

Abstract
The behavioral consequences of fetal alcohol spectrum disorders (FASD) are serious and persist throughout life. The causative mechanisms underlying FASD are poorly understood. However, much has been learned about FASD from human structural and functional studies as well as from animal models, which have provided a greater understanding of the mechanisms underlying FASD. Using animal models of FASD, it has been recently discovered that ethanol induces neuroimmune activation in the developing brain. The resulting microglial activation, production of proinflammatory molecules, and alteration in expression of developmental genes are postulated to alter neuron survival and function and lead to long-term neuropathological and cognitive defects. It has also been discovered that microglial loss occurs, reducing microglia’s ability to protect neurons and contribute to neuronal development. This is important, because emerging evidence demonstrates that microglial depletion during brain development leads to long-term neuropathological and cognitive defects. Interestingly, the behavioral consequences of microglial depletion and neuroimmune activation in the fetal brain are particularly relevant to FASD. This chapter reviews the neuropathological and behavioral abnormalities of FASD and delineates correlates in animal models. This serves as a foundation to discuss the role of the neuroimmune system in normal brain development, the consequences of microglial depletion and neuroinflammation, the evidence of ethanol induction of neuroinflammatory processes in animal models of FASD, and the development of anti-inflammatory therapies as a new strategy for prevention or treatment of FASD. Together, this knowledge provides a framework for discussion and further investigation of the role of neuroimmune processes in FASD.
1. OVERVIEW OF FETAL ALCOHOL SPECTRUM DISORDERS
Maternal consumption of alcohol during pregnancy can lead to a spectrum of defects in their offspring. The range of disorders induced by gestational alcohol exposure is designated as fetal alcohol spectrum disorders (FASD) and includes the severe disorder of fetal alcohol syndrome (FAS). FASD is associated with monumental personal, societal, and economic impacts. In the United States, a staggering 12% of pregnant women consume alcohol despite extensive public health warnings (Floyd, Weber, Denny, & O’Connor, 2009). The consequence is an incidence of FASD of at least 1 in 100 births and an incidence of FAS of 2–7 in 1000 births (May et al., 2009; Sampson et al., 1997). The direct economic cost of FASD in the United States was recently estimated at $4 billion per year (Lupton, Burd, & Harwood, 2004). Thus, FASD is a major public health problem.
FASD represents a range of mild to severe effects on the brain and is the primary cause of mental retardation (Abel & Sokol, 1986; American Academy of Pediatrics Committee on Substance Abuse and Committee on Children with Disabilities, 2000). Both structural and functional defects are produced in the developing brain by fetal alcohol exposure (Riley & McGee, 2005). The CNS pathology and cognitive and behavioral impairments commonly persist throughout life (Riley, Infante, & Warren, 2011; Streissguth, Landesman-Dwyer, Martin, & Smith, 1980). Brain structural defects in individuals with FASD are commonly identified in the corpus callosum, cerebellum, cerebral cortex, hippocampus, amygdala, thalamus, and basal ganglia (Astley et al., 2009; Norman, Crocker, Mattson, & Riley, 2009). These defects range from diminution in the size of the brain region to microstructural pathology at the level of loss of neurons and glial cells, ectopic locations of neurons and glia, or defects in neural connectivity. Clinical imaging studies of individuals with FASD indicate that the extent of brain damage correlates with the extent of cognitive deficits as well as with FAS facial dysmorphology (Astley et al., 2009; Coles & Li, 2011; Lebel, Roussotte, & Sowell, 2011; O’Hare et al., 2005; Suttie et al., 2013).
2. NORMAL BRAIN DEVELOPMENT
The fetal brain develops throughout the entire period of pregnancy and is the most vulnerable organ to alcohol pathology. The nervous system develops from neuroectoderm, which forms the neural tube (see Squire et al., 2013). Neuroepithelial cells in the wall of the neural tube proliferate to form neuroblasts. Glioblasts are produced by neuroepithelial proliferation to produce astrocytes and oligodendrocytes. Microglial cells are produced from nonneuroectodermal, mesenchymal tissue as described below (Saijo & Glass, 2011). The cranial end of the neural tube forms the prosencephalon (forebrain), mesencephalon (midbrain), and rhombencephalon (hindbrain). The cerebral cortex, hippocampus, and cerebellum develop by the proliferation of neuroepithelial cells in the innermost layers of the neural tube. Cerebral cortical and hippocampal neurons are generated from progenitor cells in the ventricular and subventricular zones of the telencephalon by proliferation, differentiation, and migration. Cerebellar Purkinje neurons are generated by the proliferation of neuroepithelial cells in the ventricular zone and migration into the developing cerebellar cortex. Cerebellar granule cells are generated by the proliferation of neuroepithelial cells in the rhombencephalon and migration to form the granular layer. Migrating neurons use radial glial cells, chemoattractant and chemorepulsive molecules, cytokines and chemokines, neurotrophins, and neurotransmitters as guides to their correct position. As described below, studies in animal models reveal that alcohol exposure leads to neuron and glial death and disruption of normal neurogenesis, differentiation, and migration of neurons.
3. FASD NEUROPATHOLOGY IN HUMANS
Because of advanced imaging capabilities and earlier and more reliable diagnosis of FASD, detailed analyses of structural anomalies in the brain of individuals with FASD are advancing. Microencephaly is the most commonly reported structural brain defect. But, magnetic resonance imaging (MRI) has revealed more specific volume reductions in the corpus callosum, cerebral cortex, cerebellum, and subcortical structures including the hippocampus, basal ganglia, amygdala, and thalamus (Astley et al., 2009; Coles & Li, 2011; Lebel et al., 2011; Norman et al., 2009; Riley et al., 2011; Riley, McGee, & Sowell, 2004). Additional studies, as described below, have uncovered both gross and microstructural brain defects that correspond to functional deficits in individuals with FASD.
The corpus callosum is the principal white matter track in the brain and is very sensitive to fetal alcohol exposure. It exhibits gross changes with partial to complete agenesis and changes in volume, length, and thickness in individuals with FASD (Yang et al., 2012). At a higher level of resolution, diffusion tensor imaging (DTI) reveals microstructural abnormalities throughout the region (Wozniak et al., 2009). There is a striking correspondence between gross and microstructural pathologies in the corpus callosum and FASD disease severity, facial dysmorphology, and functional deficits (Sowell et al., 2001; Wozniak & Muetzel, 2011). For example, reduction in the size of the corpus callosum and displacement correspond to decreased executive function, learning and memory, and impaired motor skills (Kodituwakku, Kalberg, & May, 2001; Roebuck-Spencer, Mattson, Marion, Brown, & Riley, 2004; Sowell et al., 2001).
The cerebral cortex is also highly vulnerable to fetal alcohol exposure. At the gross level, aberrant formation of the gyri and sulci occurs (De Guio et al., 2013). MRI reveals that the frontal cortex atrophies (Leigland, Ford, Lerch, & Kroenke, 2013; Sowell et al., 2008; Yang et al., 2012; Zhou et al., 2011). Structural and functional imaging also indicates deficits in the frontal, parietal, and temporal lobes (Archibald et al., 2001; Bhatara et al., 2002; Lebel et al., 2012; Riley et al., 2004). There is a reduction in the volume and altered morphology of both the cortical gray matter and white matter (Bjorkquist, Fryer, Reiss, Mattson, & Riley, 2010; Nardelli, Lebel, Rasmussen, Andrew, & Beaulieu, 2011; Riley et al., 2004). Further, there are changes throughout the subcortical region with particular losses in the hippocampus, thalamus, and globus pallidus (Riley et al., 2004; Wozniak et al., 2009). Correlation to disease severity is strong for structural and functional defects in the cerebral cortex (Norman et al., 2009; Yang et al., 2012).
Gross and fine structural defects are also produced in the cerebellum by fetal alcohol exposure. The volume of the entire cerebellum is smaller, and the reduced volume and displacement of the anterior vermis and posterior inferior regions are particularly striking (Archibald et al., 2001; Autti-Ramo et al., 2002; O’Hare et al., 2005; Sowell et al., 1996). Defects in cerebellar development have been often documented in individuals with moderate to severe FASD and correlate with disease severity (O’Hare et al., 2005; Wozniak et al., 2009). For example, DTI studies of the cerebellum have revealed defects in the cerebellar white matter tracts that correspond to behavioral deficits (Spottiswoode et al., 2011).
There is an emerging wealth of information defining functional connectivity defects within and between brain regions in individuals with FASD (Norman et al., 2013; Wozniak et al., 2013). There are increases or decreases in regional brain activity that correlate to deficits in cognitive function including executive function, learning and memory, mathematical processing, attention, and inhibition (Coles & Li, 2011). For example, there is differential activation of circuitry in regions of the cerebral cortex, corpus callosum, striatum, and cerebellum during cognitive tasks (see, e.g., Coles & Li, 2011; Diwadkar et al., 2013; Lebel, Rasmussen, Wyper, Andrew, & Beaulieu, 2010; Norman et al., 2013; Wozniak et al., 2011, 2013. These studies are particularly enlightening, because the degree of difference in brain activity correlates to the degree of cognitive impairment and disease severity.
4. FASD NEUROPATHOLOGY IN ANIMAL MODELS
Human studies confirm that findings in animal models accurately reproduce major aspects of the neuropathology and behavioral dysfunction seen in FASD. Treatment of rodents and nonhuman primates with alcohol (ethanol) has proved to be highly informative for understanding the neuroanatomical and cellular events that occur as a consequence of fetal alcohol exposure. For comparison to human gestation, it is important to align the temporal stages of brain development with the corresponding stage in human brain development. For example, rodent birth represents human midgestation for the development of several brain regions, whereas birth represents the beginning of the human third trimester for the development of other brain regions (Clancy, Darlington, & Finlay, 2001; Clancy, Finlay, Darlington, & Anand, 2007; Dobbing & Sands, 1979). Understanding parallels between human and rodent brain development is particularly important because so much of what we know about FASD neuropathology and its causes have been identified first in rodent models of these disorders.
Imaging studies of mice exposed to gestational ethanol have revealed significant parallels with human FASD including facial dysmorphology and defects in brain development. There is a highly specific pattern of regional brain malformation depending on the temporal period of exposure to ethanol (O’Leary-Moore, Parnell, Lipinski, & Sulik, 2011). For example, exposure on mouse gestational day 7 produces defects in the cerebral cortex, hippocampus, and basal ganglia in addition to abnormalities in fiber tracts (Godin et al., 2010). However, exposure on gestational day 8 produces neuronal death in the hindbrain and cranial neural crest cells, cranial nerve defects with the loss of ganglia, and abnormalities in fiber tracts (Parnell et al., 2009). The volume of the cerebral cortex, cerebellum, and hippocampus is reduced, and hypoplasia or agenesis of the corpus callosum is also present. Other exposure periods produce different unique patterns of structural and functional brain defects as demonstrated in numerous studies (e.g., O’Leary-Moore et al., 2011).
Investigation of nonhuman primates as models of FASD has provided important insights. These studies provide strong evidence that the cerebral cortex is vulnerable to ethanol-induced neuropathology. Offspring of Macaque monkeys exposed to ethanol during early pregnancy experienced significant neuronal loss in the cortex, brainstem, and subcortical structures (Farber, Creeley, & Olney, 2010; Miller, 2006, 2007; Mooney & Miller, 2001, 2009). Studies in rodents also demonstrate loss of cortical neurons in offspring following ethanol exposure during gestation or in the postnatal period (Ikonomidou et al., 2000; Miller & Potempa, 1990; Wilson, Peterson, Basavaraj, & Saito, 2011). Cortical volume, thickness, and surface area are reduced by ethanol exposure throughout gestation in rodents (Leigland et al., 2013). Proper neuron migration into the cerebral cortex is also disrupted by ethanol (Gressens, Lammens, Picard, & Evrard, 1992; Miller, 1993) apparently through inhibition of radial glia development and ethanol-induced changes in the expression of molecules central to migration (Aronne, Guadagnoli, Fontanet, Evrard, & Brusco, 2011; Miller & Robertson, 1993). Rodent models of fetal ethanol exposure further demonstrate neuroanatomical, synaptic, and electrophysiological defects in neocortical circuitry that correspond to deficits in behavior (El Shawa, Abbott, & Huffman, 2013; Inomata, Nasu, & Tanaka, 1987; Slawecki, Thomas, Riley, & Ehlers, 2004; Whitcher & Klintsova, 2008; Wilson et al., 2011).
The developing hippocampus of the rodent is vulnerable to ethanol exposure. The size of the individual neuron populations in all regions of the hippocampus is reduced by ethanol exposure during gestation or in the postnatal period (Gil-Mohapel, Boehme, Kainer, & Christie, 2010). This includes significant loss of pyramidal neurons in CA1 and CA3 and granule neurons in the dentate gyrus (Ieraci & Herrera, 2007; Ikonomidou et al., 2000; Klintsova et al., 2007; Livy, Miller, Maier, & West, 2003; Tran & Kelly, 2003; Uban et al., 2010). Developmental ethanol exposure also produces neuroanatomical and electrophysiological defects in hippocampal circuitry (Diaz et al., 2014; Everett, Licon-Munoz, & Valenzuela, 2012; Lindquist, Sokoloff, Milner, & Steinmetz, 2013; Sakata-Haga et al., 2003). Defects in the hippocampus correlate with behavioral deficits, particularly with learning and memory (Banuelos et al., 2012; El Shawa et al., 2013; Hamilton et al., 2010; Idrus, McGough, Riley, & Thomas, 2014; Kelly, Pierce, & West, 1987; Thomas, Idrus, Monk, & Dominguez, 2010; Wagner, Zhou, & Goodlett, 2014; West, Kelly, & Pierce, 1987; Zink et al., 2011).
The developing cerebellum is vulnerable to ethanol exposure. Administration of ethanol to Macaque monkeys demonstrates significant cerebellar Purkinje neuron loss in their offspring (Bonthius et al., 1996; Farber et al., 2010). In rodents, cerebellar volume is reduced by midgestational ethanol exposure (Parnell et al., 2013). The cerebellum is also vulnerable to ethanol in the postnatal period where there is volume loss as well as marked loss of Purkinje neurons, granule neurons, and deep cerebellar neurons (Goodlett & Eilers, 1997; Green, Tran, Steinmetz, & Goodlett, 2002; Hamre & West, 1993; Napper & West, 1995; Pierce, Kane, Serbus, & Light, 1997; Pierce, Serbus, & Light, 1993; Pierce, Williams, & Light, 1999). Functional development of neurons is inhibited as evidenced by impaired migration, stunted development of dendrites, reduction in the number of synapses, and impaired electrophysiological activity (Allam et al., 2013; Jiang, Kumada, Cameron, & Komuro, 2008; Kane et al., 2011; Servais et al., 2007; Smith & Davies, 1990; Valenzuela, Lindquist, & Zamudio-Bulcock, 2010). These defects contribute to the deficits in motor coordination and classical conditioning commonly observed in rodent models of FASD (Brown, Calizo, & Stanton, 2008; Goodlett, Thomas, & West, 1991; Idrus, McGough, Riley, & Thomas, 2011; Klintsova et al., 1998; Lewis, Wellmann, & Barron, 2007; Murawski, Jablonski, Brown, & Stanton, 2013; Wagner, Klintsova, Greenough, & Goodlett, 2013).
5. BEHAVIORAL CONSEQUENCES IN HUMANS WITH FASD
The negative cognitive and behavioral outcomes associated with FASD are far ranging. This reflects the extended vulnerability of the developing brain to ethanol throughout gestation. The cognitive consequences of FASD include deficits in executive function, deficits in learning and memory, impaired information processing, deficits in vigilance, delayed reaction time, restricted mathematical ability, poor speech and language skills, and impaired visual-spatial ability (Guerri, Bazinet, & Riley, 2009; Lebel et al., 2010; Mattson & Riley, 1998; Mattson & Roebuck, 2002; Mattson et al., 2013; Santhanam, Li, Hu, Lynch, & Coles, 2009). Impairment of executive function is particularly common in FASD, leading to problems with planning, set shifting, fluency, response inhibition, and working memory (Connor, Sampson, Bookstein, Barr, & Streissguth, 2000; Kodituwakku et al., 2001). Psychiatric disorders include abuse and addiction to alcohol and other drugs, internalizing and externalizing behavioral disorders, mood disturbances, and major depressive disorder (Barr et al., 2006; Fryer, McGee, Matt, Riley, & Mattson, 2007; Olson, Feldman, Streissguth, Sampson, & Bookstein, 1998; Streissguth et al., 2004; Ware et al., 2014). It is becoming increasingly clear due to functional imaging studies that these cognitive deficits are closely linked to ethanol-induced neuropathology in the cerebral cortex, hippocampus, cerebellum, and corpus callosum, as well as in other structures. Neuropathology in the cerebellum and basal ganglia is evident as delayed motor development and ataxia, including difficulty with balance, gait, and fine motor skills (Adnams et al., 2001; Connor, Sampson, Streissguth, Bookstein, & Barr, 2006; Mattson & Riley, 1998; Roebuck, Simmons, Mattson, & Riley, 1998; Roebuck-Spencer et al., 2004).
6. BEHAVIORAL CONSEQUENCES IN RODENT MODELS OF FASD
Human FASD behavioral outcomes have been modeled in rodents. Cerebral cortical, hippocampal, and cerebellar functions have been assessed with cognitive tests of learning and memory, motivation, anxiety, and locomotion, among others. Gestational exposure produces altered social behavior, impaired learning and memory, delayed development of spontaneous alternation behavior, increased anxiety, and poor motor coordination (El Shawa et al., 2013; Hamilton et al., 2010; Thomas et al., 2010). Postnatal exposure produces impaired learning and memory, altered delay discounting, and increased hyperactivity (Banuelos et al., 2012; Idrus et al., 2014; Kelly et al., 1987; Wagner et al., 2014; West et al., 1987; Zink et al., 2011). Ethanol-induced cerebellar pathology underlies deficits in classical eyeblink conditioning tasks (Brown et al., 2008; Murawski et al., 2013; Wagner et al., 2013). The cerebellum is centrally involved in motor function and ethanol impairs balance and coordination in rodents exposed to ethanol during early postnatal development (Goodlett et al., 1991; Idrus et al., 2011; Klintsova et al., 1998; Lewis et al., 2007).
The correspondence between ethanol-induced neuroanatomical and behavioral outcomes in humans and that in animals allows the use of animal models to elucidate links between structural and functional neuropathology and cognitive and motor deficits. Thus, a foundation for further investigation of the cellular and molecular mechanisms underlying FASD is established. This is important because, as there is no treatment that targets the causes underlying FASD and the long-term consequences can be so severe, there is a pressing need to develop therapeutic interventions to ameliorate the spectrum of deficits associated with the disorders.
7. OVERVIEW: ALCOHOL EFFECTS ON IMMUNE RESPONSE IN THE BRAIN
It is well established that alcohol has potent effects on immune activity in the CNS throughout the life span (Drew & Kane, 2013). Ethanol effects on immune response have begun to be evaluated in animal models of maternal consumption, resulting in FASD, models of binge drinking common to adolescents, and chronic alcohol abuse common in adults and in aged populations. Alcohol abuse at each of these stages can result in neuroinflammation, which is believed to contribute to ethanol-induced neurodegeneration. This chapter focuses primarily on the effects of ethanol on immune activation in the developing CNS.
The effects of ethanol on the peripheral immune system have been extensively investigated. Therefore, ethanol has complex effects on immune activity that depend on a variety of factors including age, gender, pattern of ethanol exposure (acute, binge, or chronic administration), the timing of evaluation following ethanol exposure, and the cells or tissues under investigation (Goral, Karavitis, & Kovacs, 2008). These studies demonstrate that ethanol can induce or alternatively suppress immune activation in the periphery, depending on the specific experimental paradigm. An example of the dichotomous effects of alcohol on peripheral immune responses that occurs in human alcoholics who are generally immunosuppressed and yet frequently exhibit elevated serum levels of proinflammatory cytokines (Goral et al., 2008). Relative to the periphery, the effects of ethanol on the CNS is understudied (Blanco & Guerri, 2007; Crews & Nixon, 2009; Crews et al., 2006).
A series of seminal studies demonstrated that ethanol increases the expression of proinflammatory cytokines and chemokines, as well as molecules such as nitric oxide and Cox-2 in the CNS (Blanco, Pascual, Valles, & Guerri, 2004; Crews et al., 2006; Davis & Syapin, 2004; Knapp & Crews, 1999; Ward et al., 1996; Zou & Crews, 2006, 2010). These studies further determined that transcription factors including NF-κB and CREB are activated by ethanol (Blanco et al., 2004; Crews et al., 2006; Davis & Syapin, 2004; Ward et al., 1996; Zou & Crews, 2006, 2010), suggesting that these molecules may play critical roles in ethanol induction of proinflammatory molecules, oxidative stress, and ethanol-induced pathology in the CNS. Ethanol has been demonstrated to alter the function and viability of CNS glia including microglia and astrocytes, as well as neurons. However, much work is required to better understand the effects of ethanol on neuroinflammation and neurodegeneration.
8. MICROGLIA
8.1. Overview
Together with astrocytes and oligodendrocytes, microglia comprise the glial cells of the CNS. While oligodendrocytes play a critical role in forming myelin, astrocytes and microglia maintain the health of neurons through the production of neurotrophic factors and the removal of potentially neurotoxic molecules from the CNS. Microglia also maintain the CNS by the removal of debris and are involved in synaptic pruning critical in the formation of functional synapses. However, in response to CNS insult, microglia can become activated, resulting in the production of inflammatory molecules that can contribute to neurodegenerative disorders.
8.2. Role in the CNS development
Distinct from other glia and neurons that are derived from neuroectoderm, microglia are believed to be of hematopoietic origin (Saijo & Glass, 2011). Microglia are generated in the primitive yolk sac and migrate to the CNS early during fetal development prior to formation of the blood–brain barrier. As myeloid lineage cells, microglia are most closely related to macrophages. Microglia and macrophages both express molecules such as CD11b and CD14 (Kettenmann, Hanisch, Noda, & Verkhratsky, 2011). Like macrophages, the differentiation of microglia is dependent of the transcription factor PU.1, as well as colony stimulating factor 1 and its receptor. The fact that PU.1-knock-out mice are devoid of both microglia and macrophages supports a common myeloid lineage for these cells (McKercher et al., 1996).
Controversy has existed regarding whether macrophages are able to migrate into the mature CNS and form microglia, thus serving as a source to replenish the microglial cell population. Bone marrow chimera studies suggested that this is the case (Eglitis & Mezey, 1997; Hickey, 1991; Hickey & Kimura, 1988; Priller et al., 2001). However, these studies are complicated by the fact that the irradiation used in these protocols compromised the blood–brain barrier. More recent parabiosis studies that involve direct connection of the circulation of the donor and recipient animals demonstrate that few donor cells enter the CNS in the absence of irradiation (Ajami, Bennett, Krieger, Tetzlaff, & Rossi, 2007; Mildner et al., 2007) and macrophages that do enter do not appear to differentiate into microglia (Ajami, Bennett, Krieger, McNagny, & Rossi, 2011). In addition, microglia appear to be long-lived cells that can proliferate in response to pathological conditions in the CNS (Fellner, Jellinger, Wenning, & Stefanova, 2011; Glass, Saijo, Winner, Marchetto, & Gage, 2010; Lawson, Perry, & Gordon, 1992; Reitz, Brayne, & Mayeux, 2011), suggesting that they are replenished from stores of microglial precursors in the CNS when needed.
8.3. Function in the healthy CNS
Microglia comprise approximately 5–20% of the cells in the CNS. The relative abundance of microglia varies regionally as does the morphology of these cells (Lawson, Perry, Dri, & Gordon, 1990). This may reflect regional differences in the function and phenotype of microglia. Microglia are more abundant in gray matter than in white matter (Rivest, 2009). In the healthy mature CNS, microglia generally exhibit a ramified appearance characterized by a small soma, limited cytoplasm, and a series of thin, highly branched processes (Fig. 3.1A). Although these branched processes extend significantly away from the soma, it is believed that the territory occupied by individual microglia does not overlap with adjacent microglia (Ransohoff & Perry, 2009). Microglia contribute to homeostasis of the healthy brain by producing growth factors critical to the survival of neurons as well as protecting neurons by removing potentially neurotoxic molecules from the parenchyma. Microglia play a critical surveillance role in the CNS. They are motile and phagocytose cellular debris including that derived from apoptotic cells (Sierra et al., 2010). Removal of cellular debris in this manner occurs in the absence of microglial activation and inflammation, thus maintaining homeostasis. Microglia also play a critical role in the formation and remodeling of synapses. Multiphoton microscopy studies indicate that microglia are dynamic in vivo, constantly sampling the microenvironment by extending and retracting processes, particularly at the synapse (Davalos et al., 2005; Nimmerjahn, Kirchhoff, & Helmchen, 2005). Microglia surveillance is particularly important in synaptic plasticity during development through activity-dependent synaptic pruning (Tremblay, Lowery, & Majewska, 2010). The role of microglia in synaptic plasticity is also supported by the documented physical association of these cells with developing and mature synapses (Dalmau, Finsen, Zimmer, Gonzalez, & Castellano, 1998; Fiske & Brunjes, 2000; Perry, Hume, & Gordon, 1985). Microglia also associate with dendritic spines, which further support a role for microglia in modulating the structure and function of synapses (Tremblay et al., 2010; Wake, Moorhouse, Jinno, Kohsaka, & Nabekura, 2009). Interestingly, microglia-derived products are critical to synapse development and function. This includes molecules classically defined as proinflammatory including major histocompatibility (MHC) class I, MHC I-binding receptors, complement proteins, and cytokines including TNF-α and IL-6 (Boulanger, 2009). The chemokine receptor CX3CR1 is expressed specifically on microglia and is critical in microglia–neuron interactions through association with the chemokine CX3CL1/fractalkine present on neurons. Interestingly, CX3CR1-deficient mice exhibit reduced microglial numbers during development, and these mice exhibit deficits in synapse formation and plasticity in the hippocampus (Paolicelli et al., 2011). This further supports a role of microglia in the modulation of neuronal circuitry in the developing brain.
Figure 3.1.
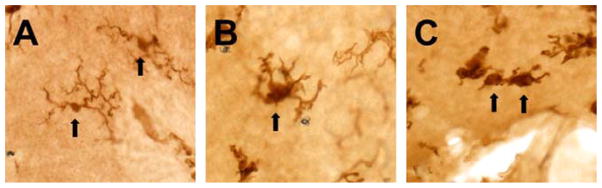
Microglial morphology exhibits a stepwise modification as the process of activation changes the functional phenotype of the cell. Microglial cells in the normal, unperturbed CNS exhibit a ramified morphology with a small soma, limited cytoplasm, and long, thin, highly branched processes (A). With activation, the cells hypertrophy and the processes retract, becoming broader and stunted with less branching (B). Highly activated cells progress toward an ameboid, hypertrophied form with few or no processes (C). These images are from the postnatal mouse cerebellum and are representative of the described morphological phenotypes of microglia. The microglial cells in these images were visualized with immunohistochemistry against the ionized calcium-binding adapter molecule (Iba-1), classically used to identify CNS microglia. Arrows indicate microglial cells with the described morphologies.
8.4. Immune response
Traditionally, the CNS has been considered to be an immune-privileged site. This is due to the absence of lymphatic drainage from the CNS, the observation that tissue grafted into the CNS is relatively protected from immune-mediated destruction, and the presence of a blood–brain barrier that limits the movement of immune cells from the periphery. However, it is now known that peripheral immune cells are capable of entering the CNS parenchyma. In addition, resident CNS cells including microglia and to a lesser extent astrocytes provide immune functions (Carson, Doose, Melchior, Schmid, & Ploix, 2006). As noted earlier, microglia provide homeostatic functions in the healthy CNS. Upon insult or aging, microglia can become activated, changing morphology from a highly ramified cell with a small soma to a hypertrophied cell with broad, stunted processes, and then to an ameboid cell (Fig. 3.1). When activated, they can produce a variety of proinflammatory molecules including cytokines, chemokines, reactive oxygen species, and nitric oxide. These molecules can be protective, for example, in ridding the CNS of infectious agents. In this regard, microglia are the primary cell type that performs innate immune functions in the CNS. However, chronically activated microglia and these same proinflammatory molecules can be toxic to parenchymal cells including neurons. Activated microglia are more aggressively phagocytic. They also express increased levels of MHC class I and II, which are central to antigen presentation during immune responses, and are, thus, capable of serving as antigen-presenting cells. In this manner, microglia are capable of presenting antigen to T cells and mediating adaptive immune responses (Ransohoff & Perry, 2009; Saijo & Glass, 2011).
Microglial activation is triggered following the interaction of Toll-like receptors (TLRs) present on the surface of these cells with conserved motifs associated with pathogens. In addition, microglia can also be activated in response to nonpathogenic insults through TLR engagement by endogenous danger signals including heat shock protein 70 and high mobility group box protein (HMGB) 1. TLR activation results in the activation of signal transduction pathways and changes in the functional phenotype of microglia including increased cell proliferation, increased production of proinflammatory molecules, increased antigen presentation, and increased phagocytic activity. Microglial activation occurs in a stepwise and not in an all or none manner (Carson et al., 2007; Colton, 2009; Hanisch & Kettenmann, 2007; Perry, Cunningham, & Holmes, 2007; Schwartz, Butovsky, Bruck, & Hanisch, 2006; Streit, Walter, & Pennell, 1999). Microglia can also undergo distinct forms of activation characterized by differential protein expression patterns and function in a manner similar to that first described as classical or alternative activation of macrophages (Ransohoff & Perry, 2009). Following the removal of inflammatory stimuli, microglia can revert back toward a quiescent form with a ramified morphological phenotype. However, studies indicate that these microglia are not fully deactivated and are primed for subsequent activation (Hanisch & Kettenmann, 2007).
9. POTENTIAL LONG-TERM CONSEQUENCES OF IMMUNE ACTIVATION IN THE DEVELOPING CNS
As discussed previously, proinflammatory molecules including cytokines and chemokines play an important role in ridding the brain of pathogens, but expression of these molecules is low in the mature brain in the absence of inflammatory insults. In contrast, in the developing brain, proinflammatory molecules are expressed at significant levels in the absence of inflammatory insults. This reflects the fact that these molecules classically recognized for their inflammatory activity have other important roles in CNS development that are independent of an immune response. For example, expression of the cytokines IL-1β and TNF-α is developmentally regulated, and these molecules are believed to play important roles in CNS development including modulation of neural cell migration, proliferation, differentiation, and death (Merrill, 1992). The cytokine IL-6 is believed to serve as a neurotrophic factor and to contribute to vascular development in the CNS (Fee et al., 2000; Gadient & Otten, 1994). MHC class I molecules have been demonstrated to mediate activity-dependent synapse formation in the developing visual system (Corriveau, Huh, & Shatz, 1998). Complement, a traditional immune molecule central to antigen presentation, is necessary for microglial sculpting of early postnatal neural circuits (Schafer et al., 2012). Clearly, altered expression of traditional immune system molecules that have important nonimmune roles in the developing brain will have significant, detrimental consequences for brain development.
Recent studies have shown that the induction of an inflammatory response in the developing rodent brain can have dramatic, lasting effects as evidenced by cognitive deficits and behavioral disorders in adulthood (Bilbo, Smith, & Schwarz, 2012; Schwarz & Bilbo, 2012). Immune activation during early development can also alter the immune response later in life (Bilbo et al., 2012; Schwarz & Bilbo, 2012). Furthermore, even transient reduction in the number of microglia can have dramatic impact on the developing brain. For example, genetic deletion of the molecule CX3CR1, which is expressed by microglia, was used to generate a transient loss of microglia in the early postnatal mouse. Transient loss of microglia resulted in deficits in synaptic pruning, leading to weak synaptic transmission and behavioral deficits in adulthood (Zhan et al., 2014). In addition, mice with deletion of CX3CR1 demonstrate impaired hippocampal synaptic plasticity and cognitive function (Rogers et al., 2011). Collectively, these studies indicate that immune activation in the developing brain has dramatic long-term effects on synapse formation and plasticity, cognition, development of behavioral disorders, and altered immune responses. Future studies are needed to determine if ethanol induction of immune activity in the developing brain contributes to the long-term sequelae associated with FASD.
10. LINK BETWEEN ETHANOL AND IMMUNE RESPONSES
10.1. Human studies
The effects of alcohol on peripheral immune responses in humans are well established. For example, serum from alcoholics has been demonstrated to exhibit increased levels of cytokines including IL-1β, IL-6, IL-12, and TNF-α (Achur, Freeman, & Vrana, 2010). Increased cytokine expression in the serum of alcoholics is believed to result in part from release of these molecules into the circulation from the alcohol-damaged liver. In addition, peripheral blood mononuclear cells derived from alcoholics express increased levels of proinflammatory cytokines (Laso, Vaquero, Almeida, Marcos, & Orfao, 2007). Even in healthy individuals, excessive acute or binge alcohol and associated hangover is marked by increased cytokine expression in peripheral blood (Kim et al., 2003). A vicious cycle of alcohol abuse develops since proinflammatory cytokines increase alcohol craving behavior and result in increased consumption (Kiefer, Jahn, Schick, & Wiedemann, 2002).
Relatively little is known regarding the effects of alcohol on the immune response in the human brain. Postmortem evaluation of adult alcoholic brains indicates increased expression of mRNAs encoding immune molecules in the prefrontal cortex of alcoholics relative to nonalcoholics (Liu et al., 2006). The expression of the transcription factor NF-κB, which activates the expression of a wide variety of neuroimmune genes, is also increased in alcoholic brain (Okvist et al., 2007; Yakovleva, Bazov, Watanabe, Hauser, & Bakalkin, 2011). NF-κB also modulates synaptic plasticity suggesting a mechanism by which neuroimmune activation may impair cognition and memory in alcoholics. Contribution of neuroimmune activation to alcohol neuropathology is further suggested through the activity of the proinflammatory chemokine CCL2/MCP-1. CCL2 expression is increased in the hippocampus, amygdala, substantia nigra, and ventral teg-mentum in alcoholics (He & Crews, 2008). Studies in transgenic mice over-expressing CCL2 in the CNS reveal that CCL2 alters synaptic transmission in the hippocampus (Nelson, Hao, Manos, Ransohoff, & Gruol, 2011) suggesting another mechanism by which neuroimmune activation may impair cognition and memory. These studies suggest that neuroimmune activity and proinflammatory molecules may alter synaptic plasticity in alcoholics.
Relatively little is known concerning the effects of alcohol on immune response in the developing human brain. It has been demonstrated that increased expression of proinflammatory cytokines including IL-1β, IL-6, and TNF-α is detected shortly after birth in the blood of neonates and mothers, when the mother chronically consumed alcohol during pregnancy (Ahluwalia et al., 2000). It was not possible in these studies to determine if these cytokines were of neonatal or maternal origin, but it is clear that the neonate is exposed to these proinflammatory molecules. Studies are needed to determine if alcohol alters the expression of proinflammatory molecules in the developing brain.
10.2. Animal studies
Animal models of FASD have played a critical role in defining the mechanisms by which ethanol damages the developing CNS. These models involve prenatal or neonatal exposure to ethanol. Prenatal exposure to ethanol is toxic to neurons in vivo including cerebral cortical, hippocampal, and cerebellar neurons as described above. Ethanol is also toxic to cultured primary neurons. For example, conditioned medium from ethanol-treated microglia was toxic to primary basal hippocampal neurons. Furthermore, TNF-α neurtralizing antibodies blocked ethanol-induced neuronal death (Boyadjieva & Sarkar, 2010). This suggests that ethanol induces the production of TNF-α and possibly other proinflammatory molecules by activated microglia, which contributes to ethanol-induced neuronal cell death.
The neonatal rodent brain is also highly susceptible to the toxic effects of ethanol with neuropathology in the cerebral cortex, cerebellum, corpus callosum, hippocampus, and other regions as detailed above. The relevance of the neonatal rodent is that it is developmentally similar to the brain during the second half to third trimester of pregnancy (Clancy et al., 2001; Dobbing & Sands, 1979). We have demonstrated that ethanol exposure of 3- to 5-day-old neonatal mice results in a significant loss of cerebellar Purkinje neurons (Kane et al., 2011) as has been well documented in a variety of FASD models (Dikranian, Qin, Labruyere, Nemmers, & Olney, 2005; Hamre & West, 1993; Pierce et al., 1999). In addition to neurons, we demonstrated that microglia are highly susceptible to ethanol-induced death. Strikingly, the surviving microglia in ethanol-treated animals exhibit an altered morphology with enlarged soma and shorter, broader, less branched processes characteristic of activated microglia, as opposed to the ramified appearance of normal microglia in vehicle-treated control animals. Our studies in this FASD model further demonstrated that agonists of the anti-inflammatory nuclear receptor PPAR-γ block microglial activation and protect neurons and microglia against ethanol cytotoxicity, suggesting a possible link between ethanol-induced microglial activation and loss of neurons in the developing brain. As discussed previously, microglia perform a variety of functions in the developing brain and their loss or activation during the period of brain development produces persistent neuropathological and negative behavioral consequences. Together, these observations suggest that loss of microglia and microglial activation in response to ethanol in the developing brain may contribute to the long-term behavioral and structural anomalies associated with FASD.
Neonatal exposure to ethanol also increases the expression of cytokines in the brain, which can persist following ethanol withdrawal (Tiwari & Chopra, 2011). In these studies, neonatal rats were exposed to ethanol on postnatal days 7–9, and cytokine expression was evaluated on postnatal day 28. IL-1β, TGF-β, and TNF-α expressions were elevated in the cerebral cortex and hippocampus. The transcription factor NF-κB, which activates the expression of a number of genes that encode proinflammatory molecules, was increased following ethanol exposure in these brain regions, as were markers of oxidative stress including catalase, superoxide dismutase, nitric oxide, glutathione, and lipid peroxidation. Importantly, treatment with the antioxidant resveratrol suppressed the production of oxidative stress in the developing CNS and this resulted in improved cognition in adult animals. Other studies demonstrated that a single dose of ethanol administered at postnatal day 7 resulted in lipid peroxidation and expression of the apoptotic marker caspase-3 in the developing cerebellum (Kumar, Singh, Lavoie, Dipette, & Singh, 2011). In another study, a single dose of ethanol administered at postnatal day 7 resulted in altered morphology and increased Iba-1 staining in microglia, suggesting microglial activation (Saito et al., 2010). Collectively, these studies suggest that microglial activation, neuroinflammation, and oxidative stress contribute to ethanol-induced degeneration in the developing CNS.
As discussed previously, microglia play critical roles in CNS development and activation of immune responses in the developing brain can result in long-term consequences in the mature brain (Schwarz & Bilbo, 2012). These studies suggest that ethanol-induced immune activation in the developing CNS may produce similar long-term consequences. In this regard, prenatal exposure of rats to ethanol resulted in an altered neuroimmune response to trauma in adults (DeVito & Stone, 2001). In these studies, rats exposed to ethanol prenatally were inflicted with a stab wound to the brain as adults in order to produce a neuroimmune response. The expression of the neuroimmune activation markers GFAP, ICAM-1, and TNF-α was reduced in animals exposed to ethanol relative to vehicle-treated controls, indicating that developmental ethanol exposure alters the neuroimmune response in adulthood. In contrast, expression of ED1, which is commonly associated with microglial activation, was increased by trauma in animals exposed to ethanol prenatally, indicating that the alteration of neuroimmune responses in adulthood due to developmental ethanol exposure is complex. Future studies are needed to determine how fetal exposure to ethanol alters immune response and long-term function of the mature brain. These studies will have important implications concerning the contribution of neuroinflammation in the developing brain to the long-term consequences of FASD.
10.3. Signaling pathways
Innate immunity provides the first line of defense against CNS insult. Resident microglia and astrocytes are the principal cells that mediate innate immune responses. These cells react to insults such as pathogens or endogenous danger signals through TLRs present on their surface. Recent studies have begun to investigate the role of TLRs in modulating ethanol activation of neuroimmune responses. Studies suggest that ethanol may cause release of HMGB1 protein, which can serve as an endogenous danger signal to activate TLR signaling and generate neuroimmune activation (Vetreno, Qin, & Crews, 2013; Whitman, Knapp, Werner, Crews, & Breese, 2013; Zou & Crews, 2014). Ethanol can activate the TLR-dependent transcription factors AP1 and NF-κB in astrocytes (Blanco, Valles, Pascual, & Guerri, 2005). Ethanol can also increase the expression of downstream TLR-dependent genes including Cox-2 and iNOS via activation of p38 MAP kinase, ERK1/2, and SAPK/JNK signaling pathways (Blanco et al., 2005). Ethanol was demonstrated to cause IL-1R and TLR4 to move into lipid rafts in astrocytes (Blanco, Perez-Arago, Fernandez-Lizarbe, & Guerri, 2008). IL-1 and TLR4 signaling is important in these processes as demonstrated by the fact that neutralizing antibodies to IL-1R and TLR4 can suppress ethanol induction of these proinflammatory molecules and signaling pathways (Blanco et al., 2005). TLR4 was also shown to be critical in ethanol induction of Cox-2 and Src phosphorylation in astrocytes (Floreani et al., 2010). These observations suggest that ethanol induction of immune responses in the CNS occurs through the movement of these receptors to lipid rafts, resulting in the activation of signaling pathways that ultimately lead to the production of proinflammatory molecules.
The role of TLR4 in modulating ethanol effects on immune responses in the CNS is further supported by studies utilizing TLR4-deficient mice. Ethanol induced the expression of Cox-2 and iNOS and activated p38 MAP kinase, ERK, and JNK signaling pathways in microglia from wild-type but not TLR4-deficient mice. Conditioned medium from microglia treated with ethanol from wild-type but not TLR4-deficient mice caused apoptosis of cortical neurons. These studies indicate that ethanol effects on microglia result in the production of molecules that are indirectly toxic to neurons in a TLR4-dependent manner (Fernandez-Lizarbe, Pascual, & Guerri, 2009). In other studies, ethanol was demonstrated to increase the expression of CD11b by microglia and GFAP by astrocytes in the frontal cortex of wild-type but not TLR4-deficient mice. Furthermore, these studies indicated that ethanol increased the expression of IL-6, TNF-α, NF-κB, and the apoptotic marker caspase-3 in a TLR4-dependent manner (Alfonso-Loeches, Pascual-Lucas, Blanco, Sanchez-Vera, & Guerri, 2010). Collectively, these studies demonstrate that TLR4 plays a critical role in ethanol-induced neuroinflammation and neurodegeneration.
Proinflammatory molecules are known to increase alcohol consumption in rodents (Blednov et al., 2005, 2012). Thus, ethanol induction of neuroinflammation may create a vicious cycle, which supports additional alcohol consumption, abuse, and addiction. As noted previously, TLR4-deficient mice exhibit decreased ethanol-induced neuroinflammation. In addition, these mice consume less alcohol than their wild-type counterparts and exhibit less alcohol-mediated anxiety and cognitive impairment than control animals (Pascual, Balino, Alfonso-Loeches, Aragon, & Guerri, 2011). These studies also indicate that alcohol decreases histone acetylation in the brain in a TLR4-dependent manner. This suggests that ethanol triggers TLR4-dependent epigenetic changes in chromatin configuration, which are linked to downstream effects on neuroinflammation and ultimately to ethanol-induced alterations in cognition and behavior. Further studies indicate that TLR4-deficient mice exhibit less sedation in response to ethanol than wild-type control animals (Wu et al., 2012). This altered behavior was not due to altered alcohol pharmacodynamics, which was similar in wild-type and TLR4-deficient mice. Furthermore, these studies demonstrated that TLR4 siRNA infused into the amygdala inhibited binge drinking behavior in alcohol-preferring rats (Wu et al., 2012). Based on our understanding of the role of GABA in alcohol consumption, it may be significant that GABAA α2 plays a crucial role in TLR4-dependent changes in alcohol consumption (Liu et al., 2011). These studies collectively support a role for TLR4 in modulating ethanol-induced neuroinflammation, alcohol consumption, and alcohol-mediated changes in cognition and behavior.
The studies outlined above clearly demonstrate a role for TLR4 in ethanol-induced neuroinflammation and neurodegeneration. Ligand binding to TLR4 can result in the activation of the MyD88-dependent or alternatively the MyD88-independent signaling pathway. In the MyD88-dependent signaling pathway, TLR4 physically interacts with MyD88, which results in the activation of NF-κB. The MyD88-independent pathway involves interaction of TLR4 with the adaptor protein TRIF. This leads to the activation of NF-κB and IRF-3, both transcription factors that activate the expression of genes encoding proinflammatory molecules. Future studies utilizing MyD88- and TRIF-deficient mice are required to determine if these TLR4-dependent effects occur through MyD88-dependent or -independent mechanisms. In this regard, studies indicate that MyD88-deficient mice exhibit altered ethanol-induced sedation and motor impairment (Wu et al., 2012); this supports a role for MyD88-dependent signaling in modulating ethanol consumption. However, additional studies indicate that ethanol increases the activation of NF-κB and IRF-3 in microglia (Fernandez-Lizarbe et al., 2009), which suggests that MyD88-independent signaling may also mediate effects of ethanol.
As mentioned above, IL-1 signaling is believed to play a significant role in ethanol-induced neuroinflammation, where IL-1β expression is increased by ethanol (Alfonso-Loeches et al., 2010; Crews, Qin, Sheedy, Vetreno, & Zou, 2013; Lippai et al., 2013; Tiwari & Chopra, 2011). Recent studies demonstrate that in addition to IL-1β, caspase-1 activity is induced by ethanol in the adult brain (Lippai et al., 2013). Both IL-1β and caspase-1 are produced as precursor molecules, which must be processed to form mature, active IL-1β and caspase-1. Caspase-1 functions to process pro-IL-1β to mature IL-1β. Inflammasomes are protein complexes which function as a part of the innate immune system and respond to pathogens as well as to endogenous danger signals including those resulting from the toxic effects of ethanol. Caspase-1-activating inflammasomes are the best-characterized inflammasomes and function to process pro-caspase-1 to mature caspase-1. The caspase-1-activating NLRP3 inflammasome is composed of a number of proteins including NLRP3 and the adaptor protein apoptosis-associated speck-like protein (ASC). Ethanol increases caspase-1 and IL-1β expression in adult brain of wild-type but not NLRP3- or ASC-deficient mice (Lippai et al., 2013). These studies suggest that caspase-1-activating inflammasomes may modulate IL-1β-dependent and ethanol-induced neuroinflammation and neurodegeneration. However, future studies are needed to determine the mechanisms by which ethanol-induced IL-1β signaling modulates neuroinflammation and neurodegeneration in the developing and mature brain.
11. THERAPIES
11.1. Early diagnosis of FASD
Early diagnosis is believed to be essential to maximize the potential for therapies to limit the devastating effects of fetal ethanol exposure. Thus, extensive effort has been put forth to identify biomarkers of gestational ethanol exposure (Joya et al., 2012; Memo, Gnoato, Caminiti, Pichini, & Tarani, 2013). Clinical identification of characteristic facial dysmorphologies is valuable, but is most applicable for identification of FASD in children (Jones et al., 2009, 2010). Identification of FASD in younger infants and children is progressing through the use of sophisticated imaging techniques (Coles & Li, 2011; Lebel et al., 2011; Norman et al., 2009). In fact, it is becoming possible to identify fetal ethanol exposure in newborns. Analysis of nonoxidative metabolites of ethanol including fatty acid ethyl esters, ethyl glucuronide, and ethyl sulfate in meconium allows identification of fetal ethanol exposure in newborns (Joya et al., 2012; Memo et al., 2013). It has recently been demonstrated that fetal ethanol exposure can be identified effectively and at low cost using newborn screening for phosphatidyl ethanol in dried blood spots (Bakhireva et al., 2014). In addition, advanced MRI, ultrasound imaging, and functional analysis show promise for prenatal diagnosis of FASD (Bookstein et al., 2005; Hepper, Dornan, & Lynch, 2012; Kfir et al., 2009; Memo et al., 2013). Mouse models of FASD are forecasting even earlier detection of fetal ethanol exposure (O’Leary-Moore et al., 2011; Sudheendran, Bake, Miranda, & Larin, 2013). Together, these findings suggest that it will be realistic to diagnose FASD in neonates and infants in the hospital or clinic in the foreseeable future, opening the possibility that early temporal windows will be available for intervention with therapeutics. It is hoped that early diagnosis of FASD will lead to the treatment of FASD infants in addition, or alternatively, to pregnant women with therapeutics (Kodituwakku & Kodituwakku, 2011; Roach & Anderson, 2008), including those exhibiting anti-inflammatory properties.
11.2. Development of novel anti-inflammatory therapies for FASD
As discussed previously, our work and that of others indicate that ethanol exposure in neonatal rodents results in potent neuroinflammation. Since neuroinflammation is believed to contribute to ethanol-induced neurodegeneration, this suggests that anti-inflammatory agents may be effective in the treatment of FASD. Our discussion will focus on the potential of the following anti-inflammatory agents—pioglitazone, minocycline, and naltrexone—for the treatment of FASD. However, it is recognized that other anti-inflammatory agents could also proved to be effective in the treatment of FASD.
11.2.1 Pioglitazone
Pioglitazone is an FDA-approved pharmaceutical agonist for PPAR-γ, a member of a nuclear receptor family of proteins that modulate neuroinflammatory responses and other neurodegenerative responses. We (Diab et al., 2002; Drew & Chavis, 2001; Storer, Xu, Chavis, & Drew, 2005a, 2005b) and others (Bernardo, Levi, & Minghetti, 2000; Kielian, Bearden, Baldwin, & Esen, 2004; Petrova, Akama, & Van Eldik, 1999) demonstrated that PPAR-γ agonists, including pioglitazone, inhibit activation of microglia. PPAR-γ agonists also inhibit the production of proinflammatory cytokines and chemokines by microglia (Cunard et al., 2002; Lovett-Racke et al., 2004; Xu, Chavis, Racke, & Drew, 2006; Xu & Drew, 2007; Xu, Storer, Chavis, Racke, & Drew, 2005). We and others also demonstrated that PPAR-γ agonists are protective in animal models of many CNS disorders characterized by neuroinflammation and neurodegeneration (Diab et al., 2004; Heneka, Landreth, & Feinstein, 2001; Mandrekar-Colucci, Sauerbeck, Popovich, & McTigue, 2013; Niino et al., 2001). Most recently, we demonstrated that PPAR-γ agonists provide protection for neurons and microglia from ethanol-induced cell death and blocked ethanol-induced activation of microglia in a mouse model of FASD (Kane et al., 2011). We further have demonstrated that pioglitazone suppresses ethanol increases in proinflammatory cytokines and chemokines in neonatal mice P.D. Drew, J.W. Johnson, J.C. Douglas, K.D. Phelan, & C.J.M. Kane (Unpublished observations). Taken together, these studies support the possibility that PPAR-γ agonists may protect microglia and neurons against ethanol neurotoxicity and prevent ethanol induction of inflammatory processes in FASD. Interestingly, PPAR-γ agonists have been demonstrated to suppress alcohol consumption and relapse to alcohol-seeking behavior (Stopponi et al., 2011), which may be related to their anti-inflammatory activity.
11.2.2 Minocycline
Minocycline is a tetracycline derivative antibiotic used in the treatment of bacterial infections. However, it is now clear that minocycline also possesses potent anti-inflammatory activity. In fact, minocycline has been demonstrated to strongly suppress microglial activation in a variety of neuroinflammatory and neurodegenerative disorders (reviewed in Garrido-Mesa, Zarzuelo, & Galvez, 2013). However, the effects of minocycline on ethanol-induced neuroinflammation have only just begun to be evaluated and have not been evaluated in models of FASD. Minocycline was shown to block ethanol-induced microglial activation in an 8-week-old male mice treated intragastrically with 5 g/kg ethanol for 10 days (Qin & Crews, 2012). Interestingly, like PPAR-γ agonists, minocycline and the related antibiotic doxycycline reduced alcohol consumption in rodents (Agrawal, Hewetson, George, Syapin, & Bergeson, 2011; McIver, Muccigrosso, & Haydon, 2012; Wu et al., 2011). Minocycline also increased the motor impairing effects of ethanol, a phenomenon associated with reduced alcohol consumption (McIver et al., 2012; Wu et al., 2011). Further, in our investigations, we discovered that ethanol increases neuroinflammation in adult, but not adolescent mice (Kane et al., 2014). This is interesting in light of the observation that minocycline decreases alcohol consumption in adult but not adolescent mice supporting the concept that minocycline suppresses alcohol consumption by suppressing neuroinflammation (Agrawal et al., 2014).
11.2.3 Naltrexone
Naltrexone is one of the few approved treatments for alcoholism. It is an opioid antagonist with suppressive effects on alcohol consumption that have been thought to be mediated primarily through opioid receptors (Maisel, Blodgett, Wilbourne, Humphreys, & Finney, 2013). However, naltrexone (and its stereoisomer naloxone) also functions as a TLR4 antagonist (Hutchinson et al., 2008), and TLR4 plays a critical role in ethanol-induced neuroinflammation and neurodegeneration as outlined above (Alfonso-Loeches et al., 2010; Blanco et al., 2005, 2008; Fernandez-Lizarbe et al., 2009; Lippai et al., 2013; Pascual et al., 2011; Wu et al., 2012). It is notable then, that in addition to suppressing alcohol consumption, naltrexone can block ethanol-induced microglial activation in adult mice (Qin & Crews, 2012). However, the effects of naltrexone on microglial activation and neuroinflammation in response to ethanol in models of FASD have not been evaluated. The PPAR-γ agonist pioglitazone was demonstrated to potentiate the suppressive effects on naltrexone on alcohol consumption (Stopponi et al., 2013). This suggests that although PPAR-γ agonists and naltrexone are both anti-inflammatory, these agents may act through distinct pathways. Collectively, these studies suggest that a combination of antiinflammatories such as pioglitazone, minocycline, and naltrexone may act cooperatively to suppress ethanol-induced neuroinflammation and neurodegeneration.
12. CONCLUSIONS
FASD is associated with neuropathology and functional deficits that persist throughout life. Animal models are providing not only a foundation for understanding the causes and consequences of FASD but also new knowledge that is leading to understanding the cellular and molecular mechanisms at play, when the fetal brain is exposed to alcohol. Investigation of alcohol’s effect on microglia and neuronal–microglial interactions has led to a model of the novel contributions of microglia to the neuropathology and functional deficits of FASD (Fig. 3.2). This is based on the recent findings in models of FASD that alcohol causes loss of not only neurons but also microglia in the developing brain, depleting the population of protective microglia. Further, alcohol also induces activation of microglia, changing their phenotype to a proinflammatory form that can lead to neuron death. These observations are particularly important given emerging evidence that the loss of microglia in the developing brain is associated with loss of synapses, loss of neuronal plasticity, reduced neurogenesis, cognitive abnormalities, behavioral deficits, and altered immune response in adulthood. These same abnormalities are produced by microglial activation and inflammation in the developing brain. This knowledge elucidates potentially critical consequences of microglial loss, microglial activation, and inflammatory processes during brain development. Thus, alcohol’s effects on microglia during fetal development may contribute to the neuropathological and behavioral abnormalities associated with FASD. The age at which FASD can be diagnosed in the clinic is progressively declining, making therapeutic intervention in infants a realistic possibility in the fore-seeable future. Based on these observations, therapeutic intervention in the ethanol-induced microglial activation and neuroinflammation with anti-inflammatory pharmaceuticals is suggested to be of potential long-term benefit to infants born with FASD. Clearly, this area of research is yielding new understanding of cellular and molecular mechanisms underlying FASD and holds promise for even greater knowledge.
Figure 3.2.
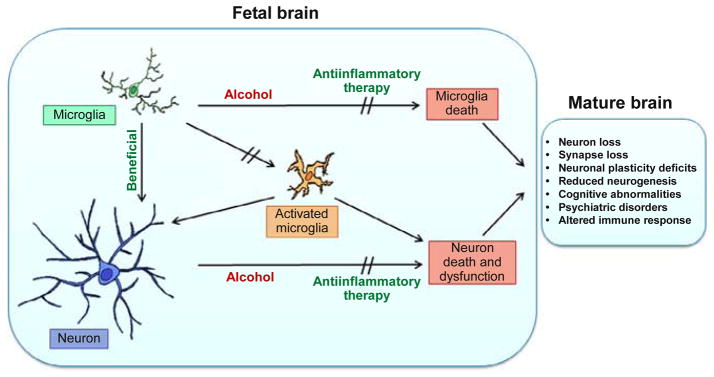
Together, direct toxic effects of alcohol on neurons and postulated indirect effects of alcohol mediated through effects on microglia lead to significant neuronal cell death and dysfunction in the developing brain. Microglia in the unperturbed fetal brain serve beneficial, protective roles for neurons. Alcohol depletes the microglial population in the developing brain through microglia cell loss. In addition, alcohol exposure induces microglial activation and expression of neuroinflammatory molecules in the developing brain. These effects of alcohol on microglia reduce the beneficial activity of microglia exerted during normal development and induce proinflammatory activity of microglia that leads to neurodegenerative processes. The direct consequences of microglial loss and neuroinflammation in the developing brain are now known to generate long-term neuroanatomical and behavioral consequences in the adult brain. These include persistent loss of neurons, loss of synapses, deficits in neuronal plasticity, cognitive abnormalities, psychiatric disorders, and altered reactivity of the immune system. These are consistent with some of the long-term consequences in individuals with FASD. Further, based on our findings, it is suggested that treatment with anti-inflammatory pharmaceuticals will prevent alcohol-induced microglial and neuronal cells’ loss, microglial activation, and neuroinflammation, leading to better long-term outcomes in FASD.
Acknowledgments
This work was supported by NIH Grants AA18834, AA18839, and AA19108.
References
- Abel EL, Sokol RJ. Fetal alcohol syndrome is now leading cause of mental retardation. Lancet. 1986;2(8517):1222. doi: 10.1016/s0140-6736(86)92234-8. [DOI] [PubMed] [Google Scholar]
- Achur RN, Freeman WM, Vrana KE. Circulating cytokines as biomarkers of alcohol abuse and alcoholism. Journal of Neuroimmune Pharmacology. 2010;5(1):83–91. doi: 10.1007/s11481-009-9185-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adnams CM, Kodituwakku PW, Hay A, Molteno CD, Viljoen D, May PA. Patterns of cognitive-motor development in children with fetal alcohol syndrome from a community in South Africa. Alcoholism, Clinical and Experimental Research. 2001;25(4):557–562. [PubMed] [Google Scholar]
- Agrawal RG, Hewetson A, George CM, Syapin PJ, Bergeson SE. Minocycline reduces ethanol drinking. Brain, Behavior, and Immunity. 2011;25(Suppl 1):S165–S169. doi: 10.1016/j.bbi.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal RG, Owen JA, Levin PS, Hewetson A, Berman AE, Franklin SR, et al. Bioinformatics analyses reveal age-specific neuroimmune modulation as a target for treatment of high ethanol drinking. Alcoholism, Clinical and Experimental Research. 2014;38(2):428–437. doi: 10.1111/acer.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahluwalia B, Wesley B, Adeyiga O, Smith DM, Da-Silva A, Rajguru S. Alcohol modulates cytokine secretion and synthesis in human fetus: An in vivo and in vitro study. Alcohol. 2000;21(3):207–213. doi: 10.1016/s0741-8329(00)00076-8. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nature Neuroscience. 2011;14(9):1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nature Neuroscience. 2007;10(12):1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. The Journal of Neuroscience. 2010;30(24):8285–8295. doi: 10.1523/JNEUROSCI.0976-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam A, Abdul-Hamjid M, Allam G, Al-hroob A, Ibraheem G, Alsubaie M. Perinatal ethyl alcohol effects on the development of cerebellar cortex in albino rat. African Journal of Pharmacy and Pharmacology. 2013;7:1293–1301. [Google Scholar]
- American Academy of Pediatrics Committee on Substance Abuse and Committee on Children with Disabilities. Fetal alcohol syndrome and alcohol-related neurodevelopmental disorders. Pediatrics. 2000;106(2 Pt 1):358–361. [PubMed] [Google Scholar]
- Archibald SL, Fennema-Notestine C, Gamst A, Riley EP, Mattson SN, Jernigan TL. Brain dysmorphology in individuals with severe prenatal alcohol exposure. Developmental Medicine and Child Neurology. 2001;43(3):148–154. [PubMed] [Google Scholar]
- Aronne MP, Guadagnoli T, Fontanet P, Evrard SG, Brusco A. Effects of prenatal ethanol exposure on rat brain radial glia and neuroblast migration. Experimental Neurology. 2011;229(2):364–371. doi: 10.1016/j.expneurol.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Astley SJ, Aylward EH, Olson HC, Kerns K, Brooks A, Coggins TE, et al. Magnetic resonance imaging outcomes from a comprehensive magnetic resonance study of children with fetal alcohol spectrum disorders. Alcoholism, Clinical and Experimental Research. 2009;33(10):1671–1689. doi: 10.1111/j.1530-0277.2009.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autti-Ramo I, Autti T, Korkman M, Kettunen S, Salonen O, Valanne L. MRI findings in children with school problems who had been exposed prenatally to alcohol. Developmental Medicine and Child Neurology. 2002;44(2):98–106. doi: 10.1017/s0012162201001748. [DOI] [PubMed] [Google Scholar]
- Bakhireva LN, Leeman L, Savich RD, Cano S, Gutierrez H, Savage DD, et al. The validity of phosphatidylethanol in dried blood spots of newborns for the identification of prenatal alcohol exposure. Alcoholism, Clinical and Experimental Research. 2014;38(4):1078–1085. doi: 10.1111/acer.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuelos C, Gilbert RJ, Montgomery KS, Fincher AS, Wang H, Frye GD, et al. Altered spatial learning and delay discounting in a rat model of human third trimester binge ethanol exposure. Behavioural Pharmacology. 2012;23(1):54–65. doi: 10.1097/FBP.0b013e32834eb07d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr HM, Bookstein FL, O’Malley KD, Connor PD, Huggins JE, Streissguth AP. Binge drinking during pregnancy as a predictor of psychiatric disorders on the structured clinical interview for DSM-IV in young adult offspring. The American Journal of Psychiatry. 2006;163(6):1061–1065. doi: 10.1176/ajp.2006.163.6.1061. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Levi G, Minghetti L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR-gamma) and its natural ligand 15-deoxy-delta12, 14-prostaglandin J2 in the regulation of microglial functions. The European Journal of Neuroscience. 2000;12(7):2215–2223. doi: 10.1046/j.1460-9568.2000.00110.x. [DOI] [PubMed] [Google Scholar]
- Bhatara VS, Lovrein F, Kirkeby J, Swayze V, II, Unruh E, Johnson V. Brain function in fetal alcohol syndrome assessed by single photon emission computed tomography. South Dakota Journal of Medicine. 2002;55(2):59–62. [PubMed] [Google Scholar]
- Bilbo SD, Smith SH, Schwarz JM. A lifespan approach to neuroinflammatory and cognitive disorders: A critical role for glia. Journal of Neuroimmune Pharmacology. 2012;7(1):24–41. doi: 10.1007/s11481-011-9299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkquist OA, Fryer SL, Reiss AL, Mattson SN, Riley EP. Cingulate gyrus morphology in children and adolescents with fetal alcohol spectrum disorders. Psychiatry Research. 2010;181(2):101–107. doi: 10.1016/j.pscychresns.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco AM, Guerri C. Ethanol intake enhances inflammatory mediators in brain: Role of glial cells and TLR4/IL-1RI receptors. Frontiers in Bioscience. 2007;12:2616–2630. doi: 10.2741/2259. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Pascual M, Valles SL, Guerri C. Ethanol-induced iNOS and COX-2 expression in cultured astrocytes via NF-kappa B. Neuroreport. 2004;15(4):681–685. doi: 10.1097/00001756-200403220-00021. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Perez-Arago A, Fernandez-Lizarbe S, Guerri C. Ethanol mimics ligand-mediated activation and endocytosis of IL-1RI/TLR4 receptors via lipid rafts caveolae in astroglial cells. Journal of Neurochemistry. 2008;106(2):625–639. doi: 10.1111/j.1471-4159.2008.05425.x. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Valles SL, Pascual M, Guerri C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. Journal of Immunology. 2005;175(10):6893–6899. doi: 10.4049/jimmunol.175.10.6893. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Bergeson SE, Walker D, Ferreira VM, Kuziel WA, Harris RA. Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behavioural Brain Research. 2005;165(1):110–125. doi: 10.1016/j.bbr.2005.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF, Harris RA. Neuroimmune regulation of alcohol consumption: Behavioral validation of genes obtained from genomic studies. Addiction Biology. 2012;17(1):108–120. doi: 10.1111/j.1369-1600.2010.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthius DJ, Bonthius NE, Napper RM, Astley SJ, Clarren SK, West JR. Purkinje cell deficits in nonhuman primates following weekly exposure to ethanol during gestation. Teratology. 1996;53(4):230–236. doi: 10.1002/(SICI)1096-9926(199604)53:4<230::AID-TERA5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Bookstein FL, Connor PD, Covell KD, Barr HM, Gleason CA, Sze RW, et al. Preliminary evidence that prenatal alcohol damage may be visible in averaged ultrasound images of the neonatal human corpus callosum. Alcohol. 2005;36(3):151–160. doi: 10.1016/j.alcohol.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64(1):93–109. doi: 10.1016/j.neuron.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Boyadjieva NI, Sarkar DK. Role of microglia in ethanol’s apoptotic action on hypothalamic neuronal cells in primary cultures. Alcoholism, Clinical and Experimental Research. 2010;34(11):1835–1842. doi: 10.1111/j.1530-0277.2010.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KL, Calizo LH, Stanton ME. Dose-dependent deficits in dual interstimulus interval classical eyeblink conditioning tasks following neonatal binge alcohol exposure in rats. Alcoholism, Clinical and Experimental Research. 2008;32(2):277–293. doi: 10.1111/j.1530-0277.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Bilousova TV, Puntambekar SS, Melchior B, Doose JM, Ethell IM. A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics. 2007;4(4):571–579. doi: 10.1016/j.nurt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: Hiding in plain sight. Immunological Reviews. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105(1):7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- Clancy B, Finlay BL, Darlington RB, Anand KJ. Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007;28(5):931–937. doi: 10.1016/j.neuro.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CD, Li Z. Functional neuroimaging in the examination of effects of prenatal alcohol exposure. Neuropsychology Review. 2011;21(2):119–132. doi: 10.1007/s11065-011-9165-y. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. Journal of Neuroimmune Pharmacology. 2009;4(4):399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor PD, Sampson PD, Bookstein FL, Barr HM, Streissguth AP. Direct and indirect effects of prenatal alcohol damage on executive function. Developmental Neuropsychology. 2000;18(3):331–354. doi: 10.1207/S1532694204Connor. [DOI] [PubMed] [Google Scholar]
- Connor PD, Sampson PD, Streissguth AP, Bookstein FL, Barr HM. Effects of prenatal alcohol exposure on fine motor coordination and balance: A study of two adult samples. Neuropsychologia. 2006;44(5):744–751. doi: 10.1016/j.neuropsychologia.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21(3):505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- Crews FT, Bechara R, Brown LA, Guidot DM, Mandrekar P, Oak S, et al. Cytokines and alcohol. Alcoholism, Clinical and Experimental Research. 2006;30(4):720–730. doi: 10.1111/j.1530-0277.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Nixon K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol and Alcoholism. 2009;44(2):115–127. doi: 10.1093/alcalc/agn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biological Psychiatry. 2013;73(7):602–612. doi: 10.1016/j.biopsych.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunard R, Ricote M, DiCampli D, Archer DC, Kahn DA, Glass CK, et al. Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. Journal of Immunology. 2002;168(6):2795–2802. doi: 10.4049/jimmunol.168.6.2795. [DOI] [PubMed] [Google Scholar]
- Dalmau I, Finsen B, Zimmer J, Gonzalez B, Castellano B. Development of microglia in the postnatal rat hippocampus. Hippocampus. 1998;8(5):458–474. doi: 10.1002/(SICI)1098-1063(1998)8:5<458::AID-HIPO6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Davis RL, Syapin PJ. Ethanol increases nuclear factor-kappa B activity in human astroglial cells. Neuroscience Letters. 2004;371(2–3):128–132. doi: 10.1016/j.neulet.2004.08.051. [DOI] [PubMed] [Google Scholar]
- De Guio F, Mangin JF, Riviere D, Perrot M, Molteno CD, Jacobson SW, et al. A study of cortical morphology in children with fetal alcohol spectrum disorders. Human Brain Mapping. 2013;35(5):2285–2296. doi: 10.1002/hbm.22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito WJ, Stone S. Prenatal exposure to ethanol alters the neuroimmune response to a central nervous system wound in the adult rat. Alcohol. 2001;25(1):39–47. doi: 10.1016/s0741-8329(01)00161-6. [DOI] [PubMed] [Google Scholar]
- Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, et al. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-delta (12,14)-prostaglandin J2 ameliorates experimental autoimmune encephalomyelitis. Journal of Immunology. 2002;168(5):2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- Diab A, Hussain RZ, Lovett-Racke AE, Chavis JA, Drew PD, Racke MK. Ligands for the peroxisome proliferator-activated receptor-gamma and the retinoid X receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. Journal of Neuroimmunology. 2004;148(1–2):116–126. doi: 10.1016/j.jneuroim.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Diaz MR, Vollmer CC, Zamudio-Bulcock PA, Vollmer W, Blomquist SL, Morton RA, et al. Repeated intermittent alcohol exposure during the third trimester-equivalent increases expression of the GABA(A) receptor delta subunit in cerebellar granule neurons and delays motor development in rats. Neuropharmacology. 2014;79:262–274. doi: 10.1016/j.neuropharm.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikranian K, Qin YQ, Labruyere J, Nemmers B, Olney JW. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Brain Research. Developmental Brain Research. 2005;155(1):1–13. doi: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Diwadkar VA, Meintjes EM, Goradia D, Dodge NC, Warton C, Molteno CD, et al. Differences in cortico-striatal-cerebellar activation during working memory in syndromal and nonsyndromal children with prenatal alcohol exposure. Human Brain Mapping. 2013;34(8):1931–1945. doi: 10.1002/hbm.22042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Human Development. 1979;3(1):79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Drew PD, Chavis JA. The cyclopentone prostaglandin 15-deoxy-delta(12,14) prostaglandin J2 represses nitric oxide, TNF-alpha, and IL-12 production by microglial cells. Journal of Neuroimmunology. 2001;115(1–2):28–35. doi: 10.1016/s0165-5728(01)00267-3. [DOI] [PubMed] [Google Scholar]
- Drew PD, Kane CJM. Neuroimmune mechanisms of glia and their interplay with alcohol exposure across the lifespan. In: Cui C, Grandison L, Noronha A, editors. Neural–immune interactions in brain function and alcohol related disorders. New York, NY: Springer; 2013. pp. 359–386. [Google Scholar]
- Eglitis MA, Mezey E. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):4080–4085. doi: 10.1073/pnas.94.8.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Shawa H, Abbott CW, III, Huffman KJ. Prenatal ethanol exposure disrupts intraneocortical circuitry, cortical gene expression, and behavior in a mouse model of FASD. The Journal of Neuroscience. 2013;33(48):18893–18905. doi: 10.1523/JNEUROSCI.3721-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett JC, Licon-Munoz Y, Valenzuela CF. Effects of third trimester-equivalent ethanol exposure on Cl− co-transporter expression, network activity, and GABAergic transmission in the CA3 hippocampal region of neonatal rats. Alcohol. 2012;46(6):595–601. doi: 10.1016/j.alcohol.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber NB, Creeley CE, Olney JW. Alcohol-induced neuroapoptosis in the fetal macaque brain. Neurobiology of Disease. 2010;40(1):200–206. doi: 10.1016/j.nbd.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fee D, Grzybicki D, Dobbs M, Ihyer S, Clotfelter J, Macvilay S, et al. Interleukin 6 promotes vasculogenesis of murine brain microvessel endothelial cells. Cytokine. 2000;12(6):655–665. doi: 10.1006/cyto.1999.0599. [DOI] [PubMed] [Google Scholar]
- Fellner L, Jellinger KA, Wenning GK, Stefanova N. Glial dysfunction in the pathogenesis of alpha-synucleinopathies: Emerging concepts. Acta Neuropathologica. 2011;121(6):675–693. doi: 10.1007/s00401-011-0833-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Pascual M, Guerri C. Critical role of TLR4 response in the activation of microglia induced by ethanol. Journal of Immunology. 2009;183(7):4733–4744. doi: 10.4049/jimmunol.0803590. [DOI] [PubMed] [Google Scholar]
- Fiske BK, Brunjes PC. Microglial activation in the developing rat olfactory bulb. Neuroscience. 2000;96(4):807–815. doi: 10.1016/s0306-4522(99)00601-6. [DOI] [PubMed] [Google Scholar]
- Floreani NA, Rump TJ, Abdul Muneer PM, Alikunju S, Morsey BM, Brodie MR, et al. Alcohol-induced interactive phosphorylation of SRC and Toll-like receptor regulates the secretion of inflammatory mediators by human astrocytes. Journal of Neuroimmune Pharmacology. 2010;5(4):533–545. doi: 10.1007/s11481-010-9213-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd RL, Weber MK, Denny C, O’Connor MJ. Prevention of fetal alcohol spectrum disorders. Developmental Disabilities Research Reviews. 2009;15(3):193–199. doi: 10.1002/ddrr.75. [DOI] [PubMed] [Google Scholar]
- Fryer SL, McGee CL, Matt GE, Riley EP, Mattson SN. Evaluation of psychopathological conditions in children with heavy prenatal alcohol exposure. Pediatrics. 2007;119(3):e733–e741. doi: 10.1542/peds.2006-1606. [DOI] [PubMed] [Google Scholar]
- Gadient RA, Otten U. Expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat brain during postnatal development. Brain Research. 1994;637(1–2):10–14. doi: 10.1016/0006-8993(94)91211-4. [DOI] [PubMed] [Google Scholar]
- Garrido-Mesa N, Zarzuelo A, Galvez J. Minocycline: Far beyond an antibiotic. British Journal of Pharmacology. 2013;169(2):337–352. doi: 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Mohapel J, Boehme F, Kainer L, Christie BR. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: Insights from different rodent models. Brain Research Reviews. 2010;64(2):283–303. doi: 10.1016/j.brainresrev.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin EA, O’Leary-Moore SK, Khan AA, Parnell SE, Ament JJ, Dehart DB, et al. Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: Effects of acute insult on gestational day 7. Alcoholism, Clinical and Experimental Research. 2010;34(1):98–111. doi: 10.1111/j.1530-0277.2009.01071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlett CR, Eilers AT. Alcohol-induced Purkinje cell loss with a single binge exposure in neonatal rats: A stereological study of temporal windows of vulnerability. Alcoholism, Clinical and Experimental Research. 1997;21(4):738–744. [PubMed] [Google Scholar]
- Goodlett CR, Thomas JD, West JR. Long-term deficits in cerebellar growth and rotarod performance of rats following “binge-like” alcohol exposure during the neonatal brain growth spurt. Neurotoxicology and Teratology. 1991;13(1):69–74. doi: 10.1016/0892-0362(91)90029-v. [DOI] [PubMed] [Google Scholar]
- Goral J, Karavitis J, Kovacs EJ. Exposure-dependent effects of ethanol on the innate immune system. Alcohol. 2008;42(4):237–247. doi: 10.1016/j.alcohol.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JT, Tran T, Steinmetz JE, Goodlett CR. Neonatal ethanol produces cerebellar deep nuclear cell loss and correlated disruption of eyeblink conditioning in adult rats. Brain Research. 2002;956(2):302–311. doi: 10.1016/s0006-8993(02)03561-8. [DOI] [PubMed] [Google Scholar]
- Gressens P, Lammens M, Picard JJ, Evrard P. Ethanol-induced disturbances of gliogenesis and neuronogenesis in the developing murine brain: An in vitro and in vivo immunohistochemical and ultrastructural study. Alcohol and Alcoholism. 1992;27(3):219–226. [PubMed] [Google Scholar]
- Guerri C, Bazinet A, Riley EP. Foetal alcohol spectrum disorders and alterations in brain and behaviour. Alcohol and Alcoholism. 2009;44(2):108–114. doi: 10.1093/alcalc/agn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DA, Akers KG, Rice JP, Johnson TE, Candelaria-Cook FT, Maes LI, et al. Prenatal exposure to moderate levels of ethanol alters social behavior in adult rats: Relationship to structural plasticity and immediate early gene expression in frontal cortex. Behavioural Brain Research. 2010;207(2):290–304. doi: 10.1016/j.bbr.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamre KM, West JR. The effects of the timing of ethanol exposure during the brain growth spurt on the number of cerebellar Purkinje and granule cell nuclear profiles. Alcoholism, Clinical and Experimental Research. 1993;17(3):610–622. doi: 10.1111/j.1530-0277.1993.tb00808.x. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nature Neuroscience. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- He J, Crews FT. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Experimental Neurology. 2008;210(2):349–358. doi: 10.1016/j.expneurol.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Landreth GE, Feinstein DL. Role for peroxisome proliferator-activated receptor-gamma in Alzheimer’s disease. Annals of Neurology. 2001;49(2):276. doi: 10.1002/1531-8249(20010201)49:2<276::aid-ana53>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Hepper PG, Dornan JC, Lynch C. Fetal brain function in response to maternal alcohol consumption: Early evidence of damage. Alcoholism, Clinical and Experimental Research. 2012;36(12):2168–2175. doi: 10.1111/j.1530-0277.2012.01832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF. Migration of hematogenous cells through the blood brain barrier and the initiation of CNS inflammation. Brain Pathology. 1991;1(2):97–105. doi: 10.1111/j.1750-3639.1991.tb00646.x. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239(4837):290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: Involvement of toll-like receptor 4 (TLR4) The European Journal of Neuroscience. 2008;28(1):20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idrus NM, McGough NN, Riley EP, Thomas JD. Administration of memantine during ethanol withdrawal in neonatal rats: Effects on long-term ethanol-induced motor incoordination and cerebellar Purkinje cell loss. Alcoholism, Clinical and Experimental Research. 2011;35(2):355–364. doi: 10.1111/j.1530-0277.2010.01351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idrus NM, McGough NN, Riley EP, Thomas JD. Administration of memantine during withdrawal mitigates overactivity and spatial learning impairments associated with neonatal alcohol exposure in rats. Alcoholism, Clinical and Experimental Research. 2014;38(2):529–537. doi: 10.1111/acer.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieraci A, Herrera DG. Single alcohol exposure in early life damages hippocampal stem/progenitor cells and reduces adult neurogenesis. Neurobiology of Disease. 2007;26(3):597–605. doi: 10.1016/j.nbd.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287(5455):1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Inomata K, Nasu F, Tanaka H. Decreased density of synaptic formation in the frontal cortex of neonatal rats exposed to ethanol in utero. International Journal of Developmental Neuroscience. 1987;5(5–6):455–460. doi: 10.1016/0736-5748(87)90023-2. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Kumada T, Cameron DB, Komuro H. Cerebellar granule cell migration and the effects of alcohol. Developmental Neuroscience. 2008;30(1–3):7–23. doi: 10.1159/000109847. [DOI] [PubMed] [Google Scholar]
- Jones KL, Hoyme HE, Robinson LK, del Campo M, Manning MA, Bakhireva LN, et al. Developmental pathogenesis of short palpebral fissure length in children with fetal alcohol syndrome. Birth Defects Research, Part A: Clinical and Molecular Teratology. 2009;85(8):695–699. doi: 10.1002/bdra.20585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KL, Hoyme HE, Robinson LK, Del Campo M, Manning MA, Prewitt LM, et al. Fetal alcohol spectrum disorders: Extending the range of structural defects. American Journal of Medical Genetics Part A. 2010;152A(11):2731–2735. doi: 10.1002/ajmg.a.33675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joya X, Friguls B, Ortigosa S, Papaseit E, Martinez SE, Manich A, et al. Determination of maternal-fetal biomarkers of prenatal exposure to ethanol: A review. Journal of Pharmaceutical and Biomedical Analysis. 2012;69:209–222. doi: 10.1016/j.jpba.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Kane CJM, Phelan KD, Douglas JC, Wagoner G, Johnson JW, Xu J, et al. Effects of ethanol on immune response in the brain: Region-specific changes in adolescent versus adult mice. Alcoholism, Clinical and Experimental Research. 2014;38(2):384–391. doi: 10.1111/acer.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane CJM, Phelan KD, Han L, Smith RR, Xie J, Douglas JC, et al. Protection of neurons and microglia against ethanol in a mouse model of fetal alcohol spectrum disorders by peroxisome proliferator-activated receptor-gamma agonists. Brain, Behavior, and Immunity. 2011;25(Suppl 1):S137–S145. doi: 10.1016/j.bbi.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly SJ, Pierce DR, West JR. Microencephaly and hyperactivity in adult rats can be induced by neonatal exposure to high blood alcohol concentrations. Experimental Neurology. 1987;96(3):580–593. doi: 10.1016/0014-4886(87)90220-2. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiological Reviews. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kfir M, Yevtushok L, Onishchenko S, Wertelecki W, Bakhireva L, Chambers CD, et al. Can prenatal ultrasound detect the effects of in-utero alcohol exposure? A pilot study. Ultrasound in Obstetrics & Gynecology. 2009;33(6):683–689. doi: 10.1002/uog.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer F, Jahn H, Schick M, Wiedemann K. Alcohol intake, tumour necrosis factor-alpha, leptin and craving: Factors of a possibly vicious circle? Alcohol and Alcoholism. 2002;37(4):401–404. doi: 10.1093/alcalc/37.4.401. [DOI] [PubMed] [Google Scholar]
- Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-alpha play a pivotal role in the host immune response in a mouse model of staphylococcus aureus-induced experimental brain abscess. Journal of Neuropathology and Experimental Neurology. 2004;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- Kim DJ, Kim W, Yoon SJ, Choi BM, Kim JS, Go HJ, et al. Effects of alcohol hangover on cytokine production in healthy subjects. Alcohol. 2003;31(3):167–170. doi: 10.1016/j.alcohol.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Cowell RM, Swain RA, Napper RM, Goodlett CR, Greenough WT. Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats. I. Behavioral results. Brain Research. 1998;800(1):48–61. doi: 10.1016/s0006-8993(98)00495-8. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Helfer JL, Calizo LH, Dong WK, Goodlett CR, Greenough WT. Persistent impairment of hippocampal neurogenesis in young adult rats following early postnatal alcohol exposure. Alcoholism, Clinical and Experimental Research. 2007;31(12):2073–2082. doi: 10.1111/j.1530-0277.2007.00528.x. [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Crews FT. Induction of cyclooxygenase-2 in brain during acute and chronic ethanol treatment and ethanol withdrawal. Alcoholism, Clinical and Experimental Research. 1999;23(4):633–643. [PubMed] [Google Scholar]
- Kodituwakku PW, Kalberg W, May PA. The effects of prenatal alcohol exposure on executive functioning. Alcohol Research & Health. 2001;25(3):192–198. [PMC free article] [PubMed] [Google Scholar]
- Kodituwakku PW, Kodituwakku EL. From research to practice: An integrative framework for the development of interventions for children with fetal alcohol spectrum disorders. Neuropsychology Review. 2011;21(2):204–223. doi: 10.1007/s11065-011-9170-1. [DOI] [PubMed] [Google Scholar]
- Kumar A, Singh CK, Lavoie HA, Dipette DJ, Singh US. Resveratrol restores NRF2 level and prevents ethanol-induced toxic effects in the cerebellum of a rodent model of fetal alcohol spectrum disorders. Molecular Pharmacology. 2011;80(3):446–457. doi: 10.1124/mol.111.071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laso FJ, Vaquero JM, Almeida J, Marcos M, Orfao A. Production of inflammatory cytokines by peripheral blood monocytes in chronic alcoholism: Relationship with ethanol intake and liver disease. Cytometry. Part B, Clinical Cytometry. 2007;72(5):408–415. doi: 10.1002/cyto.b.20169. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48(2):405–415. doi: 10.1016/0306-4522(92)90500-2. [DOI] [PubMed] [Google Scholar]
- Lebel C, Mattson SN, Riley EP, Jones KL, Adnams CM, May PA, et al. A longitudinal study of the long-term consequences of drinking during pregnancy: Heavy in utero alcohol exposure disrupts the normal processes of brain development. The Journal of Neuroscience. 2012;32(44):15243–15251. doi: 10.1523/JNEUROSCI.1161-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, Rasmussen C, Wyper K, Andrew G, Beaulieu C. Brain microstructure is related to math ability in children with fetal alcohol spectrum disorder. Alcoholism, Clinical and Experimental Research. 2010;34(2):354–363. doi: 10.1111/j.1530-0277.2009.01097.x. [DOI] [PubMed] [Google Scholar]
- Lebel C, Roussotte F, Sowell ER. Imaging the impact of prenatal alcohol exposure on the structure of the developing human brain. Neuropsychology Review. 2011;21(2):102–118. doi: 10.1007/s11065-011-9163-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigland LA, Ford MM, Lerch JP, Kroenke CD. The influence of fetal ethanol exposure on subsequent development of the cerebral cortex as revealed by magnetic resonance imaging. Alcoholism, Clinical and Experimental Research. 2013;37(6):924–932. doi: 10.1111/acer.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B, Wellmann KA, Barron S. Agmatine reduces balance deficits in a rat model of third trimester binge-like ethanol exposure. Pharmacology, Biochemistry, and Behavior. 2007;88(1):114–121. doi: 10.1016/j.pbb.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist DH, Sokoloff G, Milner E, Steinmetz JE. Neonatal ethanol exposure results in dose-dependent impairments in the acquisition and timing of the conditioned eyeblink response and altered cerebellar interpositus nucleus and hippocampal CA1 unit activity in adult rats. Alcohol. 2013;47(6):447–457. doi: 10.1016/j.alcohol.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt-Jones EA, et al. Alcohol-induced IL-1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. Journal of Leukocyte Biology. 2013;94(1):171–182. doi: 10.1189/jlb.1212659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lewohl JM, Harris RA, Iyer VR, Dodd PR, Randall PK, et al. Patterns of gene expression in the frontal cortex discriminate alcoholic from non-alcoholic individuals. Neuropsychopharmacology. 2006;31(7):1574–1582. doi: 10.1038/sj.npp.1300947. [DOI] [PubMed] [Google Scholar]
- Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL, Jr, et al. Binge alcohol drinking is associated with GABAa alpha2-regulated toll-like receptor 4 (TLR4) expression in the central amygdala. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(11):4465–4470. doi: 10.1073/pnas.1019020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: Effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicology and Teratology. 2003;25(4):447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Lovett-Racke AE, Hussain RZ, Northrop S, Choy J, Rocchini A, Matthes L, et al. Peroxisome proliferator-activated receptor alpha agonists as therapy for autoimmune disease. Journal of Immunology. 2004;172(9):5790–5798. doi: 10.4049/jimmunol.172.9.5790. [DOI] [PubMed] [Google Scholar]
- Lupton C, Burd L, Harwood R. Cost of fetal alcohol spectrum disorders. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 2004;127C(1):42–50. doi: 10.1002/ajmg.c.30015. [DOI] [PubMed] [Google Scholar]
- Maisel NC, Blodgett JC, Wilbourne PL, Humphreys K, Finney JW. Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: When are these medications most helpful? Addiction. 2013;108(2):275–293. doi: 10.1111/j.1360-0443.2012.04054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Sauerbeck A, Popovich PG, McTigue DM. PPAR agonists as therapeutics for CNS trauma and neurological diseases. ASN Neuro. 2013;5(5):e00129. doi: 10.1042/AN20130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson SN, Riley EP. A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcoholism, Clinical and Experimental Research. 1998;22(2):279–294. doi: 10.1111/j.1530-0277.1998.tb03651.x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Roebuck TM. Acquisition and retention of verbal and nonverbal information in children with heavy prenatal alcohol exposure. Alcoholism, Clinical and Experimental Research. 2002;26(6):875–882. [PubMed] [Google Scholar]
- Mattson SN, Roesch SC, Glass L, Deweese BN, Coles CD, Kable JA, et al. Further development of a neurobehavioral profile of fetal alcohol spectrum disorders. Alcoholism, Clinical and Experimental Research. 2013;37(3):517–528. doi: 10.1111/j.1530-0277.2012.01952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, Manning M, et al. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Developmental Disabilities Research Reviews. 2009;15(3):176–192. doi: 10.1002/ddrr.68. [DOI] [PubMed] [Google Scholar]
- McIver SR, Muccigrosso MM, Haydon PG. The effect of doxycycline on alcohol consumption and sensitivity: Consideration for inducible transgenic mouse models. Experimental Biology and Medicine (Maywood, NJ) 2012;237(10):1129–1133. doi: 10.1258/ebm.2012.012029. [DOI] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. The EMBO Journal. 1996;15(20):5647–5658. [PMC free article] [PubMed] [Google Scholar]
- Memo L, Gnoato E, Caminiti S, Pichini S, Tarani L. Fetal alcohol spectrum disorders and fetal alcohol syndrome: The state of the art and new diagnostic tools. Early Human Development. 2013;89(Suppl 1):S40–S43. doi: 10.1016/S0378-3782(13)70013-6. [DOI] [PubMed] [Google Scholar]
- Merrill JE. Tumor necrosis factor alpha, interleukin 1 and related cytokines in brain development: Normal and pathological. Developmental Neuroscience. 1992;14(1):1–10. doi: 10.1159/000111642. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, et al. Microglia in the adult brain arise from LY-6CHICCR2 + monocytes only under defined host conditions. Nature Neuroscience. 2007;10(12):1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Miller MW. Migration of cortical neurons is altered by gestational exposure to ethanol. Alcoholism, Clinical and Experimental Research. 1993;17(2):304–314. doi: 10.1111/j.1530-0277.1993.tb00768.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effect of prenatal exposure to ethanol on glutamate and GABA immunoreactivity in macaque somatosensory and motor cortices: Critical timing of exposure. Neuroscience. 2006;138(1):97–107. doi: 10.1016/j.neuroscience.2005.10.060. [DOI] [PubMed] [Google Scholar]
- Miller MW. Exposure to ethanol during gastrulation alters somatosensory-motor cortices and the underlying white matter in the macaque. Cerebral Cortex. 2007;17(12):2961–2971. doi: 10.1093/cercor/bhm024. [DOI] [PubMed] [Google Scholar]
- Miller MW, Potempa G. Numbers of neurons and glia in mature rat somatosensory cortex: Effects of prenatal exposure to ethanol. The Journal of Comparative Neurology. 1990;293(1):92–102. doi: 10.1002/cne.902930108. [DOI] [PubMed] [Google Scholar]
- Miller MW, Robertson S. Prenatal exposure to ethanol alters the postnatal development and transformation of radial glia to astrocytes in the cortex. The Journal of Comparative Neurology. 1993;337(2):253–266. doi: 10.1002/cne.903370206. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Miller MW. Episodic exposure to ethanol during development differentially affects brainstem nuclei in the macaque. Journal of Neurocytology. 2001;30(12):973–982. doi: 10.1023/a:1021832522701. [DOI] [PubMed] [Google Scholar]
- Mooney SM, Miller MW. Vulnerability of macaque cranial nerve neurons to ethanol is time- and site-dependent. Alcohol. 2009;43(4):323–331. doi: 10.1016/j.alcohol.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murawski NJ, Jablonski SA, Brown KL, Stanton ME. Effects of neonatal alcohol dose and exposure window on long delay and trace eyeblink conditioning in juvenile rats. Behavioural Brain Research. 2013;236(1):307–318. doi: 10.1016/j.bbr.2012.08.025. [DOI] [PubMed] [Google Scholar]
- Napper RM, West JR. Permanent neuronal cell loss in the cerebellum of rats exposed to continuous low blood alcohol levels during the brain growth spurt: A stereological investigation. The Journal of Comparative Neurology. 1995;362(2):283–292. doi: 10.1002/cne.903620210. [DOI] [PubMed] [Google Scholar]
- Nardelli A, Lebel C, Rasmussen C, Andrew G, Beaulieu C. Extensive deep gray matter volume reductions in children and adolescents with fetal alcohol spectrum disorders. Alcoholism, Clinical and Experimental Research. 2011;35(8):1404–1417. doi: 10.1111/j.1530-0277.2011.01476.x. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Hao C, Manos J, Ransohoff RM, Gruol DL. Altered hippocampal synaptic transmission in transgenic mice with astrocyte-targeted enhanced CCL2 expression. Brain, Behavior, and Immunity. 2011;25(Suppl 1):S106–S119. doi: 10.1016/j.bbi.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A, et al. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. Journal of Neuroimmunology. 2001;116(1):40–48. doi: 10.1016/s0165-5728(01)00285-5. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Norman AL, Crocker N, Mattson SN, Riley EP. Neuroimaging and fetal alcohol spectrum disorders. Developmental Disabilities Research Reviews. 2009;15(3):209–217. doi: 10.1002/ddrr.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman AL, O’Brien JW, Spadoni AD, Tapert SF, Jones KL, Riley EP, et al. A functional magnetic resonance imaging study of spatial working memory in children with prenatal alcohol exposure: Contribution of familial history of alcohol use disorders. Alcoholism, Clinical and Experimental Research. 2013;37(1):132–140. doi: 10.1111/j.1530-0277.2012.01880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare ED, Kan E, Yoshii J, Mattson SN, Riley EP, Thompson PM, et al. Mapping cerebellar vermal morphology and cognitive correlates in prenatal alcohol exposure. Neuroreport. 2005;16(12):1285–1290. doi: 10.1097/01.wnr.0000176515.11723.a2. [DOI] [PubMed] [Google Scholar]
- Okvist A, Johansson S, Kuzmin A, Bazov I, Merino-Martinez R, Ponomarev I, et al. Neuroadaptations in human chronic alcoholics: Dysregulation of the NF-kappaB system. PLoS One. 2007;2(9):e930. doi: 10.1371/journal.pone.0000930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary-Moore SK, Parnell SE, Lipinski RJ, Sulik KK. Magnetic resonance-based imaging in animal models of fetal alcohol spectrum disorder. Neuropsychology Review. 2011;21(2):167–185. doi: 10.1007/s11065-011-9164-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson HC, Feldman JJ, Streissguth AP, Sampson PD, Bookstein FL. Neuropsychological deficits in adolescents with fetal alcohol syndrome: Clinical findings. Alcoholism, Clinical and Experimental Research. 1998;22(9):1998–2012. [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Parnell SE, Holloway HT, O’Leary-Moore SK, Dehart DB, Paniaqua B, Oguz I, et al. Magnetic resonance microscopy-based analyses of the neuroanatomical effects of gestational day 9 ethanol exposure in mice. Neurotoxicology and Teratology. 2013;39:77–83. doi: 10.1016/j.ntt.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell SE, O’Leary-Moore SK, Godin EA, Dehart DB, Johnson BW, Allan Johnson G, et al. Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: Effects of acute insult on gestational day 8. Alcoholism, Clinical and Experimental Research. 2009;33(6):1001–1011. doi: 10.1111/j.1530-0277.2009.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Balino P, Alfonso-Loeches S, Aragon CM, Guerri C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain, Behavior, and Immunity. 2011;25(Suppl 1):S80–S91. doi: 10.1016/j.bbi.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nature Reviews Immunology. 2007;7(2):161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Perry VH, Hume DA, Gordon S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience. 1985;15(2):313–326. doi: 10.1016/0306-4522(85)90215-5. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: Down-regulation of inducible nitric-oxide synthase by 15-deoxy-delta12,14-prostaglandin J2. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(8):4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce DR, Kane CJM, Serbus DC, Light KE. Microencephaly and selective decreases in cerebellar Purkinje cell numbers following combined exposure to ethanol and methadone during rat brain development. Developmental Neuroscience. 1997;19(5):438–445. doi: 10.1159/000111241. [DOI] [PubMed] [Google Scholar]
- Pierce DR, Serbus DC, Light KE. Intragastric intubation of alcohol during postnatal development of rats results in selective cell loss in the cerebellum. Alcoholism, Clinical and Experimental Research. 1993;17(6):1275–1280. doi: 10.1111/j.1530-0277.1993.tb05241.x. [DOI] [PubMed] [Google Scholar]
- Pierce DR, Williams DK, Light KE. Purkinje cell vulnerability to developmental ethanol exposure in the rat cerebellum. Alcoholism, Clinical and Experimental Research. 1999;23(10):1650–1659. [PubMed] [Google Scholar]
- Priller J, Flugel A, Wehner T, Boentert M, Haas CA, Prinz M, et al. Targeting gene-modified hematopoietic cells to the central nervous system: Use of green fluorescent protein uncovers microglial engraftment. Nature Medicine. 2001;7(12):1356–1361. doi: 10.1038/nm1201-1356. [DOI] [PubMed] [Google Scholar]
- Qin L, Crews FT. Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. Journal of Neuroinflammation. 2012;9:130. doi: 10.1186/1742-2094-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: Unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nature Reviews Neurology. 2011;7(3):137–152. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EP, Infante MA, Warren KR. Fetal alcohol spectrum disorders: An overview. Neuropsychology Review. 2011;21(2):73–80. doi: 10.1007/s11065-011-9166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EP, McGee CL. Fetal alcohol spectrum disorders: An overview with emphasis on changes in brain and behavior. Experimental Biology and Medicine (Maywood, NJ) 2005;230(6):357–365. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- Riley EP, McGee CL, Sowell ER. Teratogenic effects of alcohol: A decade of brain imaging. American Journal of Medical Genetics Part C : Seminars in Medical Genetics. 2004;127C(1):35–41. doi: 10.1002/ajmg.c.30014. [DOI] [PubMed] [Google Scholar]
- Rivest S. Regulation of innate immune responses in the brain. Nature Reviews Immunology. 2009;9(6):429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- Roach D, Anderson SM. Interagency coordinating committee on fetal alcohol syndrome work group on women, drinking, and pregnancy. In: Roach D, Anderson SM, editors. Preventing alcohol tobacco, and other substance-exposed pregnancies: A community affair. Rockville MD: NIAAA NIH American Legacy Foundation; 2008. pp. 1–62. [Google Scholar]
- Roebuck TM, Simmons RW, Mattson SN, Riley EP. Prenatal exposure to alcohol affects the ability to maintain postural balance. Alcoholism, Clinical and Experimental Research. 1998;22(1):252–258. [PubMed] [Google Scholar]
- Roebuck-Spencer TM, Mattson SN, Marion SD, Brown WS, Riley EP. Bimanual coordination in alcohol-exposed children: Role of the corpus callosum. Journal of the International Neuropsychological Society. 2004;10(4):536–548. doi: 10.1017/S1355617704104116. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. The Journal of Neuroscience. 2011;31(45):16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nature Reviews Immunology. 2011;11(11):775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- Saito M, Chakraborty G, Mao RF, Paik SM, Vadasz C, Saito M. Tau phosphorylation and cleavage in ethanol-induced neurodegeneration in the developing mouse brain. Neurochemical Research. 2010;35(4):651–659. doi: 10.1007/s11064-009-0116-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata-Haga H, Sawada K, Ohta K, Cui C, Hisano S, Fukui Y. Adverse effects of maternal ethanol consumption on development of dorsal hippocampus in rat offspring. Acta Neuropathologica. 2003;105(1):30–36. doi: 10.1007/s00401-002-0606-9. [DOI] [PubMed] [Google Scholar]
- Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, et al. Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology. 1997;56(5):317–326. doi: 10.1002/(SICI)1096-9926(199711)56:5<317::AID-TERA5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Santhanam P, Li Z, Hu X, Lynch ME, Coles CD. Effects of prenatal alcohol exposure on brain activation during an arithmetic task: An fMRI study. Alcoholism, Clinical and Experimental Research. 2009;33(11):1901–1908. doi: 10.1111/j.1530-0277.2009.01028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: Is the commitment reversible? Trends in Neurosciences. 2006;29(2):68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD. The immune system and the developing brain. In: McCarthy MM, editor. Colloquium series on the developing brain. San Rafael, CA: Morgan & Claypool Life Sciences; 2012. pp. 1–118. [Google Scholar]
- Servais L, Hourez R, Bearzatto B, Gall D, Schiffmann SN, Cheron G. Purkinje cell dysfunction and alteration of long-term synaptic plasticity in fetal alcohol syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(23):9858–9863. doi: 10.1073/pnas.0607037104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawecki CJ, Thomas JD, Riley EP, Ehlers CL. Neurophysiologic consequences of neonatal ethanol exposure in the rat. Alcohol. 2004;34(2–3):187–196. doi: 10.1016/j.alcohol.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Smith DE, Davies DL. Effect of perinatal administration of ethanol on the CA1 pyramidal cell of the hippocampus and Purkinje cell of the cerebellum: An ultrastructural survey. Journal of Neurocytology. 1990;19(5):708–717. doi: 10.1007/BF01188039. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Jernigan TL, Mattson SN, Riley EP, Sobel DF, Jones KL. Abnormal development of the cerebellar vermis in children prenatally exposed to alcohol: Size reduction in lobules I V. Alcoholism, Clinical and Experimental Research. 1996;20(1):31–34. doi: 10.1111/j.1530-0277.1996.tb01039.x. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Mattson SN, Kan E, Thompson PM, Riley EP, Toga AW. Abnormal cortical thickness and brain-behavior correlation patterns in individuals with heavy prenatal alcohol exposure. Cerebral Cortex. 2008;18(1):136–144. doi: 10.1093/cercor/bhm039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell ER, Mattson SN, Thompson PM, Jernigan TL, Riley EP, Toga AW. Mapping callosal morphology and cognitive correlates: Effects of heavy prenatal alcohol exposure. Neurology. 2001;57(2):235–244. doi: 10.1212/wnl.57.2.235. [DOI] [PubMed] [Google Scholar]
- Spottiswoode BS, Meintjes EM, Anderson AW, Molteno CD, Stanton ME, Dodge NC, et al. Diffusion tensor imaging of the cerebellum and eyeblink conditioning in fetal alcohol spectrum disorder. Alcoholism, Clinical and Experimental Research. 2011;35(12):2174–2183. doi: 10.1111/j.1530-0277.2011.01566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, Berg D, Bloom FE, du Lac S, Ghosh A, Spitzer NC. Fundamental neuroscience. Oxford, UK: Elsevier, Inc; 2013. pp. 1–1426. [Google Scholar]
- Stopponi S, de Guglielmo G, Somaini L, Cippitelli A, Cannella N, Kallupi M, et al. Activation of PPARgamma by pioglitazone potentiates the effects of naltrexone on alcohol drinking and relapse in MSP rats. Alcoholism, Clinical and Experimental Research. 2013;37(8):1351–1360. doi: 10.1111/acer.12091. [DOI] [PubMed] [Google Scholar]
- Stopponi S, Somaini L, Cippitelli A, Cannella N, Braconi S, Kallupi M, et al. Activation of nuclear PPARgamma receptors by the antidiabetic agent pioglitazone suppresses alcohol drinking and relapse to alcohol seeking. Biological Psychiatry. 2011;69(7):642–649. doi: 10.1016/j.biopsych.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Storer PD, Xu J, Chavis JA, Drew PD. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: Implications for multiple sclerosis. Journal of Neuroimmunology. 2005a;161(1–2):113–122. doi: 10.1016/j.jneuroim.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Storer PD, Xu J, Chavis JA, Drew PD. Cyclopentenone prostaglandins PGA2 and 15-deoxy-delta12,14 PGJ2 suppress activation of murine microglia and astrocytes: Implications for multiple sclerosis. Journal of Neuroscience Research. 2005b;80(1):66–74. doi: 10.1002/jnr.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streissguth AP, Bookstein FL, Barr HM, Sampson PD, O’Malley K, Young JK. Risk factors for adverse life outcomes in fetal alcohol syndrome and fetal alcohol effects. Journal of Developmental and Behavioral Pediatrics. 2004;25(4):228–238. doi: 10.1097/00004703-200408000-00002. [DOI] [PubMed] [Google Scholar]
- Streissguth AP, Landesman-Dwyer S, Martin JC, Smith DW. Teratogenic effects of alcohol in humans and laboratory animals. Science. 1980;209(4454):353–361. doi: 10.1126/science.6992275. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Progress in Neurobiology. 1999;57(6):563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Sudheendran N, Bake S, Miranda RC, Larin KV. Comparative assessments of the effects of alcohol exposure on fetal brain development using optical coherence tomography and ultrasound imaging. Journal of Biomedical Optics. 2013;18(2):20506. doi: 10.1117/1.JBO.18.2.020506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttie M, Foroud T, Wetherill L, Jacobson JL, Molteno CD, Meintjes EM, et al. Facial dysmorphism across the fetal alcohol spectrum. Pediatrics. 2013;131(3):e779–e788. doi: 10.1542/peds.2012-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Idrus NM, Monk BR, Dominguez HD. Prenatal choline supplementation mitigates behavioral alterations associated with prenatal alcohol exposure in rats. Birth Defects Research, Part A: Clinical and Molecular Teratology. 2010;88(10):827–837. doi: 10.1002/bdra.20713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V, Chopra K. Resveratrol prevents alcohol-induced cognitive deficits and brain damage by blocking inflammatory signaling and cell death cascade in neonatal rat brain. Journal of Neurochemistry. 2011;117(4):678–690. doi: 10.1111/j.1471-4159.2011.07236.x. [DOI] [PubMed] [Google Scholar]
- Tran TD, Kelly SJ. Critical periods for ethanol-induced cell loss in the hippocampal formation. Neurotoxicology and Teratology. 2003;25(5):519–528. doi: 10.1016/s0892-0362(03)00074-6. [DOI] [PubMed] [Google Scholar]
- Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biology. 2010;8(11):e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uban KA, Sliwowska JH, Lieblich S, Ellis LA, Yu WK, Weinberg J, et al. Prenatal alcohol exposure reduces the proportion of newly produced neurons and glia in the dentate gyrus of the hippocampus in female rats. Hormones and Behavior. 2010;58(5):835–843. doi: 10.1016/j.yhbeh.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela CF, Lindquist B, Zamudio-Bulcock PA. A review of synaptic plasticity at Purkinje neurons with a focus on ethanol-induced cerebellar dysfunction. International Review of Neurobiology. 2010;91:339–372. doi: 10.1016/S0074-7742(10)91011-8. [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Qin L, Crews FT. Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiology of Disease. 2013;59:52–62. doi: 10.1016/j.nbd.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JL, Klintsova AY, Greenough WT, Goodlett CR. Rehabilitation training using complex motor learning rescues deficits in eyeblink classical conditioning in female rats induced by binge-like neonatal alcohol exposure. Alcoholism, Clinical and Experimental Research. 2013;37(9):1561–1570. doi: 10.1111/acer.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JL, Zhou FC, Goodlett CR. Effects of one- and three-day binge alcohol exposure in neonatal C57BL/6 mice on spatial learning and memory in adolescence and adulthood. Alcohol. 2014;48(2):99–111. doi: 10.1016/j.alcohol.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. The Journal of Neuroscience. 2009;29(13):3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RJ, Zhang Y, Crichton RR, Piret B, Piette J, de Witte P. Identification of the nuclear transcription factor NFkappaB in rat after in vivo ethanol administration. FEBS Letters. 1996;389(2):119–122. doi: 10.1016/0014-5793(96)00545-5. [DOI] [PubMed] [Google Scholar]
- Ware AL, Glass L, Crocker N, Deweese BN, Coles CD, Kable JA, et al. Effects of prenatal alcohol exposure and attention-deficit/hyperactivity disorder on adaptive functioning. Alcoholism, Clinical and Experimental Research. 2014;38(5):1439–1447. doi: 10.1111/acer.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JR, Kelly SJ, Pierce DR. Severity of alcohol-induced deficits in rats during the third trimester equivalent is determined by the pattern of exposure. Alcohol and Alcoholism. Supplement. 1987;1:461–465. [PubMed] [Google Scholar]
- Whitcher LT, Klintsova AY. Postnatal binge-like alcohol exposure reduces spine density without affecting dendritic morphology in rat MPFC. Synapse. 2008;62(8):566–573. doi: 10.1002/syn.20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman BA, Knapp DJ, Werner DF, Crews FT, Breese GR. The cytokine mRNA increase induced by withdrawal from chronic ethanol in the sterile environment of brain is mediated by CRF and HMGB1 release. Alcoholism, Clinical and Experimental Research. 2013;37(12):2086–2097. doi: 10.1111/acer.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA, Peterson J, Basavaraj BS, Saito M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcoholism, Clinical and Experimental Research. 2011;35(11):1974–1984. doi: 10.1111/j.1530-0277.2011.01549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Mueller BA, Bell CJ, Muetzel RL, Hoecker HL, Boys CJ, et al. Global functional connectivity abnormalities in children with fetal alcohol spectrum disorders. Alcoholism, Clinical and Experimental Research. 2013;37(5):748–756. doi: 10.1111/acer.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Mueller BA, Muetzel RL, Bell CJ, Hoecker HL, Nelson ML, et al. Inter-hemispheric functional connectivity disruption in children with prenatal alcohol exposure. Alcoholism, Clinical and Experimental Research. 2011;35(5):849–861. doi: 10.1111/j.1530-0277.2010.01415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Muetzel RL. What does diffusion tensor imaging reveal about the brain and cognition in fetal alcohol spectrum disorders? Neuropsychology Review. 2011;21(2):133–147. doi: 10.1007/s11065-011-9162-1. [DOI] [PubMed] [Google Scholar]
- Wozniak JR, Muetzel RL, Mueller BA, McGee CL, Freerks MA, Ward EE, et al. Microstructural corpus callosum anomalies in children with prenatal alcohol exposure: An extension of previous diffusion tensor imaging findings. Alcoholism, Clinical and Experimental Research. 2009;33(10):1825–1835. doi: 10.1111/j.1530-0277.2009.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Coller JK, Rice KC, et al. Inhibiting the TLR4-MYD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. British Journal of Pharmacology. 2012;165(5):1319–1329. doi: 10.1111/j.1476-5381.2011.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Robertson SA, Coller JK, et al. Attenuation of microglial and IL-1 signaling protects mice from acute alcohol-induced sedation and/or motor impairment. Brain, Behavior, and Immunity. 2011;25(Suppl 1):S155–S164. doi: 10.1016/j.bbi.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Xu J, Chavis JA, Racke MK, Drew PD. Peroxisome proliferator-activated receptor-alpha and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. Journal of Neuroimmunology. 2006;176(1–2):95–105. doi: 10.1016/j.jneuroim.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Xu J, Drew PD. Peroxisome proliferator-activated receptor-gamma agonists suppress the production of IL-12 family cytokines by activated glia. Journal of Immunology. 2007;178(3):1904–1913. doi: 10.4049/jimmunol.178.3.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Storer PD, Chavis JA, Racke MK, Drew PD. Agonists for the peroxisome proliferator-activated receptor-alpha and the retinoid X receptor inhibit inflammatory responses of microglia. Journal of Neuroscience Research. 2005;81(3):403–411. doi: 10.1002/jnr.20518. [DOI] [PubMed] [Google Scholar]
- Yakovleva T, Bazov I, Watanabe H, Hauser KF, Bakalkin G. Transcriptional control of maladaptive and protective responses in alcoholics: A role of the NF-kappaB system. Brain, Behavior, and Immunity. 2011;25(Suppl 1):S29–S38. doi: 10.1016/j.bbi.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Phillips OR, Kan E, Sulik KK, Mattson SN, Riley EP, et al. Callosal thickness reductions relate to facial dysmorphology in fetal alcohol spectrum disorders. Alcoholism, Clinical and Experimental Research. 2012;36(5):798–806. doi: 10.1111/j.1530-0277.2011.01679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nature Neuroscience. 2014;17(3):400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- Zhou D, Lebel C, Lepage C, Rasmussen C, Evans A, Wyper K, et al. Developmental cortical thinning in fetal alcohol spectrum disorders. NeuroImage. 2011;58(1):16–25. doi: 10.1016/j.neuroimage.2011.06.026. [DOI] [PubMed] [Google Scholar]
- Zink M, Ferbert T, Frank ST, Seufert P, Gebicke-Haerter PJ, Spanagel R. Perinatal exposure to alcohol disturbs spatial learning and glutamate transmission-related gene expression in the adult hippocampus. The European Journal of Neuroscience. 2011;34(3):457–468. doi: 10.1111/j.1460-9568.2011.07776.x. [DOI] [PubMed] [Google Scholar]
- Zou JY, Crews FT. CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cellular and Molecular Neurobiology. 2006;26(4–6):385–405. doi: 10.1007/s10571-006-9045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JY, Crews FT. Induction of innate immune gene expression cascades in brain slice cultures by ethanol: Key role of NF-kappaB and proinflammatory cytokines. Alcoholism, Clinical and Experimental Research. 2010;34(5):777–789. doi: 10.1111/j.1530-0277.2010.01150.x. [DOI] [PubMed] [Google Scholar]
- Zou JY, Crews FT. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS One. 2014;9(2):e87915. doi: 10.1371/journal.pone.0087915. [DOI] [PMC free article] [PubMed] [Google Scholar]