 0.6 mg/mL lectin binding capacity was measured. The ability to observe faster, flow-rate independent separations, which is one advantage of utilizing membranes, is restricted by the inherently slow nature of the binding reaction occurring along the polymer tentacles on the membrane surface. Nonetheless, regenerated cellulose membranes have other advantages such as easy scale-up and lower pressure drop relative to resins and gels that make it a desirable platform even at equivalent throughput. Furthermore, there still exists potential for tuning and optimizing the glycopolymer architecture on this platform by altering the ATRP grafting and ligand reaction conditions that may increase the capacity. Membrane platforms also offer other advantages highlighted by previous authors, such as better resolution than resin media under high loading conditions40, which improves product purity.
4. Conclusions
Affinity membranes for the adsorption of the lectin conA were fabricated by reacting dGluc to epoxy groups grown from the surface of regenerated cellulose through surface-initiated ATRP. Membranes were characterized with ATR-FTIR to confirm ligand incorporation. Static binding capacity was quantified and found to be comparable to that of other substrates. Limitations in characterization prohibited the quantification of ligand incorporation, which would elucidate if experimental grafting densities were high enough to achieve optimal binding under the “glycoside cluster effect.” Binding capacity measurements in conjunction with an analysis of transport phenomena indicate that the binding kinetics represent the limiting factor for achieving high productivities. However, the membrane platform offers easy scalability, low column pressure drop, and the opportunity for continuous processing.
Supplementary Material
Acknowledgments
Funding for this work was provided by the National Institutes of Health under award 1 R15 GM094676-01. We thank Kimberly Ivey of the Clemson University School of Materials Science and Engineering for assistance with FTIR measurements and data analysis.
References
- 1.Sharon N. J Biol Chem. 2007;282:2753–2764. doi: 10.1074/jbc.X600004200. [DOI] [PubMed] [Google Scholar]
- 2.Kaneko Y, Nimmerjahn F, Ravetch JV. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 3.Agrawal B, Goldstein I. J Biochim Biophys Acta. 1967;147:262–271. [PubMed] [Google Scholar]
- 4.Batista-Viera F, Janson J-C, Carlsson J. In Protein Purification: Principles, High Resolution Methods, and Applications. 3. Wiley; Hoboken, NJ: 2011. pp. 221–258. [Google Scholar]
- 5.Clemmitt R, Chase H. Biotechnol Bioeng. 2003;82:506–516. doi: 10.1002/bit.10596. [DOI] [PubMed] [Google Scholar]
- 6.Reichelt S, Elsner C, Prager A, Naumov S, Kuballa J, Buchmeiser MR. Analyst. 2012;137:2600–2607. doi: 10.1039/c2an16087k. [DOI] [PubMed] [Google Scholar]
- 7.Babac C, Yavuz H, Galaev IY, Pişkin E, Denizli A. React Funct Polym. 2006;66:1263–1271. [Google Scholar]
- 8.Khan TA, Husain Q, Naeem A. Eur J Appl Sci. 2010;2:70–76. [Google Scholar]
- 9.Ueda E, Gout P, Morganti L. J Chromatogr A. 2003;988:1–23. doi: 10.1016/s0021-9673(02)02057-5. [DOI] [PubMed] [Google Scholar]
- 10.Beyersmann D. Toxicol Lett. 2002;127:63–68. doi: 10.1016/s0378-4274(01)00484-2. [DOI] [PubMed] [Google Scholar]
- 11.Gutierrez R, Martín del Valle E, Galan M. Sep Purif Rev. 2007;36:71–111. [Google Scholar]
- 12.Mahalingam A, Geonnotti AR, Balzarini J, Kiser PF. Mol Pharmaceut. 2011;8:2465–2475. doi: 10.1021/mp2002957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nascimento KS, Cunha AI, Nascimento KS, Cavada BS, Azevedo AM, Aires-Barros MR. J Mol Recognit. 2012;25:527–541. doi: 10.1002/jmr.2200. [DOI] [PubMed] [Google Scholar]
- 14.Guo W, Ruckenstein E. J Membr Sci. 2001;182:227–234. [Google Scholar]
- 15.Perçin I, Yavuz H, Aksöz E, Denizli A. Biotechnol Prog. 2012;28:756–761. doi: 10.1002/btpr.1552. [DOI] [PubMed] [Google Scholar]
- 16.Hu MX, Wan LS, Xu ZK. J Membr Sci. 2009;335:111–117. [Google Scholar]
- 17.Ogata Y, Seto H, Murakami T, Hoshino Y, Miura Y. Membranes. 2013;3:169–181. doi: 10.3390/membranes3030169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye X, Huang X, Xu Z. Colloid Surface B. 2014;115:340–348. doi: 10.1016/j.colsurfb.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 19.Yang Q, Strathmann M, Rumpf A, Schaule G, Ulbricht M. ACS Appl Mater Inter. 2010;2:3555–3562. doi: 10.1021/am1007276. [DOI] [PubMed] [Google Scholar]
- 20.Yang Q, Ulbricht M. Macromolecules. 2011;44:1303–1310. [Google Scholar]
- 21.Yang Q, Hu M, Dai Z, Tian J, Xu Z. Langmuir. 2006;22:9345–9349. doi: 10.1021/la0610598. [DOI] [PubMed] [Google Scholar]
- 22.Singh N, Wang J, Ulbricht M, Wickramasinghe SR, Husson SM. J Membr Sci. 2008;309:64–72. [Google Scholar]
- 23.Bhut BV, Wickramasinghe SR, Husson SM. J Membr Sci. 2008;325:176–183. [Google Scholar]
- 24.Bhut BV, Husson SMJ. Membr Sci. 2009;337:215–223. [Google Scholar]
- 25.Wang J, Faber R, Ulbricht M. J Chromatogr A. 2009;1216:6490–6501. doi: 10.1016/j.chroma.2009.07.042. [DOI] [PubMed] [Google Scholar]
- 26.Jain P, Vyas MK, Geiger JH, Baker GL, Bruening ML. Biomacromolecules. 2010;11:1019–1026. doi: 10.1021/bm9014792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menkhaus TJ, Varadaraju H, Zhang L, Schneiderman S, Bjustrom S, Liu L, Fong H. Chem Commun. 2010;46:3720–3722. doi: 10.1039/c001802c. [DOI] [PubMed] [Google Scholar]
- 28.Anuraj N, Bhattacharjee S, Geiger JH, Baker GL, Bruening ML. J Membr Sci. 2012;389:117–125. doi: 10.1016/j.memsci.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhut BV, Weaver J, Carter AR, Wickramasinghe SR, Husson SM. Biotechnol Bioeng. 2011;108:2645–2653. doi: 10.1002/bit.23221. [DOI] [PubMed] [Google Scholar]
- 30.Bhut BV, Weaver J, Carter AR, Wickramasinghe SR, Husson SM. Biotechnol Bioeng. 2011;108:2654–2660. doi: 10.1002/bit.23222. [DOI] [PubMed] [Google Scholar]
- 31.Chenette HCS, Robinson JR, Hobley E, Husson SM. J Membr Sci. 2012;423–424:43–52. doi: 10.1016/j.memsci.2012.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei Y, Ma J, Wang C. J Membr Sci. 2013;427:197–206. [Google Scholar]
- 33.Himstedt HH, Qian X, Weaver JR, Wickramasinghe SRJ. Membr Sci. 2013;447:335–344. [Google Scholar]
- 34.Wang J, Sproul RT, Anderson LS, Husson SM. Polymer. 2014;55:1404–1411. [Google Scholar]
- 35.Ran J, Wu L, Zhang Z, Xu T. Prog Polym Sci. 2014;39:124–144. [Google Scholar]
- 36.Bruening ML, Dotzauer DM, Jain P, Ouyang L, Baker GL. Langmuir. 2008;24:7663–7673. doi: 10.1021/la800179z. [DOI] [PubMed] [Google Scholar]
- 37.Ting SS, Chen G, Stenzel MH. Polym Chem. 2010;1:1392–1412. [Google Scholar]
- 38.Martins SIFS, Van Boekel MAJS. Food Chemistry. 2005;92:437–488. [Google Scholar]
- 39.Vogel JH, Nguyen H, Giovannini R, Ignowski J, Garger S, Salgotra A, Tom J. Biotechnol Bioeng. 2012;109:3049–3058. doi: 10.1002/bit.24578. [DOI] [PubMed] [Google Scholar]
- 40.Bhut BV, Christensen KA, Husson SM. J Chromatogr A. 2010;1217:4946–4957. doi: 10.1016/j.chroma.2010.05.049. [DOI] [PubMed] [Google Scholar]
- 41.Klein E. J Membr Sci. 2000;179:1–27. [Google Scholar]
- 42.Clegg RM, Loontiens FG, Van Landschoot A, Jovin TM. Biochemistry. 1981;20:4687–4692. doi: 10.1021/bi00519a025. [DOI] [PubMed] [Google Scholar]
- 43.Knudsen HL, Fahrner RL, Xu Y, Norling LA, Blank GS. J Chromatogr A. 2001;907:145–154. doi: 10.1016/s0021-9673(00)01041-4. [DOI] [PubMed] [Google Scholar]
- 44.Liu HC, Fried JR. AICHE J. 1994;40:40–49. [Google Scholar]
- 45.Boi C, Cattoli F, Facchini R, Sorci M, Sarti GC. J Membr Sci. 2006;273:12–19. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.