

Abstract
Multidrug regimens can sometimes treat recalcitrant diseases when single-drug therapies fail. Recapitulating complex multidrug administration from controlled release films for localized delivery remains challenging because their release kinetics are frequently intertwined and an initial burst release of each drug is usually uncontrollable. Herein we demonstrate kinetic control over protein release by crosslinking Layer-by-Layer films during the assembly process. We used biodegradable and naturally derived components and relied on copper-free click chemistry for bioorthogonal covalent crosslinks throughout the film that entrap, but do not modify the embedded protein. We found that this strategy restricted the interdiffusion of protein while maintaining its activity. By depositing a barrier layer and a second protein-containing layer atop this construct, we generated well-defined sequential protein release with minimal overlap that follows their spatial distribution within the film.
Keywords: staged release, drug delivery, polyelectrolyte multilayers, controlled release, click chemistry
The development of chemical and biological therapeutics has profoundly improved the lifestyles and life expectancies of people worldwide, but single-drug treatments can sometimes be ineffective for especially recalcitrant diseases that have developed drug resistances or have temporal progression through different phases. For these cases, combination therapies with spatiotemporally optimized multi-drug regimens can profoundly improve biological effect. In fact, the sequential treatments of erlotinib prior to doxorubicin[1], siRNA followed by a small molecule[2], and antibiotics in sequence[3] have shown significant improvements over simultaneous administration..
Spatiotemporal treatment is especially important during the administration of growth factors[4]. In the complex and multimodal process of wound healing, the judicious introduction of factors in a specific sequence can help drive the wound through the different phases of proper remediation[5]. Studies have shown that the simultaneous introduction of multiple factors can be ineffective or even inhibitory[6], whereas temporally discrete, sequential administration can markedly improve results[6i, 6j, 7]. In addition, sustained growth factor administration is essential to improving biological response because of rapid elimination; basic fibroblast growth factor and vascular endothelial growth factor have half-lives of 3 min[8] and 50 min[9], respectively. As growth factors and other signaling molecules can elicit a variety of responses, their indiscriminate systemic or bolus application can be deleterious. For these reasons, among others, it is desirable to deliver such drugs from an implant or scaffold located in close proximity to the target site with pre-programmed release kinetics, thus minimizing the concentration-related side effects typically associated with systemic delivery and eliminating the need for additional, potentially invasive procedures to administer more drug, which would likely improve patient compliance and therapeutic outcome[10].
Recapitulating a multi-drug dosing regimen with a biodegradable, controlled release formulation remains a challenge, as drug release kinetics frequently have significant overlap, especially during the early phases of release. Some approaches have utilized combinations of different hydrophobic polyesters (e.g., poly[lactic-co-glycolic acid] or PLGA, poly[ε-caprolactone], and poly[3-hydroxybutyrate-co-3-hydroxyvalerate]) in strategic arrangements[11], as well as their combinations with hydrogels[12]. Others have simply used scaffolds based on modified alginate[13] or gelatin[6h, 14] to manipulate release kinetics. The most common outcome is the acceleration or deceleration in drug elution, but it still remains difficult to achieve well defined multi-therapeutic release kinetics without some level of simultaneous release, often with an initial burst release. For the release of growth factors, biologic drugs, and more broadly, any synergistic therapeutic systems that require complex time dependent release, we sought to design ultrathin film coatings that could exhibit truly staggered and sustained release profiles for multiple therapeutics, as well as the ability to control loading.
Using the Layer-by-Layer (LbL) assembly approach, we and several others have shown the capability of incorporating high loadings of growth factors into thin films with controlled release and unique biological effect[15]. This is a desirable approach because films can be assembled from benign aqueous solutions with minimal risk of inactivating sensitive biologics. In addition, its modularity in generating stacked composite films, such as a VEGF-film deposited atop a BMP2-film, has shown that one can easily achieve simultaneous co-release of both growth factors[15d]. The interdiffusion that occurs during film assembly creates a thoroughly blended nanoscale film that can be highly desirable in some situations, but is also detrimental when aiming to exert more precise control over release behavior. Striking a balance between the chemical and/or physical means of controlling interdiffusion, while maintaining significant loadings of active drug, desirable release kinetics, and facile assembly conditions makes it an extremely challenging problem.
A number of researchers, in addition to us, have sought to control interdiffusion within LbL films using different film components and types of architectures, each with varying degrees of success. Early pioneering work found that barrier layers of linearly-growing (PAH/SPS)n could inhibit interdiffusion during film assembly by separating exponentially-growing (poly-L-lysine/hyaluronic acid), or (PLL/HA)n, films into multiple “compartments”[16]. Analogously, PLGA barriers deposited from aerosolized chloroform solutions also compartmentalized (PLL/HA)n films[17]. The degradable nature of PLGA allowed for localized, cell-based film degradation, but also remains subject to a localized pH decrease typical of PLGA, which can lower protein activity. Additionally, the exposure to harsh solvents and complex processing steps needed for its fabrication provides reasons to pursue fully water-based nanolayer assembly approaches. Further investigations into different types of LbL barrier layers showed that electrostatically-crosslinked (polyvinylbenzyl ammonium chloride/SPS) or (PBA/SPS) films were ineffective at preventing the mixing of two dyes, while the increased tortuosity presented by the clay platelets in (PBA/laponite) films slowed mixing, and thermally-crosslinked, covalent barriers of (PAH/PAA) fully inhibited dye diffusion[18].
Expanding on the concept of compartmentalization, use of fully hydrolytically degradable components could facilitate true control of drug release rates and generate interesting, tunable release behaviors. By thermally-crosslinking even a single bilayer of (polyallylamine/polyacrylic acid), we found it could act as a barrier and thus delay the release of a polysaccharide from a hydrolytically-degradable film buried underneath[19]. While providing the initial proof that sequential release was possible, the crosslinking required heating to 215°C for 20 min, which would denature biologics and yield undesired side reactions with other components in the film. In another approach, we found that graphene oxide sheets were able to also act as a barrier layer by modulating the release of an underlying model protein, ovalbumin[20]. Again this demonstrated the possibility of using a barrier layer to influence the film's release kinetics, but relied on a non-degradable graphene oxide layer that is not at this time generally regarded as safe (GRAS)[21] and may present possible protein-denaturing effects[22]. More recently, we have used laponite clays to achieve time lag between release of a small molecule and a growth factor. Unfortunately, even with these approaches, constant interdiffusion during assembly leads to significant phase mixing that makes it difficult to achieve well-defined sequencing, particularly with multiple proteins. For in vivo delivery systems, it would be ideal to deliver multiple drugs with minimally overlapping release profiles from a completely biocompatible and biodegradable film without the use of non-aqueous solvents, heat or other process conditions that can severely lower the activity of biologic drugs.
We posited that controlling interdiffusion in the film during the actual assembly process would allow us to judiciously embed therapeutics in different regions of the film at will. In a surface-erosion model, as previously demonstrated for hydrolytically degradable LbL films[23] the therapeutic's location and depth in the film would govern its release order and kinetics; thus, when incorporating a hydrolytically degradable component throughout the film, deposition of a sacrificial barrier layer could putatively delay the onset of release and enable truly sequential release behavior. Herein, we describe our approach to introduce crosslinking in situ (i.e., as the film is deposited) using copper-free click functionalities in a hydrolytically degradable LbL film. The bio-orthogonality of the click reaction ensured no unwanted side-reactions (e.g., with the embedded protein). We also found that the protein was effectively isolated to its designated region in the film, and that the subsequent addition of a degradable barrier layer effectively suppressed the onset of release, with the extent of suppression scaling with barrier thickness. With the addition of a second protein-containing layer, the film demonstrated exquisite control over release kinetics and allowed for sequential release.
In an earlier report studying the use of LbL assembled multilayered films[23c], we found that we could generate protein-loaded thin films using completely naturally-derived materials whose degradation products are generally recognized as safe (GRAS) by the FDA. These films were able to controllably sustain the release of protein over multiple days under physiological conditions. The growth behavior of these films[23c] and many other protein-containing LbL assembled films[24] has revealed exponential increases in film thickness as a function of layers deposited. This phenomenon has been well documented for certain LbL systems and has been explained by an “in-and-out” diffusion hypothesis that suggests the diffusivity of weakly charged polymeric species (i.e., proteins, polysaccharides, weak polyelectrolytes) in the film contributes significantly to this growth behavior; the diffusion and absorption of excess polyelectrolytes into and out of the film during assembly causes this exponential film growth[24d]. It is also hypothesized that there is a “diffusional zone” with finite thickness throughout which interdiffusion can readily occur over the timeframe of the adsorption step[24e].
In single protein films, we found that interdiffusion facilitates loading and blending on the nano-scale[15a, 15b, 25]. When combining two separately assembled protein-containing films into a composite film, with VEGF-loaded films stacked atop BMP-2-loaded films, both proteins released simultaneously due to interdiffusion, despite their sequence of deposition[15d]; they each have their own unique release profiles, but both simultaneously begin releasing upon hydration. We hypothesized that by kinetically freezing the interlayer diffusion during film assembly via covalent crosslinks, we would be able to dictate the sequence of their release based on the order of deposition. As schematically represented in Figure 1, the assembly of one film on top of another typically leads to film blending, where the drug is distributed throughout the film (Figures 1A-B, upper panel). With crosslinking that limits interdiffusion, the drug would remain in the region to which it was deposited (Figures 1A-B, lower panel). The resultant surface erosion would reflect this drug distribution (Figures 1C-D) with immediate or delayed release for diffusive or non-diffusive systems, respectively. The in-situ generation of crosslinks by copper-free click chemistry would not only lower the diffusivity of large and intermediate sized biomacromolecules but also the other polyelectrolyte components within the LbL film matrix, thus lowering their mixing during assembly. Copper-assisted click crosslinking has previously been shown to facilitate LbL film assembly[26] especially as “click capsules”[27], but herein we endeavored to generate such crosslinks without the need of copper or any post-treatment.
Figure 1.

Illustration of the proposed assembly and degradation process of multilayer films without (upper panel) and with (lower panel) crosslinking, where the therapeutic (green spheres) is loaded into films composed of polycations (blue) and degradable polyanions (red). Each film undergoes typical LbL film assembly (A), however those films with crosslinking retain their stratified structure while non-crosslinked films are highly interdiffused (B). Surface erosion either degrades a blended film where the therapeutic is distributed throughout the film, or a stratified film with the therapeutic sequestered to where it was deposited (C). The release profiles reflect the effect of crosslinking, and hence interdiffusion, on kinetics of drug release (D).
To this end, we used a poly(β-L-malic acid) (PMLA) based LbL film architecture. PMLA is a bioresorbable, degradable polyanion with the added advantage of presenting available acid groups for side group derivatization. It is well tolerated in vitro and in vivo without toxicity or immunogenicity[28]. We functionalized separate batches of PMLA to contain either pendant azide or dibenzocyclooctyne (DBCO) functionalities (Scheme 1). The azide-DBCO click reaction is driven via the release of ring-strain without needing a copper catalyst and has been shown to be bioorthogonal and biocompatible[29].Through N,N′-dicyclohexylcarbodiimide/N-hydroxysuccinimide mediated amide coupling (Figure S1), we achieved azide (PMLA-az) and DBCO (PMLA-DBCO) functionalization, which were confirmed by FTIR (Figure S2), with degrees of functionalization of 25.5 mol% and 28.9 mol%, respectively, as determined by NMR. While assembling tetralayer films of (chitosan/PMLA-az/protein/PMLA-DBCO)n would putatively minimize interdiffusion, we also envisioned that the hydrolytic degradation of the PMLA ester backbone would impart controlled release behavior.
Scheme 1.

Chemical structures of poly(β-L-malic acid) (PMLA) and its derivatives functionalized with an azide moiety (PMLA-Az) and dibenzocyclooctyne moiety (PMLA-DBCO).
For an understanding of the growth behavior in our films, we examined the thicknesses of different film architectures at 20 tetralayer intervals. For convenience and brevity, we adopt the nomenclature outlined in Table 1. As shown in Figure S3, using click crosslinking reduces the thickness of 20 tetralayer films from 514 ± 12 nm to 258 ± 18 nm for Lys and Lysx-linked films, respectively. Subsequent deposition of n-Barrier Film revealed a linear growth (R2 = 0.992) with 11.7 nm deposited per tetralayer, or ∼3 nm per layer for the combined Lysx-linked + n-Barrier film. This is in striking contrast to our previous data, in which the growth behavior of (chitosan/PMLA/protein/PMLA)n films increases exponentially with up to ∼69.2 nm (R2 = 0.9999) deposited per tetralayer[23c], suggesting a significant suppression of exponential growth and interdiffusion.
Table 1. Film Nomenclature.
| Film Architecture | Number of Tetralayers | Abbreviation |
|---|---|---|
| (Chitosan/PMLA/Lysozyme/PMLA)n | 20 | Lys Film |
| (Chitosan/PMLA-az/Lysozyme/PMLA-DBCO)n | 20 | Lysx-linked Film |
| (Chitosan/PMLA-az/Chitosan/PMLA-DBCO)n | 20 | Barrier Film |
| (Chitosan/PMLA-az/Lysozyme/PMLA-DBCO)n | 20 + | Lysx-linked+ n |
| + (Chitosan/PMLA-az/Chitosan/PMLA-DBCO)n | 0 → 60 | Barrier Film |
In our above-described strategy, we deposited an initial 20 tetralayers of crosslinked protein-containing film (chitosan/PMLA-az/lys/PMLA-DBCO)20, followed by additional crosslinked film devoid of protein (chitosan/PMLA-az/chitosan/PMLA-DBCO)n to act as a sacrificial barrier layer. Lysozyme has one of the greater diffusivities among proteins in LbL films[30] and it is critical to confirm that it is segregated to the underlying layers with suppressed interdiffusion. To this end, we tracked the lysozyme profile through the film with X-ray Photoelectron Spectroscopy (XPS) by monitoring the sulfur signal as a function of probe depth. Coupling C60+ ion sputtering with XPS allowed us to intermittently etch ∼6.6 nm of film from the surface and obtain surface-specific elemental information, analogous to an earlier strategy that tracked polymer interdiffusion in multilayer films[31]. By monitoring the sulfur content, an element uniquely characteristic of lysozyme in the film, we found its peak emerging above background after ∼40 cycles (Figure 2A). When examining the sulfur peak intensity as a function of probe depth (Figure 2B), the S2p signal remained stagnant until reaching ∼328 nm into the film, at which point the intensity significantly increased for an additional 80 nm before reaching a plateau. This step change reveals a gradual yet well defined transition from protein-free to protein-rich sections in the film, demonstrating the confinement of lysozyme to the portion of film beneath the barrier layer.
Figure 2.

Depth profiling XPS analysis of sulfur content using a C60+ ion bombardment of Lys x-linked + 20 Barrier Layer Films. Stacked spectra in the S2p region after 1, 20, 40, 50, 60, and 80 cycles corresponding to probe depths of approximately 7 nm, 131 nm, 263 nm, 328 nm, 394 nm, and 525 nm, respectively (A). Integrated S2p area counts after every sputter cycle is shown as a function of probe depth from the film surface (B).
Stratification in the film architecture via crosslinking should not only isolate lysozyme to its designed region in the film but also minimize its loss as additional barrier layers are deposited. Figure S4 shows that in addition to the reduced thickness, there is a dramatic decrease in lysozyme loading when comparing Lys (30.1 ± 1.1 μg/cm2) and Lysx-linked films (6.4 ± 0.1 μg/cm2). Their loading densities of 586 μg/mm3 and 247 μg/mm3, respectively, also suggests that the fewer available carboxylates on PMLA-az and PMLA-DBCO and the limited interdiffusion from crosslinking lowers the extent of lysozyme complexation and film-incorporation. Comparing Lysx-linked films with Lysx-linked + n barrier layer films, we find that the additional barrier layers deposited do not significantly affect the total lysozyme loading.
Based on our analysis thus far, we have found reduced interdiffusion through in situ crosslinking and have sequestered lysozyme beneath a degradable barrier layer. Seeking the proof of principle for sequential release, we evaluated the effect of crosslinking and thickness of barrier layers on the kinetics of lysozyme release. Herein, and reported previously[23c], we found that LbL assembled Lys films sustain the release of lysozyme for up to two days (Figure 3A). By introducing crosslinking, we suppressed the initial burst release from Lysx-linked films and slightly extended the duration of release to three days. In both cases, release was initiated at the start of incubation. As progressively thicker barrier layer films of 20, 40, and 60 tetralayers were deposited, we found that the start of lysozyme release was correspondingly suppressed up to ∼0.5, ∼1, and ∼1.5 days, respectively (Figure 3A, arrows). Transformation of lysozyme release profiles to their rates as shown in Figure 3B further illustrates the effect that both crosslinking and barrier films have on the release kinetics; not only is the onset of release suppressed, its rate and period of release is also dramatically shifted. This heralds the possibility of pre-programmable release behavior without the need of external intervention.
Figure 3.

The effect of crosslinking and barrier layer thickness on the lysozyme release (a) and rate of fractional lysozyme release (b) into PBS, pH 7.4 at 37°C.
For downstream biomedical applications, biocompatibility is vital; we found analogous (chitosan/PMLA)n films, without click functionality, to be non-cytotoxic[23c], and the addition of click functionality through amide linkages should have minimal, if any, impact on cell viability. To prove this, we incubated a weeks worth of release solutions from Lysx-linked + 60 barrier films with NIH3T3 cells and quantified their effect on cellular metabolic activity (Figure S5). Cells incubated with these release media (in cell culture medium) showed no difference in viability compared to cell culture medium alone, thus further demonstrating this as a biocompatible and biodegradable method for generating controllable protein release.
We next tested the ability to release two therapeutics in sequential fashion from these thin films through the deposition of an additional protein-containing layer on top of a Lysx-linked + n barrier film. As shown in Figure 4A, a triple-stacked composite film undergoing surface erosion would first release the protein from the upper layers (BMP2), then progress through the sacrificial barrier layer, and eventually release the buried protein in the lower layers (lysozyme). We deposited a rapidly releasing (chitosan/PMLA/BMP2/PMLA)20 film on top of Lysx-linked + 60 barrier films, as schematically represented in Figure 4A, and studied its release behavior. Shown in Figure 4B, we found that BMP2 is rapidly released upon hydration with more than 90% of its 9.1 ± 0.7 ng/cm2 eluting in the first 12 hours. Then, 20 hours later, 1.0 ± 0.3 μg/cm2 lysozyme elutes for an additional 40 hours. Surprisingly the lysozyme loading for these films was reduced after BMP2 film deposition, which we suspect may be due to the effects of the BMP2 excipients (e.g., glycine, glutamic acid, sucrose, and polysorbate 80) whose preservative effects by reducing intermolecular interactions[32] can also disrupt LbL film interactions. Despite their impact, substantial protein remains in the film.
Figure 4.
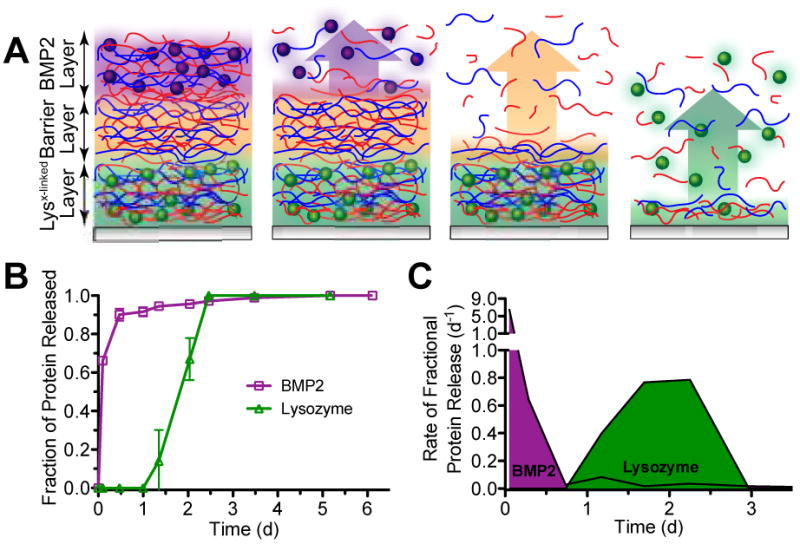
Characteristics of sequential release from composite multilayer films with a schematic view of the proposed film architecture and surface-based erosion (A). Protein release profiles (B) and their rates of fractional release (C) into PBS, pH 7.4 at 37°C.
Overall we have developed a kinetically discrete protein delivery platform, where release of the temporally second therapeutic (lysozyme in the present case) is initially suppressed and does not coincide with release of the first therapeutic (BMP2 in the present case). This is even more evident when examining the rate of fractional release, shown in Figure 4C. We find two distinct schedules of release with each demonstrating unique release behaviors resulting in spatiotemporal separation of BMP2 and lysozyme for their sequential delivery. This is markedly different than many other dual release formulations, which purport “sequential release” behavior, but in fact have an uncontrolled initial co-release of the second therapeutic with the first.
We have designed a biodegradable and biocompatible thin film localized delivery formulation with kinetically discrete and controlled drug release. LbL assembly allowed us to use benign conditions to incorporate significant quantities of active protein and with copper-free click chemistry, bio-orthogonal crosslinking during film assembly significantly reduced interdiffusion to maintain film stratification. Depth-dependent elemental analysis of these films revealed sequestration of lysozyme to its designed region, beneath a barrier layer, and release studies showed that the combination of crosslinking and barrier layers suppressed the initial burst release and effectively delayed the onset of release with increasing barrier layer thickness. Depositing an additional protein-containing LbL film on top of this construct yielded a sequential release behavior as dictated by logical film construction. This demonstration of spatiotemporally discrete protein delivery reveals the possibility of localized, non-overlapping multi-therapeutic administration from a biodegradable thin film that can be tuned for a broad variety of biomedical applications.
Supplementary Material
Footnotes
This research was supported (in part) by the U.S. Army Research Office under contract W911NF-13-D-0001 and the Air Force under contract W911NF-07-D-0004. This work made use of the MIT MRSEC Shared Experimental Facilities supported by the National Science Foundation under award number DMR-0819762.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
Contributor Information
Bryan B. Hsu, Koch Institute for Integrative Cancer Research and the Institute for Soldier Nanotechnologies, Massachusetts Institute for Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 USA, Department of Chemistry, Massachusetts Institute for Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 USA
Kelsey S. Jamieson, Department of Chemical Engineering, Massachusetts Institute for Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 USA
Samantha R. Hagerman, Department of Chemical Engineering, Massachusetts Institute for Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 USA
Eggehard Holler, Nanomedicine Research Center; Department of Neurosurgery, Cedars Sinai Medical Center, 8700 Beverly Boulevard, Los Angeles, CA 90048 USA
Julia Y. Ljubimova, Nanomedicine Research Center; Department of Neurosurgery, Cedars Sinai Medical Center, 8700 Beverly Boulevard, Los Angeles, CA 90048 USA
Paula T. Hammond, Email: hammond@mit.edu, Koch Institute for Integrative Cancer Research and the Institute for Soldier Nanotechnologies, Massachusetts Institute for Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 USA, Department of Chemical Engineering, Massachusetts Institute for Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 USA.
References
- 1.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Cell. 2012;149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacDiarmid JA, Amaro-Mugridge NB, Madrid-Weiss J, Sedliarou I, Wetzel S, Kochar K, Brahmbhatt VN, Phillips L, Pattison ST, Petti C, Stillman B, Graham RM, Brahmbhatt H. Nat Biotech. 2009;27:643–651. doi: 10.1038/nbt.1547. [DOI] [PubMed] [Google Scholar]
- 3.Vaira D, Zullo A, Vakil N, Gatta L, Ricci C, Perna F, Hassan C, Bernabucci V, Tampieri A, Morini S. Ann Intern Med. 2007;146:556–563. doi: 10.7326/0003-4819-146-8-200704170-00006. [DOI] [PubMed] [Google Scholar]
- 4.a Lee K, Silva EA, Mooney DJ. Journal of the Royal Society Interface. 2011;8:153–170. doi: 10.1098/rsif.2010.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen FM, Zhang M, Wu ZF. Biomaterials. 2010;31:6279–6308. doi: 10.1016/j.biomaterials.2010.04.053. [DOI] [PubMed] [Google Scholar]; c Mehta M, Schmidt-Bleek K, Duda GN, Mooney DJ. Adv Drug Del Rev. 2012;64:1257–1276. doi: 10.1016/j.addr.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Wound Repair Regen. 2008;16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 6.a De Marchis F, Ribatti D, Giampietri C, Lentini A, Faraone D, Scoccianti M, Capogrossi MC, Facchiano A. Blood. 2002;99:2045–2053. doi: 10.1182/blood.v99.6.2045. [DOI] [PubMed] [Google Scholar]; b Facchiano A, De Marchis F, Turchetti E, Facchiano F, Guglielmi M, Denaro A, Palumbo R, Scoccianti M, Capogrossi MC. J Cell Sci. 2000;113:2855–2863. doi: 10.1242/jcs.113.16.2855. [DOI] [PubMed] [Google Scholar]; c Russo K, Ragone R, Facchiano AM, Capogrossi MC, Facchiano A. J Biol Chem. 2002;277:1284–1291. doi: 10.1074/jbc.M108858200. [DOI] [PubMed] [Google Scholar]; d Zellin G, Beck S, Hardwick R, Linde A. Bone. 1998;22:613–620. doi: 10.1016/s8756-3282(98)00059-3. [DOI] [PubMed] [Google Scholar]; e Vonau RL, Bostrom MPG, Aspenberg P, Sams AE. Clin Orthop Relat Res. 2001:243–251. doi: 10.1097/00003086-200105000-00032. [DOI] [PubMed] [Google Scholar]; f Ripamonti U, Crooks J, Petit JC, Rueger DC. Eur J Oral Sci. 2001;109:241–248. doi: 10.1034/j.1600-0722.2001.00041.x. [DOI] [PubMed] [Google Scholar]; g Marden LJ, Fan RSP, Pierce GF, Reddi AH, Hollinger JO. J Clin Invest. 1993;92:2897–2905. doi: 10.1172/JCI116912. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Raiche AT, Puleo DA. Biomaterials. 2004;25:677–685. doi: 10.1016/s0142-9612(03)00564-7. [DOI] [PubMed] [Google Scholar]; i Tengood JE, Kovach KM, Vescovi PE, Russell AJ, Little SR. Biomaterials. 2010;31:7805–7812. doi: 10.1016/j.biomaterials.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Tengood JE, Ridenour R, Brodsky R, Russell AJ, Little SR. Tissue Engineering Part A. 2011;17:1181–1189. doi: 10.1089/ten.tea.2010.0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabo JK, Aumann TD, Kilpatrick TJ, Cate HS. Plos One. 2013;8 doi: 10.1371/journal.pone.0063415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edelman ER, Nugent MA, Karnovsky MJ. Proc Natl Acad Sci U S A. 1993;90:1513–1517. doi: 10.1073/pnas.90.4.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazarous DF, Shou M, Scheinowitz M, Hodge E, Thirumurti V, Kitsiou AN, Stiber JA, Lobo AD, Hunsberger S, Guetta E, Epstein SE, Unger EF. Circulation. 1996;94:1074–1082. doi: 10.1161/01.cir.94.5.1074. [DOI] [PubMed] [Google Scholar]
- 10.Richter A, Anton SF, Koch P, Dennett SL. Clin Ther. 2003;25:2307–2335. doi: 10.1016/s0149-2918(03)80222-9. [DOI] [PubMed] [Google Scholar]
- 11.a Chen RR, Silva EA, Yuen WW, Mooney DJ. Pharm Res. 2007;24:258–264. doi: 10.1007/s11095-006-9173-4. [DOI] [PubMed] [Google Scholar]; b Chapanian R, Amsden BG. J Controlled Release. 2010;143:53–63. doi: 10.1016/j.jconrel.2009.11.025. [DOI] [PubMed] [Google Scholar]; c Yilgor P, Hasirci N, Hasirci V. Journal of Biomedical Materials Research Part A. 2010;93A:528–536. doi: 10.1002/jbm.a.32520. [DOI] [PubMed] [Google Scholar]; d Bonani W, Motta A, Migliaresi C, Tan W. Langmuir. 2012;28:13675–13687. doi: 10.1021/la302386u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Yilgor P, Tuzlakoglu K, Reis RL, Hasirci N, Hasirci V. Biomaterials. 2009;30:3551–3559. doi: 10.1016/j.biomaterials.2009.03.024. [DOI] [PubMed] [Google Scholar]; b Yonet-Tanyeri N, Rich MH, Lee M, Lai MH, Jeong JH, DeVolder RJ, Kong H. Biomaterials. 2013;34:8416–8423. doi: 10.1016/j.biomaterials.2013.07.026. [DOI] [PubMed] [Google Scholar]; c Shin SH, Lee J, Lim KS, Rhim T, Lee SK, Kim YH, Lee KY. J Controlled Release. 2013;166:38–45. doi: 10.1016/j.jconrel.2012.12.020. [DOI] [PubMed] [Google Scholar]; d Nelson DM, Ma Z, Leeson CE, Wagner WR. Journal of Biomedical Materials Research Part A. 2012;100A:776–785. doi: 10.1002/jbm.a.34015. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kempen DHR, Lu L, Heijink A, Hefferan TE, Creemers LB, Maran A, Yaszemski MJ, Dhert WJA. Biomaterials. 2009;30:2816–2825. doi: 10.1016/j.biomaterials.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 13.a Ruvinov E, Leor J, Cohen S. Biomaterials. 2011;32:565–578. doi: 10.1016/j.biomaterials.2010.08.097. [DOI] [PubMed] [Google Scholar]; b Hao X, Silva EA, Mansson-Broberg A, Grinnemo KH, Siddiqui AJ, Dellgren G, Wardell E, Brodin LA, Mooney DJ, Sylven C. Cardiovasc Res. 2007;75:178–185. doi: 10.1016/j.cardiores.2007.03.028. [DOI] [PubMed] [Google Scholar]; c Freeman I, Cohen S. Biomaterials. 2009;30:2122–2131. doi: 10.1016/j.biomaterials.2008.12.057. [DOI] [PubMed] [Google Scholar]
- 14.a Wang H, Zou Q, Boerman OC, Nijhuis AWG, Jansen JA, Li Y, Leeuwenburgh SCG. J Controlled Release. 2013;166:172–181. doi: 10.1016/j.jconrel.2012.12.015. [DOI] [PubMed] [Google Scholar]; b Kim S, Kang Y, Krueger CA, Sen M, Holcomb JB, Chen D, Wenke JC, Yang Y. Acta Biomater. 2012;8:1768–1777. doi: 10.1016/j.actbio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a Macdonald ML, Rodriguez NM, Shah NJ, Hammond PT. Biomacromolecules. 2010;11:2053–2059. doi: 10.1021/bm100413w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Macdonald ML, Samuel RE, Shah NJ, Padera RF, Beben YM, Hammond PT. Biomaterials. 2011;32:1446–1453. doi: 10.1016/j.biomaterials.2010.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shah NJ, Hong J, Hyder MN, Hammond PT. Adv Mater. 2012;24:1445–1450. doi: 10.1002/adma.201104475. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shah NJ, Macdonald ML, Beben YM, Padera RF, Samuel RE, Hammond PT. Biomaterials. 2011;32:6183–6193. doi: 10.1016/j.biomaterials.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Shah NJ, Hyder MN, Moskowitz JS, Quadir MA, Morton SW, Seeherman HJ, Padera RF, Spector M, Hammond PT. Science Translational Medicine. 2013;5:191ra183. doi: 10.1126/scitranslmed.3005576. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Crouzier T, Ren K, Nicolas C, Roy C, Picart C. Small. 2009;5:598–608. doi: 10.1002/smll.200800804. [DOI] [PubMed] [Google Scholar]; g De Cock LJ, De Koker S, De Vos F, Vervaet C, Remon JP, De Geest BG. Biomacromolecules. 2010;11:1002–1008. doi: 10.1021/bm9014649. [DOI] [PubMed] [Google Scholar]; h Dierich A, Le Guen E, Messaddeq N, Stoltz JF, Netter P, Schaaf P, Voegel JC, Benkirane-Jessel N. Adv Mater. 2007;19:693–+. [Google Scholar]; i Itoh Y, Matsusaki M, Kida T, Akashi M. Biomacromolecules. 2008;9:2202–2206. doi: 10.1021/bm800321w. [DOI] [PubMed] [Google Scholar]; j Mao ZW, Ma L, Zhou J, Gao CY, Shen JC. Bioconj Chem. 2005;16:1316–1322. doi: 10.1021/bc049755b. [DOI] [PubMed] [Google Scholar]; k Muller S, Koenig G, Charpiot A, Debry C, Voegel JC, Lavalle P, Vautier D. Adv Funct Mater. 2008;18:1767–1775. [Google Scholar]; l van den Beucken JJJP, Walboomers XF, Boerman OC, Vos MRJ, Sommerdijk NAJM, Hayakawa T, Fukushima T, Okahata Y, Nolte RJM, Jansen JA. J Controlled Release. 2006;113:63–72. doi: 10.1016/j.jconrel.2006.03.016. [DOI] [PubMed] [Google Scholar]; m Vodouhe C, Schmittbuhl M, Boulmedais F, Bagnard D, Vautier D, Schaaf P, Egles C, Voegel JC, Ogier J. Biomaterials. 2005;26:545–554. doi: 10.1016/j.biomaterials.2004.02.057. [DOI] [PubMed] [Google Scholar]; n Facca S, Cortez C, Mendoza-Palomares C, Messadeq N, Dierich A, Johnston APR, Mainard D, Voegel JC, Caruso F, Benkirane-Jessel N. Proc Natl Acad Sci U S A. 2010;107:3406–3411. doi: 10.1073/pnas.0908531107. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Peterson AM, Pilz-Allen C, Kolesnikova T, Mohwald H, Shchukin D. Acs Applied Materials & Interfaces. 2014;6:1866–1871. doi: 10.1021/am404849y. [DOI] [PubMed] [Google Scholar]
- 16.Garza JM, Schaaf P, Muller S, Ball V, Stoltz JF, Voegel JC, Lavalle P. Langmuir. 2004;20:7298–7302. doi: 10.1021/la049106o. [DOI] [PubMed] [Google Scholar]
- 17.Garza JM, Jessel N, Ladam G, Dupray V, Muller S, Stoltz JF, Schaaf P, Voegel JC, Lavalle P. Langmuir. 2005;21:12372–12377. doi: 10.1021/la051465b. [DOI] [PubMed] [Google Scholar]
- 18.Peralta S, Habib-Jiwan JL, Jonas AM. Chemphyschem. 2009;10:137–143. doi: 10.1002/cphc.200800443. [DOI] [PubMed] [Google Scholar]
- 19.Wood KC, Chuang HF, Batten RD, Lynn DM, Hammond PT. Proc Natl Acad Sci U S A. 2006;103:10207–10212. doi: 10.1073/pnas.0602884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong J, Shah NJ, Drake AC, DeMuth PC, Lee JB, Chen J, Hammond PT. Acs Nano. 2012;6:81–88. doi: 10.1021/nn202607r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a Wang K, Ruan J, Song H, Zhang J, Wo Y, Guo S, Cui D. Nanoscale Research Letters. 2011;6 doi: 10.1007/s11671-010-9751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang XY, Yin JL, Peng C, Hu WQ, Zhu ZY, Li WX, Fan CH, Huang Q. Carbon. 2011;49:986–995. [Google Scholar]
- 22.Alava T, Mann JA, Théodore C, Benitez JJ, Dichtel WR, Parpia JM, Craighead HG. Anal Chem. 2013;85:2754–2759. doi: 10.1021/ac303268z. [DOI] [PubMed] [Google Scholar]
- 23.a Smith RC, Riollano M, Leung A, Hammond PT. Angew Chem Int Ed. 2009;48:8974–8977. doi: 10.1002/anie.200902782. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vazquez E, Dewitt DM, Hammond PT, Lynn DM. J Am Chem Soc. 2002;124:13992–13993. doi: 10.1021/ja026405w. [DOI] [PubMed] [Google Scholar]; c Hsu BB, Hagerman SR, Jamieson K, Veselinovic J, O'Neill N, Holler E, Ljubimova JY, Hammond PT. Submitted. 2014 doi: 10.1021/bm5001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a Picart C, Lavalle P, Hubert P, Cuisinier FJG, Decher G, Schaaf P, Voegel JC. Langmuir. 2001;17:7414–7424. [Google Scholar]; b Lavalle P, Gergely C, Cuisinier FJG, Decher G, Schaaf P, Voegel JC, Picart C. Macromolecules. 2002;35:4458–4465. [Google Scholar]; c Picart C, Mutterer J, Richert L, Luo Y, Prestwich GD, Schaaf P, Voegel JC, Lavalle P. Proc Natl Acad Sci U S A. 2002;99:12531–12535. doi: 10.1073/pnas.202486099. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lavalle P, Picart C, Mutterer J, Gergely C, Reiss H, Voegel JC, Senger B, Schaaf P. J Phys Chem B. 2004;108:635–648. [Google Scholar]; e Porcel C, Lavalle P, Ball V, Decher G, Senger B, Voegel JC, Schaaf P. Langmuir. 2006;22:4376–4383. doi: 10.1021/la053218d. [DOI] [PubMed] [Google Scholar]; f Decher G, Schlenoff JB. 2nd. Wiley-VCH; Weinheim, Germany: 2012. [Google Scholar]
- 25.a Macdonald M, Rodriguez NM, Smith R, Hammond PT. J Controlled Release. 2008;131:228–234. doi: 10.1016/j.jconrel.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shukla A, Fleming KE, Chuang HF, Chau TM, Loose CR, Stephanopoulos GN, Hammond PT. Biomaterials. 2010;31:2348–2357. doi: 10.1016/j.biomaterials.2009.11.082. [DOI] [PubMed] [Google Scholar]
- 26.Such GK, Quinn JF, Quinn A, Tjipto E, Caruso F. J Am Chem Soc. 2006;128:9318–9319. doi: 10.1021/ja063043+. [DOI] [PubMed] [Google Scholar]
- 27.a Such GK, Tjipto E, Postma A, Johnston APR, Caruso F. Nano Lett. 2007;7:1706–1710. doi: 10.1021/nl070698f. [DOI] [PubMed] [Google Scholar]; b Liang K, Such GK, Zhu Z, Dodds SJ, Johnston APR, Cui J, Ejima H, Caruso F. Acs Nano. 2012;6:10186–10194. doi: 10.1021/nn3039353. [DOI] [PubMed] [Google Scholar]; c Ochs CJ, Such GK, Yan Y, van Koeverden MP, Caruso F. Acs Nano. 2010;4:1653–1663. doi: 10.1021/nn9014278. [DOI] [PubMed] [Google Scholar]
- 28.Ljubimova JY, Portill-Arias J, Patil R, Ding H, Inoue S, Markman JL, Rekechenetskiy A, Konda B, Gangalum PR, Chesnokova A, Ljubimov AV, Black KL, Holler E. J Drug Targeting. 2013;21:956–967. doi: 10.3109/1061186X.2013.837470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a Jewett JC, Bertozzi CR. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Debets MF, van Berkel SS, Schoffelen S, Rutjes FPJT, van Hest JCM, van Delft FL. Chem Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]
- 30.Uhlig K, Madaboosi N, Schmidt S, Jager MS, Rose J, Duschl C, Volodkin DV. Soft Matter. 2012;8:11786–11789. [Google Scholar]
- 31.Gilbert JB, Rubner MF, Cohen RE. Proc Natl Acad Sci U S A. 2013;110:6651–6656. doi: 10.1073/pnas.1222325110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.a Liao YH, Brown MB, Quader A, Martin GP. Pharm Res. 2002;19:1854–1861. doi: 10.1023/a:1021497625645. [DOI] [PubMed] [Google Scholar]; b Chi EY, Krishnan S, Randolph TW, Carpenter JF. Pharm Res. 2003;20:1325–1336. doi: 10.1023/a:1025771421906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.