

Abstract
Heparan sulfate is a polysaccharide that plays essential physiological functions in the animal kingdom. Heparin, a highly sulfated form of heparan sulfate, is a widely prescribed anticoagulant drug worldwide. The heparan sulfate and heparin isolated from natural sources are highly heterogeneous mixtures differing in polysaccharide chain lengths and sulfation patterns. The access to structurally defined heparan sulfate and heparin is critically important to probe the contribution of specific sulfated saccharide structures to biological functions as well as for the development of the next generation of heparin-based anticoagulant drugs. The synthesis of heparan sulfate and heparin, using a purely chemical approach, has proven extremely difficult, especially for targets larger than octasaccharides having a high degree of site-specific sulfation. A new chemoenzymatic method has emerged as an effective alternative approach. This method utilizes recombinant heparan sulfate biosynthetic enzymes combined with unnatural uridine diphosphate-monosaccharide donors. Recent examples demonstrate the successful synthesis of ultra-low molecular weight heparin, low-molecular weight heparin and bioengineered heparin with unprecedented efficiency. The new method opens the opportunity to develop improved heparin-based therapeutics.
Introduction
Heparan sulfate (HS) is a polysaccharide-based natural product, widely expressed on the mammalian cell surfaces and in the extracellular matrix. Large body of evidence demonstrate that HS plays essential roles in a number of biological processes, including embryonic development, inflammatory responses, bacterial/viral infection and blood coagulation.1 The wide range of biological functions has attracted considerable interest in the development of new medicines using HS as a structural scaffold. Heparin, a specialized, highly sulfated form of HS, is a widely used anticoagulant to prevent and treat arterial and venous thrombosis.2,3 HS consists of a disaccharide repeating unit of either iduronic acid (IdoA) or glucuronic acid (GlcA) and glucosamine (GlcN) residues, each capable of carrying sulfate groups (Fig. 1). The presence of sulfate groups and the location of IdoA and GlcA residues dictate the functional selectivity of HS. The challenge in developing HS-based drugs centers on the synthesis of HS oligosaccharides and polysaccharides having desired size and sulfation patterns. HS oligosaccharides have been synthesized by a purely chemical approach; however, this synthesis is difficult, due to the required use of complex protecting/deprotecting procedures. The syntheses of diversified HS structures are further complicated because they require the preparation of a large number of precursor compounds. Therefore, HS oligosaccharide synthesis can be completed by skilled synthetic chemists in a small number of highly specialized labs.4-13 In recent years, a chemoenzymatic synthesis has emerged using glycosyltransferases, epimerase and sulfotransferases.14,15 Compared to chemical synthesis, the chemoenzymatic approach offers shorter synthetic routes, excellent recovery yields, and utilizes a few common precursors for the preparation of diverse HS oligosaccharide structures. This article reviews the recent development in the chemoenzymatic synthesis method and its progress towards the synthesis of HS oligosaccharide targets having diverse sulfation patterns.
Fig 1.

Structures of the disaccharide repeating unit of HS and the structure of fondaparinux.
Heparin
Discovered in 1916, heparin has been the drug of choice to treat thrombotic disorders for nearly 90 years.16 The discovery of heparin contributed significantly to the development of many advanced medical and surgical procedures.17 Three forms of heparin are approved by the US Food and Drug Administration (FDA): Unfractionated heparin ((UFH) average molecular weight (MWavg) 16,000 Da), low-molecular weight heparin (MWavg 3,500-6,000 Da) and fondaparinux (MW 1,508 Da). UFH is a safe intravenous drug for the treatment of renal-impaired patients and its effects can be reversed using the cationic-polypeptide drug protamine;18 however, it shows a 1-6% incidence of heparin-induced thrombocytopenia (HIT), a life-threatening complication.19 LMWHs are subcutaneously administered and have a longer half-life than UFH, permitting their outpatient use and self-administration. 16 However, LMWH can only be used in renal-impaired patients at the reduced doses20 and is incompletely neutralized with protamine, thereby increasing the risks of bleeding. Fondaparinux, a synthetic pentasaccharide, is subcutaneously bioavailable and has reduced risks of HIT and osteoporosis.21 However, it is primarily excreted through the kidney and, thus, is not suitable for renal-impaired patients, and it lacks an antidote.20
Both UFH and LMWH are considered to be animal-sourced products. 22 UFH is isolated from porcine intestine or bovine lung or intestine, while LMWHs are a depolymerized product of heparin from porcine intestine. A worldwide contamination of heparin in 2007 affected the purity and safety of both UFH and LMWHs associated with over 200 deaths in the US. 23,24 This incident revealed the fragility of the heparin supply chain. Although fondaparinux is fully synthetic,11 and its supply does not depend on the animal sources, the drug does not display all clinical benefits as those of UFH and LMWH. A cost-effective method to prepare a synthetic heparin and LMWH that can be manufactured under FDA’s guidelines will eliminate the needs of animal sourced materials.24 Furthermore, the synthetic heparins could be engineered to introduce additional beneficial pharmacological properties that do not exist in UFH and LMWHs from natural sources.16
Heparin and HS have similar disaccharide repeating units; however, heparin has higher sulfation levels than HS, containing 2.6 sulfo groups per disaccharide compared to 0.6 sulfo groups in HS. In addition, nearly 90% of the disaccharide units in heparin contain IdoA, while only 20% of the disaccharide units in HS contain IdoA. HS can be isolated from many cell types, whereas heparin is an exclusive product of mast cells.
Enzymes involved in the chemoenzymatic synthesis
The chemoenzymatic synthesis of HS requires several classes of biosynthetic enzymes, some comprised of multiple isoforms.25 Glycosyltransferases are used to build the HS backbone structure consisting of a disaccharide repeating unit of GlcA and N-acetylglucosamine (GlcNAc).26 An epimerase (C5-epi) is responsible for converting a GlcA residue to an IdoA residue.27 Sulfo groups are transferred from 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to oligosaccharide by specialized HS sulfotransferases. For example, 2-O-sulfotransferase (2-OST) specifically transfers a sulfo group to the 2-position of an IdoA residue and, to lesser extent, to the 2-position of a GlcA residue.28,29 The 6-O-sulfotransferase (6-OST) and the 3-O-sulfotransferase (3-OST) transfer sulfo groups to the 6-position and 3-position of glucosamine residues. 29,30 N-deacetylase/N-sulfotransferase (NDST) converts a GlcNAc residue to a N-sulfoglucosamine (GlcNS) residue.31 NDST is unique displaying two activities, N-deacetylase activity removing N-acetyl group from a GlcNAc residue, and N-sulfotrasnferase activity transferring a sulfo group to a GlcN residue, forming a GlcNS residue.32,33
The enzymes used for the chemoenzymatic synthesis of heparin are predominantly mammalian proteins, presenting challenges for making these recombinant proteins accessible in large quantities. Currently, most of these HS biosynthetic enzymes, with the exception of NDST, EXT-1 and EXT-2, have been expressed in E. coli with sufficient efficiency for high milligram-scale synthesis. 34 Only the N-sulfotransferase (NST) domain of the NDST can be effectively expressed in E. coli and, thus, this protein lacks N-deacetylase activity. Two bacterial glycosyltransferases known as KfiA (an N-acetyl glucosaminyl transferase from E. coli K5 strain) and pmHS2 (heparosan synthase 2 from Pasteurella multicida) are used as substitute EXT1 and EXT2, to build HS backbone. Both KfiA and pmHS2 enzymes can be readily expressed in E. coli.35,36
Design of sugar nucleotides for the chemoenzymatic synthesis of HS
Chemoenzymatic synthesis of HS requires a cascade of enzymatic modifications. The N-sulfation of glucosamine is essential for the down-stream enzymatic modifications to occur.25 The synthesis of the HS backbone containing GlcNS residues represented a major roadblock due to the lack of efficient recombinant NDST. An unnatural sugar nucleotide,37,38 UDP-GlcNTFA (uridine diphosphate N-trifluoroacetylated glucosamine), was introduced to overcome this problem. The UDP-GlcNTFA was first reportedly used in the synthesis of carbohydrates of O-linked glycoprotein by “core-2” GlcNAc transferase with modest yield (56%).39 For HS backbone synthesis, UDP-GlcNTFA appears to be an excellent substrate for KfiA with nearly quantitative conversion in the preparation of heparin oligosaccharide backbone.40 The GlcNTFA residue is then converted to a GlcNS residue by removing the trifluoroacetyl group under mild alkaline conditions followed by N-sulfotransferase modification (Fig. 3). The use of UDP-GlcNTFA provides a means to introduce a GlcNS at precisely the desired position within an oligosaccharide. This strategy is now used to synthesize structurally homogeneous ultra-low molecular weight heparins, low-molecular weight heparins14,15 and heterogeneous heparin-like polysaccharides.41,42
Fig 3. Building HS backbone containing GlcNS residues using a non-natural sugar nucleotide.

KfiA transfers a GlcNAc residue to the primer substrate, where the sugar nucleotide donor is UDP-GlcNAc, a natural substrate for KfiA. KfiA also transfers a GlcNTFA residue to the primer substrate, where the sugar nucleotide donor is UDP-GlcNTFA, an unnatural sugar nucleotide. The trifluoroacetyl group can be removed under mild base conditions to form an N-unsubstituted glucosamine (GlcN) residue. The GlcN residue is then converted to GlcNS by N-sulfotransferase (NST). The removal of trifluoroacetyl group can be achieved by triethylamine and methanol under room temperature, or by 0.1 M LiOH at 4°C. The synthesis of UDP-GlcNTFA is completed in two-step synthesis using N-acetylhexosamine 1-kinase and glucosamine-1-phosphate acetyltransferase/N-acetylglucosamine-1-phosphate uridyltransferase (GlmU).
A novel strategy to utilize a series of unnatural sugar nucleotides to synthesize heparin-like oligosaccharide was recently proposed by Chen and coworkers. 43 Here, the 6-hydroxyl group of GlcN was substituted by an azido group to synthesize sugar nucleotides, including UDP-[6-azido]GlcNAc or UDP-[6-azio]GlcNTFA. Chen and coworkers demonstrated the synthesis of oligosaccharides, which carry the azido groups at the 6-position of the GlcN residue, in excellent yields using pmHS2 enzyme. The azido group was then converted to an amino group followed by N-sulfation to generate 6-N-sulfo GlcN residues. Ultimately, HS-analog oligosaccharides, containing N-sulfate groups at both the 2- and the 6-positions of GlcN were synthesized, mimicking the 2-N-sulfo, 6-O-sulfo GlcN residues in natural HS and heparins. Because this synthesis only involves the use of glycosyltransferase and chemoselective N-sulfation to produce sulfated saccharides, the approach eliminates the potential complexity in introducing a sulfate group at a desired site by relying on the substrate specificity of the O-sulfotransferases. Because of this unnatural 6-N-sulfation these products might be resistant to heparanase and lysosomal degradation, increasing the biological half-life of oligosaccharides.
Design of the sequence of enzymatic modifications
The chemoenzymatic synthesis of heparin requires a series of enzymatic modification steps. The design of an appropriate modification sequence is closely related to the substrate specificities of the biosynthetic enzymes. In vivo, a HS biosynthetic pathway is responsible for installing the multiple layers of sulfation on the polysaccharide backbone by following a specific order. The cells probably utilize the modification sequence to achieve the control of HS polysaccharide structure. HS biosynthetic enzymes have evolved to only react with select substrates having unique sulfation patterns, while other saccharide substrates lacking these sulfation patterns remain unmodified. Such a feature assures the biosynthesis of HS follows a preprogrammed modification sequence. 44 For example, 2-O-sulfation step catalyzed by 2-OST occurs only after N-sulfation step, but prior to the 6-O-sulfation step. The ternary crystal structure of 2-OST reveals that three amino acid residues, Arg-80, Lys-350 and Arg-190, from 2-OST directly interact with the N-sulfo group of GlcNS residue flanked on both end of the IdoA residue. Mutation any of three residues that are interacted with the N-sulfo group resulted in the lost of sulfotransferase activity. Likewise, substituting the N-sulfo group with an N-acetyl group or hydrogen on the GlcN residue of the substrate abolishes its reactivity to 2-OST modification. 44 In contrast, two amino acid residues, Tyr-173 and Pro-82 near that active site of 2-OST are probably used by the enzyme to exclude the 6-O-sulfo group, suggesting that 6-O-sulfation occurs after 2-O-sulfation in the process of biosynthesizing highly sulfated domains consisting of -IdoA2S-GlcNS6S-, commonly found in heparin. The structural and biochemical evidence clearly demonstrates the essential roles of N-sulfation and 6-O-sulfation in regulating the 2-O-sulfation in the HS biosynthetic pathway.
Synthesis of oligosaccharides with repeating –IdoA2S-GlcNS- units
The synthesis of a domain with repeating –IdoA2S-GlcNS- disaccharide units is critically important for preparing heparin and the high sulfation domains of HS. In heparin, more than 80% of the disaccharides are composed of -IdoA2S-GlcNS6S-.3 Attempts to synthesize a domain with more than one -IdoA2S-GlcNS- motif results in a complex mixture. 45 Complex product mixtures are acceptable when preparing bioengineered heparin46 as a generic replacement for animal-sourced UFHs, since these products are polydisperse and microheterogenous. 3 The challenge comes in synthesizing structurally defined LMWHs or ULMWHs containing repeating –IdoA2S-GlcNS6S-motifs. The difficulty in installing repeating –IdoA2S-GlcNS6S units stems from a unique property of C5-epi. Unlike other HS biosynthetic enzymes, C5-epi is a two-way catalyst: it transforms a GlcA to an IdoA residue, and also converts an IdoA to a GlcA residue (Fig. 4A). 47 Therefore, the final product is a mixture of GlcA and IdoA after C5-epi modification. A recent study revealed that C5-epi is a one-way catalyst towards substrates having a unique distribution of GlcNAc and GlcNS residues. 48 The results indicate that C5-epi can display both two-way and one-way catalytic modes by recognizing a sequence motif designated as the C5-epi recognition pentasaccharide domain (Fig. 4B). 48 This pentasaccharide domain consists of an epimerization site (EPS) and a mode of reaction recognition site (MRRS). The MRRS is located three residues away from the non-reducing end side of the EPS (Fig. 4B). The N-substitution status of the MRRS residue, controlled in biosynthesis by NDST, 49 dictates the mode of action of C5-epi. A GlcNAc residue directs C5-epi to display an irreversible reaction mode, while a GlcNS or GlcN residue (or the MRRS site being unoccupied) drives C5-epi to display a reversible reaction mode. The presence of GlcNS at the -1 residue is essential for rendering the susceptibility to C5-epi modification.
Fig 4. The reaction catalyzed by C5-epi and its substrate specificity.

A shows the reaction catalyzed by C5-epi. A trisaccharide segment is shown. C5-epi removes the proton from C5 of the GlcA residue to form a putative carbanion intermediate. Conversely, C5-epi can catalyze the reverse reaction, converting an IdoA residue to a GlcA residue. B shows the substrate specificity of C5-epi. The designated epimerization site (EPS) is at the residue 0. The residue -1 must be a GlcNS residue to serve as an EPS. If residue -1 is a GlcNAc residue, the site is not reactive to C5-epi (see reaction e). The N-substitution status at the mode of reaction recognition site (MRRS) determines the mode of reaction of C5-epi. If the MRRS site (residue -3) is a GlcNS or GlcN residue or unoccupied, C5-epi displays a reversible reaction mode (see reaction a, c and b). If a GlcNAc residue is at the MRRS site, C5-epi displays an irreversible reaction mode (see reaction d). The grey shaded box indicates the pentasaccharide domain recognized by C5-epi.
An improved knowledge on the substrate specificity of C5-epi offers an alternative strategy for synthesizing IdoA2S-GlcNS repeating units to avoid forming by-products (Fig. 5). Seven steps (Fig. 5, Steps a, b, a, b, c, a, and d, transformation of 1 to 8) are required to synthesize the first IdoA2S residue from a monosaccharide starting material. Each repetition of these steps adds an additional IdoA2S residues, transforming 8 to 12, 13 and 14, containing two, three and four IdoA2S-GlcNS repeats, respectively. The construction of a C5-epi recognition pentasaccharide module with the structure of GlcNTFA-GlcA-GlcNS-GlcA-GlcNS- is the centerpiece of this strategy (Fig. 4, compound 7). This pentasaccharide module allows C5-epi to exhibit the irreversible reaction mode, leading to a single product. The resultant IdoA residue in the pentasaccharide domain is immediately 2-O-sulfated to form an IdoA2S residue (Fig. 5, compound 8). Subsequent construction of the C5-epi recognition pentasaccharide module involves three steps (Fig. 5, Step c, b and a) to introduce additional -IdoA2S-GlcNS- motifs. For example, compound 8 will be elongated to an octasaccharide with a structure of GlcNTFA-GlcA-GlcNS-GlcA-GlcNS-IdoA2S-GlcNS-GlcApnp (compound 11), containing the C5-epi recognition pentasaccharide domain (highlighted in bold). The modifications by C5-epi and 2-OST (Step d and e) will result in an octasaccharide of GlcNTFA-GlcA-GlcNS-IdoA2S-GlcNS-IdoA2S-GlcNS-GlcApnp (compound 12). The 2-O-sulfation to form an IdoA2S residue is essential because it prevents further action of C5-epi on this residue and accelerates the C5-epimerization reaction.47 These steps can be further repeated to form deca- (13) and dodecasaccharide (14) to introduce three and four repeats of IdoA2S-GlcNS disaccharides as described in a recent publication. 15
Fig 5. Route to synthesize oligosaccharides with IdoA2S-GlcNS repeats.
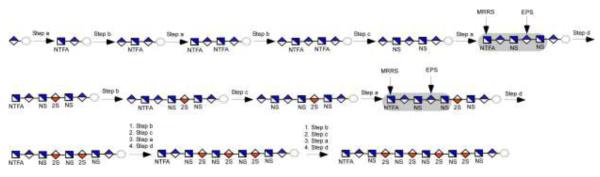
The key in this strategy is to construct a C5-epi recognition pentasaccharide module to utilize the irreversible reaction. The C5-epi recognition pentasaccharide module is highlighted in a grey box. The synthesis initiates from GlcA-pnp (1, p-nitrophenyl glucuronide). The monosaccharide is converted to a pentasaccharide 5 in four enzymatic steps. Hexasaccharide 7 consists of the C5-epi recognition pentasaccharide module. The synthesis of 8 from 7 is achieved by two enzymatic steps (step d) using both C5-epi and 2-OST. Hexasaccharide 8 contains a single IdoA2S-GlcNS disaccharide unit. The construction of another C5-epi recognition pentasaccharide module involves four enzymatic steps, b, c, a and d (transformation of 8 to 12). Repeating those steps can reach to 13 and 14.
Using the chemoenzymatic method to design heparin drugs
Two specific examples for the utilization of chemoenzymatic approach to prepare heparin oligosaccharides with anticoagulant activity have been recently demonstrated. In the first example, this method was used to synthesize a homogenous heptasaccharide (Fig. 6). The synthesis, initiated from a disaccharide (GlcA-AnMan, AnMan represents 2,5-anhydromannitol), was completed in nine enzymatic steps with 43% recovery yield. 14 The synthesis includes the elongation of the starting disaccharide to a heptasaccharide using KfiA and pmHS2. The heptasaccharide backbone is then converted to final product by a series of reactions, including conversion of the GlcNTFA residue to GlcNS (Fig. 6, step c), epimerization and 2-O-sulfation (Fig. 6, step e and f), 6-O-sulfation (Fig. 6, step g) and 3-O-sulfation (Fig. 6, step h). The product has very similar anticoagulant potency to that of fondaparinux as measured by in vitro and ex vivo experiments. Synthesis of fondaparinux using a purely chemical approach requires a much longer route to complete. 11,50
Fig 6.

Route for the chemoenzymatic synthesis of ultra-low molecular weight heparin.
The second example is to design the structures of LMWHs with desired pharmacological properties (Fig. 7). Desirable properties of LMWHs include: a homogeneous structure prepared from non-animal ingredients, safe for kidney-impaired patients, and has reversible anticoagulant activity. Toward these goals, five different oligosaccharides were synthesized. 15 Among these were 6-mer ULMW heparin target compound, very similar to fondaparinux, as well as a 10-mer and 12-mer that were in the size range of LMWHs. Because of the size and structural complexity of LMWHs, a more efficient chemoenzymatic method was required. A commercially available aglycone, 1-O-(para-nitrophenyl) glucuronide (GlcA-pNP), was used as a starting material. The presence of pNP motif provides a UV chromophore for monitoring the product during the synthesis and purification. Nonsulfated oligosaccharides carrying this pNP motif bind to conventional C18-columns, allowing easy purification of intermediates from proteins and unreacted cofactors. Moreover, this pNP group can be removed at the end of the synthesis or reduced to afford an amino group51 for attaching the resulting oligosaccharide to a support. A unique enzymatic modification sequence was employed on the GlcA-pNP to synthesize the oligosaccharides consisting of the repeating disaccharide unit of –IdoA2S-GlcNS-, having high purity (Fig. 5). The final products were purified by anion exchange chromatography to obtain a purity of > 98%. In a mouse model, oligosaccharides larger than 10-mer were metabolized in the liver, suggesting that these synthetic LMWHs will be safe for use in renal-impaired patients. Most interestingly, the anticoagulant activity of the synthetic LMWH 12-mer was reversed by protamine, an FDA approved drug to neutralize unfractionated heparin. The protamine reversibility of this 12-mer could potential reduce the risk of bleeding in its clinical use.
Fig 7. Structure of synthetic low-molecular weight heparins.

The synthetic LMWH 12-mer has two 3-O-sulfo groups.
Conclusions
Over the past five years, there has been steady improvement in the chemoenzymatic synthesis of HS and heparin oligosaccharides, focused on making synthesis more flexible for the preparation of different targets with increased purity and increased synthetic scale. To date, more than 32 heparin oligosaccharides have been synthesized (Table 1). The scale of synthesis has increased from 10 μg as reported in 200352 to 1g, a 100,000-fold increase.15 This improvement in synthesis is attributable to improved enzyme expression, higher efficiency synthesis for enzyme cofactors and an improved understanding of the substrate specificities of HS biosynthetic enzymes. The availability of these newly synthesized compounds has improved heparin and HS research. For example, synthetic heparin oligosaccharides prepared by chemoenzymatic synthesis were used to solve the co-crystal structure of 3-OST-1/PAP/heptasaccharide and 2-OST/heptasaccharides/3′-phosphoadenosine-5′-phosphate.44,53 These crystal structures helped us understand how sulfotransferases distinguish the specific saccharide sequences to exhibit substrate specificity. The results from these studies will, in turn, further improve oligosaccharide the synthesis using these enzymes.
Table 1.
Summary of synthesized HS oligosaccharides using the chemoenzymatic approach
| Compounds | Structure | Comments | Reference |
|---|---|---|---|
| 16-19 | GlcA-GlcNR1-GlcA-GlcNR2-GlcA-GlcNAc-GlcA-AnMan (16, R1=R2=Ac; 17, R1=SO3H, R2=Ac; 18, R1=Ac, R2=SO3H; 19, R1=R2=SO3H) |
Demonstration of the control of size and N- sulfation |
40 |
| 20 and 21 | GlcA-GlcNS6S-(GlcA-GlcNS6S)n-GlcA-AnMan (20, n = 3; 21, n = 4) |
Demonstration of the control of complete 6-O- sulfation |
40 |
| 22 and 23 | GlcNR’6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S-GlcA- AnMan (22, R’=SO3H; 23, R’=Ac) |
Demonstration of the synthesis of complete heparin fragment |
14 |
| 24 | GlcNAc6S-GlcA-GlcNS6S-IdoA2S-GlcNS6S-GlcA- AnMan |
Demonstration of the application in 3-OST-1 crystal structure study |
53 |
| 25 and 26 | GlcA-GlcNAc-GlcA-GlcNR’6S-GlcA-AnMan (25, R’=SO3H; 26, R’=Ac) |
Demonstration of the control partial 6-O- sulfation |
56 |
| 27-30 | GlcNS-(GlcA-GlcNS)m-(GlcA-GlcNAc)2-(GlcA-GlcNS)2- GlcA-AnMan (27, m = 2; 28, m = 3; 29, m = 4; 30, m = 5) |
Demonstration of the synthesis of large oligosaccharide up to heneicosaccharide |
45 |
| 31-36 | GlcA-GlcNR16S-GlcA-GlcNR26S-GlcA-GlcNR36S-GlcA- GlcNR46S-GlcA-pNP (31, R1=R2=R3=R4=SO3H; 32, R1=Ac; R2=R3=R4=SO3H; 33, R1=R2=Ac, R3=R4=SO3H; 34, R1=R4=Ac, R2=R3=SO3H; 35, R1=R2=R3=SO3H, R4=Ac; 36, R1=R2=Ac, R2=R2=SO3H) |
Demonstration of the synthesis of a small library of N-sulfated and 6-O-sulfated nonasaccharide library |
57 |
| 37-40 | GlcNR-GlcA-GlcNS-(IdoA2S-GlcNS)m-GlcA-pNP (37, R =−SO3H, m =1;38, R =−SO3H; m=2; 39, R = −SO3H; m =3; 40, R = −Ac, m =4) |
Synthesis of oligosaccharides with multiple –IdoA2S-GlcNS repeats |
15 |
| 41-44 | GlcNR6S-GlcA-GlcNS3S6S-(IdoA2S-GlcNS6S)m-GlcA- pNP (41, R =−SO3H, m =1;42, R =−SO3H; m=2; 43, R = − SO3H; m =3; 44, R = −Ac, m =4) |
Synthesis of low- molecular weight heparins |
15 |
| 45 | GlcNAc6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S- IdoA2S-GlcNS6S- IdoA2S-GlcNS3S6S- IdoA2S- GlcNS6S-GlcA-pNP |
Synthesis of low- molecular weight heparins |
15 |
| 46-48 | GlcNR16R-GlcA-GlcNS6R-GlcA2S-GlcNS6R-GlcA-pNP (46, R=R1= −H; 47, R=−H, R1= −SO3H; 48, R=R1=−SO3H) |
Synthesis of GlcA2S- containing oligosaccharides |
58 |
The next challenge in anticoagulant drug development is to synthesize homogeneous LMWHs that meet all of the current clinical needs. The synthesis of the LMWH 12-mer requires 22 synthetic steps with the overall yield about 10%. This chemoenzymatic synthesis is significantly shorter than the chemical synthesis of fondaparinux that requires 50 steps. 54 Fondaparinux is now synthesized in kg-scale, and has been a profitable drug for ten years. 55 Further development of the chemoenzymatic approach should result in cost-effective products, accelerating the modernization of LMWH drugs as well as defined HS oligosaccharides for biomedical researches.
Supplementary Material
{kind=link}
Fig 2. Enzymes involved in the biosynthesis of HS.

Panel A shows the elongation reactions to prepare nonsulfated and unepimerized saccharide backbone. Both EXT1 and EXT2 proteins are involved in the elongation reactions. Panel B shows the reactions involved in polysaccharide modifications. The sites of reactions are highlighted in grey boxes. Abbreviations: EXT1; exostosin glycosyltransferase 1; EXT1; exostosin glycosyltransferase 2; NDST, N-deacetylase/N-sulfotransferase; C5-epi, C5-epimerase; 2-OST, 2-O-sulfotransferase; 6-OST, 6-O-sulfotransferase; 3-OST, 3-O-sulfotransferase; PAPS, 3′-phosphoadenosine 5′-phosphosulfate.
Acknowledgments
This work is supported in part by NIH grants HL094463, GM102137, HL62244, and HL096972.
References
- 1.Bishop J, Schuksz M, Esko JD. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 2.Kahn SR, Lim W, Dunn AS, Cushman M, Dentali F, Akl EA, Cook DJ, Balekian AA, Klein RC, Le H, Schulman S, Murad MH. Chest. 2012;141(suppl):e195S–e226S. doi: 10.1378/chest.11-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linhardt RJ. J. Med. Chem. 2003;46:2551–2564. doi: 10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- 4.Arungundram S, Al-Mafraji K, Asong J, Leach FE, III, Amster IJ, Venot A, JE T, Boons GJ. J. Am. Chem. Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu Y-P, Lin S-Y, Huang C-Y, Zulueta MML, Liu J-Y, Chang W, Hung S-C. Nat Chem. 2011;3:557–563. doi: 10.1038/nchem.1073. [DOI] [PubMed] [Google Scholar]
- 6.de Paz JL, Noti C, Seeberger PH. J. Am. Chem. Soc. 2006;128:2766–2767. doi: 10.1021/ja057584v. [DOI] [PubMed] [Google Scholar]
- 7.Polat T, Wong CH. J Am Chem Soc. 2007;129:12795–12800. doi: 10.1021/ja073098r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Xu Y, Yang B, Tiruchinapally G, Sun B, Liu R, Dulaney S, Liu J, Huang X. Chem. Eur. J. 2010;16:8365–8375. doi: 10.1002/chem.201000987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansen SU, Miller GJ, Cole C, Rushton G, Avizienyte E, Jayson GC, Gardiner JM. Nat Commun. 2013;4:2016. doi: 10.1038/ncomms3016. doi: 10.1038/ncomms3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinay P. Nature. 1999;398:377–378. doi: 10.1038/18783. [DOI] [PubMed] [Google Scholar]
- 11.Petitou M, Duchaussoy P, Lederman I, Choay J, Jacquinet JC, Sinaÿ P, Torri G. Carbohydr. Res. 1987;167:67–75. doi: 10.1016/0008-6215(87)80268-9. [DOI] [PubMed] [Google Scholar]
- 12.Canales A, Angulo J, Ojeda R, Bruix M, Fayos R, Lozano R, Giménez-Gallego G, Martín-Lomas M, Nieto PM, Jiménez-Barbero JJ. J. Am. Chem. Soc. 2005;127:5778–5779. doi: 10.1021/ja043363y. [DOI] [PubMed] [Google Scholar]
- 13.Schworer R, Zubkova OV, Turnbull JE, Tyler PC. Chem. Eur. J. 2013;19:6817–6823. doi: 10.1002/chem.201204519. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa SA, Linhardt RJ, Liu J. Science. 2011;334:498–501. doi: 10.1126/science.1207478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Y, Cai C, Chandarajoti K, Hsieh P, Lin Y, Pham TQ, Sparkenbaugh EM, Sheng J, Key NS, Pawlinski RL, Harris EN, Linhardt RJ, Liu J. Nat Chem Biol. 2014;10:248–250. doi: 10.1038/nchembio.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linhardt RJ, Liu J. Curr Opin Pharmacol. 2012;12:217–219. doi: 10.1016/j.coph.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcum JA. J. Hist Med Allied Sci. 2000;55:37–66. doi: 10.1093/jhmas/55.1.37. [DOI] [PubMed] [Google Scholar]
- 18.Hirsh J, O’Donnell MO, Eikelboom JW. Circulation. 2007;116:552–560. doi: 10.1161/CIRCULATIONAHA.106.685974. [DOI] [PubMed] [Google Scholar]
- 19.Arepally G, Ortel TL. N. Eng. J. Med. 2006;355:809–817. doi: 10.1056/NEJMcp052967. [DOI] [PubMed] [Google Scholar]
- 20.Harder S. J. Clin. Pharmacol. 2012;52:964–975. doi: 10.1177/0091270011409231. [DOI] [PubMed] [Google Scholar]
- 21.Weitz JI, Linkins LA. Expert Opin. Investig. Drugs. 2007;16:271–282. doi: 10.1517/13543784.16.3.271. [DOI] [PubMed] [Google Scholar]
- 22.Bhaskar U, Sterner E, Hickey AM, Onishi A, Zhang F, Dordick JS, Linhardt RJ. Appl. Microbiol. Biotechnol. 2012;93:1–16. doi: 10.1007/s00253-011-3641-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turnbull J. Science. 2011;334:462–463. doi: 10.1126/science.1211605. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Zhang Z, Linhardt RJ. Nat Prod Rep. 2009;26:313–321. doi: 10.1039/b819896a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esko JD, Selleck SB. Ann. Rev. Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 26.McCormick C, Leduc Y, Martindale D, Mattison K, Esford LE, Dyer AP, Tufaro F. Nat. Genet. 1998;19:158–161. doi: 10.1038/514. [DOI] [PubMed] [Google Scholar]
- 27.Li J.-p., Hagner-McWhirter A, Kjellen L, Palgi J, Jalkanen M, Lindahl U. J. Biol. Chem. 1997;272:28158–28163. doi: 10.1074/jbc.272.44.28158. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi M, Habuchi H, Habuchi O, Saito M, Kimata K. J. Biol. Chem. 1996;271:7645–7653. doi: 10.1074/jbc.271.13.7645. [DOI] [PubMed] [Google Scholar]
- 29.Habuchi H, Kobayashi M, Kimata K. J. Biol. Chem. 1998;273:9208–9213. doi: 10.1074/jbc.273.15.9208. [DOI] [PubMed] [Google Scholar]
- 30.Shworak NW, Liu J, Fritze LMS, Schwartz JJ, Zhang L, Logeart D, Rosenberg RD. J. Biol. Chem. 1997;272:28008–28019. doi: 10.1074/jbc.272.44.28008. [DOI] [PubMed] [Google Scholar]
- 31.Orellana A, Hirschberg CB, Wei Z, Swiedler SJ, Ishihara M. J. Biol. Chem. 1994;269:2270–2276. [PubMed] [Google Scholar]
- 32.Kakuta Y, Sueyoshi T, Negishi M, Pedersen LC. J. Biol. Chem. 1999;274:10673–10676. doi: 10.1074/jbc.274.16.10673. [DOI] [PubMed] [Google Scholar]
- 33.Duncan MB, Liu M, Fox C, Liu J. Biochem. Biophys. Res. Commun. 2006;339:1232–1237. doi: 10.1016/j.bbrc.2005.11.142. [DOI] [PubMed] [Google Scholar]
- 34.Peterson SP, Frick A, Liu J. Nat Prod Rep. 2009;26:61–627. doi: 10.1039/b803795g. [DOI] [PubMed] [Google Scholar]
- 35.Chen M, Bridges A, Liu J. Biochemistry. 2006;45:12358–12365. doi: 10.1021/bi060844g. [DOI] [PubMed] [Google Scholar]
- 36.Sismey-Ragatz AE, D.E. G, Otto NJ, Rejzek M, Field RA, Deangelis PL. J. Biol. Chem. 2007;282:28321–28327. doi: 10.1074/jbc.M701599200. [DOI] [PubMed] [Google Scholar]
- 37.Weïwer M, Sherwood T, Green DE, DeAngelis PL, Liu J, Linhardt RJ. J Org Chem. 2008;73:7631–7637. doi: 10.1021/jo801409c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masuko S, Bera S, Green DE, Weiwer M, DeAngelis PL, Linhardt RJ. J. Org. Chem. 2012;77:1449–1456. doi: 10.1021/jo202322k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sala RF, MacKinnon SL, Palcic MM, Tanner ME. Carbohydr. Res. 1998;306:127–136. doi: 10.1016/s0008-6215(97)10033-7. [DOI] [PubMed] [Google Scholar]
- 40.Liu R, Xu Y, Chen M, Weïwer M, Zhou X, Bridges AS, DeAngelis PL, Zhang Q, Linhardt RJ, Liu J. J Biol Chem. 2010;285:34240–34249. doi: 10.1074/jbc.M110.159152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sterner E, Masuko S, Li G, Li L, Green D, Otto NJ, Xu Y, DeAngelis PL, Liu J, Dordick JS, Linhardt RJ. J. Biol. Chem. 2014;289:9754–9765. doi: 10.1074/jbc.M113.546937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chandarajoti K, Xu Y, Sparkenbaugh E, Key NS, Pawlinski R, Liu J. Glycobiology. 2014 doi: 10.1093/glycob/cwu016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y, Li Y, Yu H, Sugiarto G, Thon V, Hwang J, Ding L, Hie L, Chen X. Angew. Chem. Int. Ed. 2013;52:11852–11856. doi: 10.1002/anie.201305667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu C, Sheng J, Krahn JM, Perera L, Xu Y, Hsieh P, Duo W, Liu J, Pedersen LC. J. Biol. Chem. 2014;289:13407–13418. doi: 10.1074/jbc.M113.530535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Y, Pempe E, Liu J. J. Biol. Chem. 2012;287:29054–29061. doi: 10.1074/jbc.M112.358523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Z, Yang B, Zhang Z, Ly M, Takieddin M, Mousa SA, Liu J, Dordick JS, Linhardt RJ. Appl Biochem Biotech. 2011;91:91–99. doi: 10.1007/s00253-011-3231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J-P, Gong F, Darwish KE, Jalkanen M, Lindahl U. J. Biol. Chem. 2001;276:20069–20077. doi: 10.1074/jbc.M011783200. [DOI] [PubMed] [Google Scholar]
- 48.Sheng J, Xu Y, Dulaney SB, Huang X, Liu J. J. Biol. Chem. 2012;287:20996–21002. doi: 10.1074/jbc.M112.359885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheng Z, Liu R, Xu Y, Liu J. J Biol Chem. 2011;286:19768–19776. doi: 10.1074/jbc.M111.224311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinay P. Carbohydr. Res. 1984;132:C5–C9. [Google Scholar]
- 51.Cai C, Li L, Harvey D, Liu J, Linhardt RJ. Tetrahedron Lett. 2013;54:4471–4474. doi: 10.1016/j.tetlet.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balagurunathan K, Lech MZ, Beeler DL, Wu ZL, Rosenberg RD. Nat. Biotechnol. 2003;21:1343–1346. doi: 10.1038/nbt885. [DOI] [PubMed] [Google Scholar]
- 53.Moon AF, Xu Y, Woody S, Krahn JM, Linhardt RJ, Liu J, Pedersen LC. Proc. Natl. Acad. Sci. USA. 2012;109:5256–5270. doi: 10.1073/pnas.1117923109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petitou M, Jacquinet JC, Choay J, Lormeau JC, Nassr M. 4,818,816 US Patent. 1989
- 55.Driguez P-A, Potier P, Trouileux P. Nat Prod Rep. 2014 doi: 10.1039/c4np00012a. in press. [DOI] [PubMed] [Google Scholar]
- 56.Liu R, Liu J. Biochemistry. 2011;50:4382–4391. doi: 10.1021/bi200072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peterson SB, Liu J. J. Biol. Chem. 2012;287:34836–34843. doi: 10.1074/jbc.M112.390161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsieh P, Xu Y, Keire DA, Liu J. Glycobiology. 2014 doi: 10.1093/glycob/cwu032. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.