

The crystal structure of human P-cadherin EC1-EC2 in a closed conformation is reported at 1.62 Å resolution, showing a double conformation of Trp2 in the binding pocket and a novel monomeric packing arrangement.
Keywords: cadherin, dimerization mechanism, cell adhesion
Abstract
Cadherins are a large family of calcium-dependent proteins that mediate cellular adherens junction formation and tissue morphogenesis. To date, the most studied cadherins are those classified as classical, which are further divided into type I or type II depending on selected sequence features. Unlike other members of the classical cadherin family, a detailed structural characterization of P-cadherin has not yet been fully obtained. Here, the high-resolution crystal structure determination of the closed form of human P-cadherin EC1-EC2 is reported. The structure shows a novel, monomeric packing arrangement that provides a further snapshot in the yet-to-be-achieved complete description of the highly dynamic cadherin dimerization pathway. Moreover, this is the first multidomain cadherin fragment to be crystallized and structurally characterized in its closed conformation that does not carry any extra N-terminal residues before the naturally occurring aspartic acid at position 1. Finally, two clear alternate conformations are observed for the critical Trp2 residue, suggestive of a transient, metastable state. The P-cadherin structure and packing arrangement shown here provide new and valuable information towards the complete structural characterization of the still largely elusive cadherin dimerization pathway.
1. Introduction
Cadherins constitute a large family of transmembrane calcium-dependent cell-adhesion proteins that play a crucial role in tissue morphogenesis and stability, and that provide tissues with specific mechanical properties such as elasticity and the ability to withstand mechanical stress (Gumbiner, 2005 ▶; Sivasankar, 2013 ▶). Loss of cadherin-mediated adhesion has been implicated in many different steps of tumour progression such as invasion and migration, and is strongly related to cell–cell detachment and metastasis (Cowin et al., 2005 ▶). The different family members are structurally organized into an elongated extracellular portion formed by a variable number of immunoglobulin-like (Ig-like) domains (ECs) that are linked to each other by small connecting sequences, a single transmembrane pass and an intracellular portion that dynamically associates with cytoplasmic molecules called catenins. Three calcium ions are found at the interface between each pair of Ig-like domains in the extracellular portion of the protein, coordinated by conserved aspartic acids. Calcium ions have been shown to provide rigidity to the protein and to be essential for protein activity (Ciatto et al., 2010 ▶). Among the cadherin family members, those classified as classical share a conserved cytoplasmic domain and an ectodomain composed of five Ig-like ECs.
Placental cadherin (P-cadherin) is a prominent member of the classical cadherin subfamily. Originally found to be highly expressed in mouse placenta during pregnancy (Nose & Takeichi, 1986 ▶), the protein was subsequently found not to be expressed in human placenta (Shimoyama et al., 1989 ▶). Although transient P-cadherin expression can be observed in several types of tissues during development, the protein is permanently found only at the level of cell–cell junctions in adult epithelial tissues, partially co-localizing with E-cadherin (Hirai et al., 1989 ▶; Wakita et al., 1998 ▶; Shimoyama & Hirohashi, 1991 ▶; Fujita et al., 1992 ▶; Wu et al., 1993 ▶). Mutations in the gene encoding P-cadherin (CDH3) have been found to be associated with hypotrichosis and juvenile macular dystrophy, a rare congenital disease that causes progressive retinal degeneration and leads to early blindness (Sprecher et al., 2001 ▶). Several studies have also indicated a clear, albeit often conflicting, role of P-cadherin in different types of cancer, such as malignant melanoma and breast cancer (Paredes et al., 2007 ▶, 2012 ▶). In fact, whereas it appears to inhibit cell detachment and metastasis in melanoma patients (Van Marck et al., 2005 ▶), P-cadherin expression is considered to be a marker of tumour aggressiveness in oestrogen receptor (ER)/progesterone receptor (PgR)/HER2-triple negative breast cancers, usually representing a poor prognosis factor in the triple-negative subgroup of patients (Paredes et al., 2005 ▶). Furthermore, P-cadherin upregulation is observed in gastric, lung, colorectal and pancreatic cancer patients (Imai et al., 2008 ▶; Taniuchi et al., 2005 ▶). Hence, efforts to develop therapeutic agents against P-cadherin have recently intensified (Yoshioka et al., 2012 ▶; Zhang et al., 2010 ▶; Park et al., 2012 ▶).
Over the last two decades, a detailed analysis of the structure–function relationships in some of the most prominent members of the large cadherin family has been perfomed using a wealth of biophysics and cell-biology approaches, paving the way to our current understanding of the basic molecular mechanisms behind cadherin-mediated adhesion and homophilic dimerization (Leckband & Sivasankar, 2012 ▶; Vendome et al., 2011 ▶). As a result of structural and mutational studies conducted mostly using type I cadherins such as E-cadherin, N-cadherin and C-cadherin, as well as the type II cadherins cadherin-11, cadherin-8 and MN-cadherin, a strand-exchange mechanism has been clearly identified as the ‘endpoint’ of a highly dynamical homodimerization process (Nagar et al., 1996 ▶; Pertz et al., 1999 ▶; Boggon et al., 2002 ▶; Parisini et al., 2007 ▶; Patel et al., 2006 ▶; Shapiro et al., 1995 ▶). This mechanism consists of the opening of the N-terminal adhesion arm and the insertion of the side chain of the conserved tryptophan at position 2 (Trp2) into a highly conserved acceptor pocket in the extracellular domain 1 (EC1) of the partner molecule that protrudes from the surface of the neighbouring cell. Whereas this trans homophilic interaction occurs at the level of the EC1 of two cadherins binding each other from opposing cells to form strand dimers, further stabilizing contacts, mostly hydrophobic in nature, are found in all strand-swap structures between the EC1 of a cadherin and the EC2 of the neighbouring molecule protruding from the same cell to form the so-called cis interaction. It is believed that this lateral assembly supports and enhances cadherin adhesion properties (Harrison et al., 2011 ▶). Overall, the system forms a dynamic zipper-like structure at the cell–cell adhesion junction. The adhesion strength is mostly owing to protein avidity resulting from a diffuse network of relatively weak pairwise interactions between individual cadherins, with the K d value for homophilic adhesion of type I cadherins being in the submillimolar range (Ciatto et al., 2010 ▶; Häussinger et al., 2004 ▶).
The strand-dimer conformation, with its zipper-like packing arrangement, is not the only dimeric form that has been functionally and structurally characterized. Nonclassical T-cadherin, which lacks the Trp2-containing strand and is therefore unable to promote strand-swap dimer formation, has been found to dimerize at the Ca2+-binding site between the EC1 and EC2 domains, forming what is usually referred to as the X-dimer (Ciatto et al., 2010 ▶), a structural arrangement that is topologically identical to that found in E-cadherin mutants which are unable to form the strand-swap dimer (Harrison et al., 2010 ▶; Nagar et al., 1996 ▶; Pertz et al., 1999 ▶). Recently, the X-dimer of P-cadherin has also been identified and characterized by a combination of mutational, spectroscopic and thermodynamic approaches (Kudo et al., 2012 ▶, 2014 ▶). Like the strand-swap dimer, the X-dimer arrangement also provides adhesive force between cadherin molecules (Rakshit et al., 2012 ▶) and it is believed to be a crucial intermediate state in the full cadherin dimerization trajectory. The X-dimer interface has in fact been shown to lower the energy barrier associated with strand swapping (Li et al., 2013 ▶). While it is possible that different pathways can lead to strand-exchange formation, Li and coworkers clearly demonstrated that the X-dimer-dependent pathway is by far the major dimerization trajectory. Interestingly, variable levels of cross-reactivity between highly homologous cadherins belonging to the same subfamily have been described in the context of both strand-swap and X-dimer formation (Kudo et al., 2014 ▶; Katsamba et al., 2009 ▶; Vendome et al., 2014 ▶).
All previous structural determinations have led to the implicit assumption that at high protein concentrations such as those usually reached in crystallization experiments, strand-swap dimers would preferentially form in the crystal, leaving the fundamental question as to how this highly dynamic system would shuttle between two forms characterized by essentially similar free energies, the monomeric and the adhesive configuration, still unanswered. The existence of a dynamic equilibrium between the monomeric and the adhesive forms is clearly supported by both mutagenesis (Harrison et al., 2005 ▶) and NMR data (Sivasankar, 2013 ▶; Harrison et al., 2011 ▶; Häussinger et al., 2002 ▶), the latter having also demonstrated the presence of a further equilibrium involving two different monomeric forms of the protein, the closed and the open monomer, characterized by a different conformation of the arm (Miloushev et al., 2008 ▶). When two cadherin molecules from adjacent cells approach each other, strand-dimer formation occurs, supporting the notion that the closed protein conformation showing intramolecular docking of the adhesion arm is energetically less stable than the open form displaying intermolecular docking (Vendome et al., 2011 ▶). Interestingly, when classical cadherin fragments carry one or more extra residues at their N-terminus, the closed conformation is favoured over the strand-swap conformation owing to steric hindrance (Nagar et al., 1996 ▶; Pertz et al., 1999 ▶). In fact, since all classical cadherins are expressed as inactive precursor proteins and become active upon proteolytic cleavage of their prodomain sequence (Ozawa & Kemler, 1990 ▶; Reinés et al., 2012 ▶), it is believed that any extra protein sequence N-terminal to Asp1 would mimic the prodomain function, thus resulting in the protein being devoid of its adhesive potential. Quite surprisingly, the human P-cadherin EC1-EC2 structure reported here, corresponding to the natural, mature sequence of the protein, is found in its closed, monomeric conformation. It is possible that this novel packing arrangement represents yet another intermediate in the arguably rather flat potential energy landscape along the full cadherin dimerization pathway. It has in fact become increasingly clear that within adherens junctions several possible adhesive conformations are likely to coexist simultaneously, although their interconversion mechanism is still largely elusive (He et al., 2003 ▶). It is therefore important to try to characterize at the molecular level as many different steps as possible in the complex cadherin adhesion pathway. This would in fact clearly help elucidate not only the complete cadherin dimerization trajectory but also fundamental aspects such as the molecular bases of the homophilic binding specificity among the different members of the cadherin family.
2. Materials and methods
2.1. Cloning, expression and purification of human P-cadherin EC1-EC2
A DNA fragment encoding EC1-EC2 of human P-cadherin (residues 1–213) was obtained by PCR from the full-length sequence of the human P-cadherin extracellular portion and cloned into a pET-3a expression vector from Novagen using the NdeI and BlpI restriction sites.
The 213-amino-acid sequence was fused at its N-terminus to a 6×His tag, a spacer peptide (SSGHI) and the enterokinase-recognition site (DDDDK). Overnight protein expression at room temperature in the BL21(DE3)pLysS Escherichia coli strain (Invitrogen) afforded soluble protein in high yield. The cell lysate in Tris-buffered saline solution (TBS; pH 7.4) with 1 mM CaCl2 was purified on an Ni–NTA column and then passed through a Sephacryl 100 HR HiPrep 26/60 size-exclusion column (GE Healthcare), affording a sample of nearly 100% purity as detected by Coomassie staining. The protein sample was then dialyzed in TBS buffer containing 20 mM CaCl2, digested at room temperature with enterokinase (New England Biolabs) and subsequently passed over an Ni–NTA column to remove all traces of the cleaved 6×His tag as well as any residual uncleaved protein. The flowthrough was then collected and further purified using a Sephacryl 100 HR HiPrep 26/60 size-exclusion column (GE Healthcare) using TBS with 1 mM CaCl2 as the mobile phase and finally brought to a concentration of 14 mg ml−1 for crystallization experiments.
2.2. Crystallization and data collection
Crystals of the protein were obtained by the vapour-diffusion method at room temperature. A 1 µl drop of the protein sample was mixed with an equal volume of a 16%(w/v) PEG 12 000, 0.2 M CaCl2, 0.1 M HEPES pH 7.4, 10%(v/v) dimethyl sulfoxide (DMSO) solution. Crystals were then cooled in a chemically identical solution supplemented with 20%(v/v) glycerol for cryoprotection. A 1.62 Å resolution data set was collected from a single P-cadherin EC1-EC2 crystal of 0.15 × 0.10 × 0.10 mm in size at a wavelength of 1.000 Å on the X06DA-PXIII beamline at the Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland. Diffraction images were processed and scaled using XDS (Kabsch, 2010 ▶). Data-collection and refinement statistics are shown in Table 1 ▶.
Table 1. Data-collection, structure-solution and refinement statistics.
Values in parentheses are for the outer shell.
| Space group | C121 |
| Unit-cell parameters | |
| a () | 75.38 |
| b () | 40.95 |
| c () | 72.41 |
| () | 98.05 |
| Molecules per asymmetric unit | 1 |
| Wavelength () | 1.00 |
| Exposure time (s) | 0.1 |
| Total rotation range () | 200 |
| Rotation range per image () | 0.1 |
| Resolution range () | 37.321.62 (1.681.62) |
| Unique reflections | 27755 |
| Multiplicity | 2.0 (1.7) |
| Completeness (%) | 98.8 (99.3) |
| R merge † (%) | 4.1 (58.8) |
| Mean I/(I) | 13.64 (1.97) |
| Reflections (work/test) | 26366/1388 |
| Wilson B factor (2) | 20.4 |
| Average mosaicity () | 0.33 |
| R ‡/R free (%) | 16.3/20.2 |
| R merge | 0.041 (0.604) |
| Ramachandran plot (%) | |
| Core | 97.99 |
| Allowed | 2.01 |
| Generously allowed | 0.00 |
| Disallowed | 0.00 |
| No. of atoms | |
| Protein | 1705 |
| Calcium | 3 |
| Water | 337 |
| Average B factor (2) | |
| Protein | 28.12 |
| Ligand | 21.07 |
| Water | 39.74 |
| R.m.s.d. from ideal values | |
| Bond lengths () | 0.012 |
| Bond angles () | 1.434 |
R
merge = 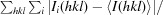
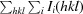 , where I
i(hkl) is the observed intensity and I(hkl) is the average intensity from observations of symmetry-related reflections.
, where I
i(hkl) is the observed intensity and I(hkl) is the average intensity from observations of symmetry-related reflections.
R = 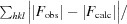
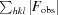 . A subset of the data (5%) was excluded from the refinement and used to calculate R
free.
. A subset of the data (5%) was excluded from the refinement and used to calculate R
free.
2.3. Structure determination
The structure was determined by molecular replacement using MOLREP (Vagin & Teplyakov, 2010 ▶) from the CCP4 package (Winn et al., 2011 ▶) with the human E-cadherin D1-D2 crystal structure (PDB entry 2o72; Parisini et al., 2007 ▶) as the search probe. Model building and refinement were carried out using REFMAC5 (Murshudov et al., 2011 ▶) and PHENIX (Adams et al., 2010 ▶). Water molecules were added both automatically using WATERTIDY from the CCP4 package (Winn et al., 2011 ▶) and manually from visual inspection of the electron-density map. All figures in the paper were generated using PyMOL (http://www.pymol.org) or Chimera (Pettersen et al., 2004 ▶). The refinement converged to a final R and R free of 16.3 and 20.2%, respectively. The final crystallographic coordinates are available in the RCSB PDB as entry 4oy9.
3. Results and discussions
The closed-form crystal structure of human P-cadherin EC1-EC2 reported here has been determined at 1.62 Å resolution, which is the highest resolution ever attained for a multidomain classical cadherin EC fragment. It consists of the two N-terminal extracellular domains (EC1-EC2) of human P-cadherin (Fig. 1 ▶ a). Each domain displays a typical cadherin domain topology comprising seven β-strands arranged in two sheets packing against each another to form an Ig-like domain fold. To date, only a few cadherin structures have been determined in their closed conformation, among which only mouse E-cadherin EC1-EC2 (PDB entry 1ff5; Pertz et al., 1999 ▶), human E-cadherin EC1 in complex with internalin from Listeria monocytogenes (PDB entry 1o6s; Schubert et al., 2002 ▶), human E-cadherin EC1 in complex with two different portions of the NK cell receptor KLRG1 (PDB entries 3ff7 and 3ff8; Li et al., 2009 ▶) and human E-cadherin EC1-EC2 bound to botulinum neurotoxin A complex (PDB code 4qd2; Lee et al., 2014 ▶) show a structurally defined and fully refined N-terminal adhesion arm. These five E-cadherin structures have so far provided the only available representations of a closed-conformation cadherin, despite these fragments having, with the exception of PDB entry 4qd2, additional amino acids N-terminal to the naturally occurring terminus and therefore being essentially forced into a conformation devoid of its natural homophilic dimerization potential. More importantly, since a role of the partner molecule (internalin, KLRG1 or botulinum neurotoxin A complex) in forcing the intermolecularly docked conformation of E-cadherin in the corresponding heterodimeric complexes cannot be completely ruled out, we can certainly consider the human P-cadherin EC1-EC2 structure reported here as the first true closed-conformation cadherin species crystallized to date. Nevertheless, it is interesting to compare the extent to which the arm contacts the main body of the protein in all of the closed cadherin structures and, indirectly, to catch a glimpse of the dynamic propensity of the adhesion arm. In the structure with PDB code 3ff7, there are two independent heterodimeric complexes in the asymmetric unit, both showing hydrogen-bonding contacts between the arm and the main body of the cadherin: CO(Lys25)⋯NH(Val3), 2.72 Å, and CO(Lys25)⋯NH(Val3), 2.82 Å (2.73 and 2.87 Å, respectively, in the second subunit). Likewise, in the structure with PDB code 3ff8 a very similar picture emerges from the two independent heterodimeric complexes that are present in the asymmetric unit: CO(Lys25)⋯NH(Val3), 2.75 Å, and CO(Lys25)⋯NH(Val3), 2.72 Å (2.73 and 2.77 Å, respectively, in the second subunit). Similarly, in the structure with PDB code 4qd2 the CO(Lys25)⋯NH(Val3) distances in the two independent heterodimeric complexes in the asymmetric unit are 2.93 and 2.82 Å. In the structure with PDB code 1o6s, the Asp1 residue was mutated to Ser. However, an almost identical hydrogen-bonding pattern can be observed: CO(Lys25)⋯NH(Val3), 2.97 Å, and CO(Ser1)⋯NH(Asn27), 2.81 Å. In all of these cases the data are suggestive of a relatively tight interaction between the arm and the main body of the protein, most likely facilitated by the intimate wrapping of the partner molecules. Since PDB entry 1ff5 is the other only available closed cadherin structure that is not involved in heterodimer formation, it appears to provide the most appropriate model for the comparison of the relevant structural features with the structure reported here, despite a substantial difference in resolution between the two structures (1.62 versus 2.93 Å), the presence of an extra residue (Met) at the N-terminus in PDB entry 1ff5 and the fact that PDB entry 1ff5 is in the X-dimer configuration. Overall, the r.m.s.d. between the mature human P-cadherin EC1-EC2 structure in its closed conformation shown here and the mouse E-cadherin EC1-EC2 structure carrying one extra residue at the N-terminus (PDB entry 1ff5) is less than 1 Å for main-chain atoms, suggesting that, globally, the two structures can be largely superimposed (Fig. 1 ▶ b). An essentially hydrophobic stabilizing interface is formed between the adhesion arm and the bulk of the EC1 of the protein, once again matching the closed-conformation mouse E-cadherin structure owing to the high sequence homology between the arm residues of the two molecules (DWVVAPI versus DWVIPPI). Here, in fact, the hydrogen-bonding interactions that were holding the cadherin monomers of the three known heterodimeric complexes mentioned before in a tight closed conformation are no longer present [CO(Lys25)⋯NH(Val3), 6.52 Å, and CO(Asp1)⋯NH(Asn27), 7.56 Å, in the P-cadherin EC1-EC2 structure reported here and CO(Lys25)⋯NH(Val3), 6.14 Å, and CO(Asp1)⋯NH(Asn27), 6.87 Å, in the structure with PDB code 1ff5 (6.70 and 6.47 Å, respectively, in the second subunit)] and only the large hydrophobic contact area between the arm and the main body of the protein remains to stabilize this conformation.
Figure 1.

(a) Crystal structure of human P-cadherin EC1-EC2 in its closed conformation. Three calcium ions (shown in red) are found at the interface between the extracellular domains 1 and 2. (b) Superposition of the human P-cadherin EC1-EC2 structure (shown in green) with the closed-conformation mouse E-cadherin EC1-EC2 structure (PDB entry 1ff5; shown in yellow).
The crucial Trp2 residue forms the same contacts inside the conserved acceptor pocket as those previously observed in all (either strand-swapped or closed-conformation) classical cadherins. In brief, along with the hydrogen bond between the N∊H atom of the Trp2 side chain and the main-chain carbonyl group of Asp90 (2.48 Å), further stabilization is provided not only by a number of van der Waals contacts between the Trp2 side chain and hydrophobic residues in the pocket, but also by the stacking arrangement of the negatively charged carboxyl group of the Glu89 side chain, the π-electron cloud of the indole moiety of Trp2 and the slightly electron-rich side chain of Met92 (Fig. 2 ▶). In turn, both Glu89 and Met92 are highly conserved residues that are held in place by a number of stabilizing contacts with the surrounding amino acids, such as Ser26, which forms a hydrogen-bonding interaction with Glu89, and several hydrophobic residues that stabilize Met92 inside the pocket. Interestingly, Ser26 is totally conserved in all classical cadherins but one (cadherin-8, where it is mutated to the highly homologous Thr residue). Furthermore, in all cadherin structures, either open or closed, Met92 is always inside the hydrophobic pocket underneath the Trp2 residue, with the only exceptions being the three heterodimeric structures (PDB entries 3ff7, 3ff8 and 1o6s) discussed above, where the Met92 side chain appears to have moved out of the pocket in order to maximize its hydrophobic interactions with the surrounding residues of both the cadherin and the partner molecule. The distance between the Glu89 Cβ and Met92 Cβ atoms is 6.71 Å. Considering the sum of the van der Waals radii of the atoms, the Trp2 side chain is clearly tightly sandwiched between the rigid ‘roof’ and ‘floor’ of the pocket. Altogether, this is suggestive of an essentially invariable size and shape of the adhesive pocket in all classical cadherins, which explains the lack of mobility and the fixed orientation of the Trp2 side chain inside the pocket across all known cadherin structures.
Figure 2.

Stabilizing contacts with the Trp2 double conformation inside the hydrophobic acceptor pocket. Two hydrogen bonds are formed between the N atom of the indole in its double conformation and the carbonyl O atoms of Asp90 and Lys25. The stacking arrangement of electron-rich residues involving Glu89, Trp2 and Met92 (shown in the two insets at a 90° rotation from each other) further contributes to the overall stabilization of the system.
Despite these close similarities to the structure with PDB code 1ff5, a careful inspection of the electron-density map around the tryptophan side chain of the high-resolution P-cadherin EC1-EC2 structure reported here revealed the presence of further, unaccounted-for, strong electron density in plane with the Trp2 indole moiety, suggesting the possibility of a second, alternate configuration of the whole indole ring (Fig. 3 ▶). In fact, during the refinement process this second electron-density zone could be easily and convincingly assigned to a flipped conformation of the whole ring, whereby a second hydrogen-bonding stabilizing contact could be formed between the indole N∊H atom and the carbonyl O atom of the Lys25 residue (2.58 Å; Figs. 2 ▶ and 3 ▶). The two alternate conformations of the ring are almost completely coplanar and could be perfectly modelled by assigning each of the two forms an occupancy value of 0.5 during refinement. The six-membered benzene rings are perfectly superimposed in the two conformers. The second indole conformation in the adhesion pocket is also stabilized by the same stacking arrangement formed by the surrounding residues and described above. Owing to the rather limited space available inside the pocket for the indole ring to complete a 180° rotation around its main axis, the second conformer can only be formed when the protein is in solution as a result of a dynamic equilibrium whereby the ring exits the pocket first, flips around outside the pocket and reinserts into the cavity in the two possible alternative conformations, with the N∊H atom pointing either towards the Asp90 or the Lys25 carbonyl O atom. The existence of a dynamic opening and closing movement of the arm for the monomeric form of the protein had been previously suggested by NMR experiments, as a small population of open-arm monomers could be detected in solution (Miloushev et al., 2008 ▶). The extremely conserved geometry of the pocket in all available cadherin structures, the short and therefore rather strong hydrogen-bonding contact formed by the N∊H atom of the indole and the carbonyl O atom of Asp90 and the tight geometrical constraints that are forced upon the tryptophan side chain by the Glu89 and Met92 residues, whose positions are in turn further stabilized by several other conserved interactions, are clearly incompatible with an internal reorientation of the indole, which to complete a 180° rotation inside the pocket would require a large sweeping radius that is clearly not sterically possible. Overall, the structure demonstrates the existence of a concerted motion whereby Trp2 moves from its original orientation inside the pocket to the outside and then inside again in a flipped conformation as a result of the constant opening and closing movement of the arm. Therefore, prior to nucleation and crystal growth, a mixed population of molecules with the two alternative Trp2 side-chain orientations is present in solution, resulting in the two conformations observed in the crystal.
Figure 3.

Trp2 double-conformation assignment during refinement. (a) OMIT maps of the crucial adhesion arm provided a clear indication of the closed conformation of the protein in the crystal, with the Trp2 residue fitting into the protein’s own acceptor pocket. (b, c, d) The electron-density map corresponding to the Trp2 side chain was found to correspond to a double conformation of the indole moiety (50% occupancy each). Strong residual electron density was in fact evident when only one of the two conformations was refined in the model. The two flipped conformations are essentially coplanar, with the six-membered ring almost perfectly coinciding in the two orientations. The maps are contoured at the 1.0 and 1.5σ levels.
In the crystal, the Asp1 residue projects from the protein and does not make any contacts that may help to stabilize the protein in its closed conformation (Fig. 1 ▶). This is likely to represent the initial event in the arm-opening process. By detaching itself from the main body of the protein, the side chain of Asp1 frees the carbonyl O atom of Lys25, with which it was previously forming a stabilizing interaction, as observed in the structure with PDB code 1ff5. In fact, even in the 1ff5 structure the two independent molecules in the asymmetric unit show two significantly different Asp1⋯Lys25 interaction modes, thus providing further evidence of the transient nature of these contacts. Moreover, as clearly demonstrated by NMR studies, the Trp2 side chain is not held firmly in the hydrophobic pocket and the N-terminus of the protein is extremely flexible in solution (Häussinger et al., 2004 ▶). Häussinger and coworkers have already suggested an important role for the carbonyl O atom of Lys25 in forming a strong, albeit transient, hydrogen bond to the amide N atom of Val3, both intramolecularly (closed form) and intermolecularly (strand-exchange form), following Trp2 insertion in the pocket. In our structure, this hydrogen-bonding interaction is not present; the distance between the amide N atom of Val3 and the carbonyl O atom of Lys25 is over 6 Å. Instead, the structure displays a rather loose and mostly hydrophobic interaction between the arm and the main body of the protein as well as the weak and transient anchoring effect provided by the Trp2 residue in the two different orientations inside the pocket. The Val3⋯Lys25 hydrogen bond therefore appears to be present only in the strand-swapped or in the tightly closed forms of the protein, breaking as the protein shuttles between the two ‘endpoint’ conformations.
Unlike the closed-conformation mouse E-cadherin EC1-EC2 structure determined previously (PDB entry 1ff5), which having an extra residue at the N-terminus is devoid of strand-exchange dimerization potential and packs to form an X-dimer in the crystal, the P-cadherin structure shown here retains a monomeric state and displays a novel antiparallel packing arrangement in the crystal, possibly representing an intermediate state in the cadherin dimerization trajectory. The main axis of the two antiparallel proteins are at an angle of 4.0° (interaxial distance of 24.2 Å), i.e. very different from the angle observed between the two partner molecules in both the strand dimer (65.7°; interaxial distance of 20.3 Å) and the X-dimer (52.9°; interaxial distance of 17.2 Å) (Fig. 4 ▶ a). It is worth noting that the disordered Asp1 residue in the structure makes water-mediated contacts with the carbonyl O atoms of the Ile139 and Tyr140 amino acids that belong to the rather long BC-loop ranging from residue 134 to residue 143 in domain 2 of the next-neighbour protein (Fig. 5 ▶). In turn, the loop is shifted with respect to the conformation adopted in all other available cadherin structures. Interestingly, the loop shifts downwards towards the N-terminal portion of the partner molecule, where it forms water-mediated contacts with the Asp1 and Val3 main-chain atoms. While we cannot exclude that these are just crystal contacts, it is possible that in the context of the highly dynamic cadherin dimerization mechanism the shift of the loop may be caused by transient adhesion forces coupling the early stages in the opening of the adhesion arm with the relative orientation of the two adjacent proteins. The loop may in fact act as a spacer, defining the correct distance between two monomers and eventually accompanying their reciprocal movement towards an adhesive conformation, possibly playing a role in selectivity. In the structure reported here, the relative orientation of two next-neighbour monomeric molecules is suggestive of a mutual transient engagement of the Asp1 residue of each molecule with the BC-loop of domain 2 of the partner molecule, a reciprocal recognition mechanism that is reminiscent of the strand-swap dimer and that may subsequently lead two neighbouring cadherin molecules to slide relative to each other, either towards the X-dimer or the strand-swap configuration (Fig. 4 ▶ b). The first and the last residues in the loop (Asp136 and Asn143, respectively), which are conserved in all type I cadherins, are both involved in the coordination of calcium ions at the interface between EC1 and EC2. The side chain of Asp136 simultaneously coordinates two distinct Ca2+ ions, while the main-chain O atom of Asn143 coordinates only one of them. The Ca2+ ions that are coordinated by the highly conserved first and last residues of the loop appear to act as a hinge in the observed rotational movement of the loop, further supporting the notion that the Ca2+ ions play an important role in conferring adhesive properties to cadherin molecules (Vunnam & Pedigo, 2011 ▶; Cailliez & Lavery, 2005 ▶; Prakasam et al., 2006 ▶). Previous studies of E-cadherin and T-cadherin have highlighted the important role of this loop (Ciatto et al., 2010 ▶; Harrison et al., 2010 ▶), showing how X-dimer formation is in fact mediated by polar amino acids in the loop sequence. In these studies, mutations such as Y142R in E-cadherin and D140K in T-cadherin have been shown to interfere with X-dimer formation and slow down strand-swap dimerization. Since a sequence alignment of all classical cadherins shows length and sequence variability in the loop (Fig. 6 ▶), we hypothesize that the loop may also be an important structural element in conferring homophilic binding specificity between different family members by acting as a structural checkpoint at different stages along the full cadherin dimerization pathway.
Figure 4.

(a) The relative orientation of next-neighbour molecules in different cadherin forms and packing arrangements. (I) The packing arrangement of the closed-conformation human P-cadherin EC1-EC2 monomer. (II) The closed-conformation mouse E-cadherin EC1-EC2 X-dimer (PDB entry 1ff5). (III) The human E-cadherin EC1-EC2 strand-swap dimer (PDB entry 2o72). (b) Schematic representation of a putative model of a multistep cadherin dimerization pathway. Upon enzymatic cleavage, the cadherin prodomain is removed to afford a mature cadherin protein primed for adhesion. In the early stages of the dimerization process, an equilibrium between open and closed monomers exists. Cellular polarization events lead to an increase in the protein concentration, ultimately triggering a concerted and highly dynamic cadherin-recognition process. Transient, metastable interactions may allow molecules from opposing cells to orient themselves in an antiparallel fashion, as shown by the P-cadherin EC1-EC2 structure reported here, and to subsequently slide into a dimeric/adhesive form. The dimerization process can then dynamically revert to again provide monomeric cadherin as a result of mechanical forces. The lengths of the arrows in this scheme are purely indicative and do not reflect the relative likelihoods of the different pathways.
Figure 5.

(a) Two next-neighbouring P-cadherin EC1-EC2 monomers. The relative orientation of the two symmetry-related molecules allows water-mediated contacts between the N-terminal portion of the adhesion arm of each molecule and the BC-loop of domain 2 in the partner molecule, an arrangement that is reminiscent of the mutual recognition process seen in strand-swap dimers, albeit at different angular orientations. (b) Detailed representation of the water-mediated interactions between main-chain atoms of the adhesion arm of the reference molecule (shown in green) and main-chain atoms of the BC-loop in the EC2 of a partner molecule (shown in purple). This feature is symmetrical in the two interacting molecules.
Figure 6.

Sequence alignment of the BC-loop of domain 2 of type I classical cadherins from different species. While the BC-loop of domain 2 shows low sequence identity and different lengths among different cadherins, orthologous sequences of the same cadherin often appear to be fully conserved in the same loop region.
In summary, the high-resolution human P-cadherin EC1-EC2 crystal structure reported here shows a wealth of novel structural details that provide new insights into the highly dynamic cadherin dimerization pathway and allow the identification of key structural elements that are involved in the stabilization of what appears to be an intermediate configuration in the cadherin dimerization process. Although the mature protein, devoid of any extra residues at the N-terminus that could mimic the original prodomain, is primed for adhesion, the structure is found in its closed conformation. It is important to note that this is the first crystal structure of a multi-domain cadherin fragment that is found in its monomeric form and the only one that is in its closed conformation without being either complexed with another protein substrate or mutated to prevent strand-swap dimerization. Therefore, the monomeric structure reported here provides for the first time, and at the one of the highest resolutions ever attained for any cadherin structure, an unbiased representation of the closed configuration of a cadherin molecule. The structural comparison of different cadherins in their various activation stages can clearly contribute to the complete elucidation of the molecular bases of the cadherin binding mechanism and homospecificity and help to clarify how the entropic cost of dimerization may be overcome in a system in which the closed and the adhesive forms of the protein have essentially similar free energies.
Supplementary Material
PDB reference: human P-cadherin EC1-EC2, 4oy9
Acknowledgments
We thank the X06DA-PXIII beamline personnel at the Paul Scherrer Institute, Villigen, Switzerland for help with data collection. The European Union (‘Marie Curie’ FP7 IRG grant No. 268231 to EP) is gratefully acknowledged for financial support.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Boggon, T. J., Murray, J., Chappuis-Flament, S., Wong, E., Gumbiner, B. M. & Shapiro, L. (2002). Science, 296, 1308–1313. [DOI] [PubMed]
- Cailliez, F. & Lavery, R. (2005). Biophys. J. 89, 3895–3903. [DOI] [PMC free article] [PubMed]
- Ciatto, C., Bahna, F., Zampieri, N., VanSteenhouse, H. C., Katsamba, P. S. & Ahlsen, G. (2010). Nature Struct. Mol. Biol. 1, 339–347. [DOI] [PMC free article] [PubMed]
- Cowin, P., Rowlands, T. M. & Hatsell, S. J. (2005). Curr. Opin. Cell Biol. 17, 499–508. [DOI] [PubMed]
- Fujita, M., Furukawa, F., Fujii, K., Horiguchi, Y., Takeichi, M. & Imamura, S. (1992). Arch. Dermatol. Res. 284, 159–166. [DOI] [PubMed]
- Gumbiner, B. M. (2005). Nature Rev. Mol. Cell Biol. 6, 622–634. [DOI] [PubMed]
- Harrison, O. J., Bahna, F., Katsamba, P. S., Jin, X., Brasch, J., Vendome, J., Ahlsen, G., Carroll, K. J., Price, S. R., Honig, B. & Shapiro, L. (2010). Nature Struct. Mol. Biol. 17, 348–357. [DOI] [PMC free article] [PubMed]
- Harrison, O. J., Corps, E. M. & Kilshaw, P. J. (2005). J. Cell Sci. 118, 4123–4130. [DOI] [PubMed]
- Harrison, O. J. et al. (2011). Structure, 19, 244–256. [DOI] [PMC free article] [PubMed]
- Häussinger, D., Ahrens, T., Aberle, T., Engel, J., Stetefeld, J. & Grzesiek, S. (2004). EMBO J. 23, 1699–1708. [DOI] [PMC free article] [PubMed]
- Häussinger, D., Ahrens, T., Sass, H.-J., Pertz, O., Engel, J. & Grzesiek, S. (2002). J. Mol. Biol. 324, 823–839. [DOI] [PubMed]
- He, W., Cowin, P. & Stokes, D. L. (2003). Science, 302, 109–113. [DOI] [PubMed]
- Hirai, Y., Nose, A., Kobayashi, S. & Takeichi, M. (1989). Development, 105, 263–270. [DOI] [PubMed]
- Imai, K., Hirata, S., Irie, A., Senju, S., Ikuta, Y., Yokomine, K., Harao, M., Inoue, M., Tsunoda, T., Nakatsuru, S., Nakagawa, H., Nakamura, Y., Baba, H. & Nishimura, Y. (2008). Clin. Cancer Res. 14, 6487–6495. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Katsamba, P., Carroll, K., Ahlsen, G., Bahna, F., Vendome, J., Posy, S., Rajebhosale, M., Price, S., Jessell, T. M., Ben-Shaul, A., Shapiro, L. & Honig, B. H. (2009). Proc. Natl Acad. Sci. USA, 106, 11594–11599. [DOI] [PMC free article] [PubMed]
- Kudo, S., Caaveiro, J. M. M., Goda, S., Nagatoishi, S., Ishii, K., Matsuura, T., Sudou, Y., Kodama, T., Hamakubo, T. & Tsumoto, K. (2014). Biochemistry, 53, 1742–1752. [DOI] [PubMed]
- Kudo, S., Caaveiro, J. M. M., Miyafusa, T., Goda, S., Ishii, K., Matsuura, T., Sudou, Y., Kodama, T., Hamakubo, T. & Tsumoto, K. (2012). Mol. Biosyst. 8, 2050–2053. [DOI] [PubMed]
- Leckband, D. & Sivasankar, S. (2012). Curr. Opin. Cell Biol. 24, 620–627. [DOI] [PMC free article] [PubMed]
- Lee, K., Zhong, X., Gu, S., Kruel, A. M., Dorner, M. B., Perry, K., Rummel, A., Dong, M. & Jin, R. (2014). Science, 344, 1405–1410. [DOI] [PMC free article] [PubMed]
- Li, Y., Altorelli, N. L., Bahna, F., Honig, B., Shapiro, L. & Palmer, A. G. III (2013). Proc. Natl Acad. Sci. USA, 110, 16462–16467. [DOI] [PMC free article] [PubMed]
- Li, Y., Hofmann, M., Wang, Q., Teng, L., Chlewicki, L. K., Pircher, H. & Mariuzza, R. (2009). Immunity, 31, 35–46. [DOI] [PMC free article] [PubMed]
- Miloushev, V. Z., Bahna, F., Ciatto, C., Ahlsen, G., Honig, B., Shapiro, L. & Palmer, A. G. (2008). Structure, 16, 1195–1205. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nagar, B., Overduin, M., Ikura, M. & Rini, J. (1996). Nature (London), 380, 360–364. [DOI] [PubMed]
- Nose, A. & Takeichi, M. (1986). J. Cell Biol. 103, 2649–2658. [DOI] [PMC free article] [PubMed]
- Ozawa, M. & Kemler, R. (1990). J. Cell Biol. 111, 1645–1650. [DOI] [PMC free article] [PubMed]
- Paredes, J., Albergaria, A., Oliveira, J. T., Jerónimo, C., Milanezi, F. & Schmitt, F. C. (2005). Clin. Cancer Res. 11, 5869–5877. [DOI] [PubMed]
- Paredes, J., Correia, A. L., Ribeiro, A. S., Albergaria, A., Milanezi, F. & Schmitt, F. C. (2007). Breast Cancer Res. 9, 214. [DOI] [PMC free article] [PubMed]
- Paredes, J. et al. (2012). Biochim. Biophys. Acta, 1826, 297–311. [DOI] [PubMed]
- Parisini, E., Higgins, J. M. G., Liu, J., Brenner, M. B. & Wang, J. (2007). J. Mol. Biol. 373, 401–411. [DOI] [PMC free article] [PubMed]
- Park, J., Park, E., Han, S.-W., Im, S.-A., Kim, T.-Y., Kim, W.-H., Oh, D. Y. & Bang, Y. J. (2012). Invest. New Drugs, 30, 1404–1412. [DOI] [PubMed]
- Patel, S. D., Ciatto, C., Chen, C. P., Bahna, F., Rajebhosale, M., Arkus, N., Schieren, I., Jessell, T. M., Honig, B., Price, S. R. & Shapiro, L. (2006). Cell, 124, 1255–1268. [DOI] [PubMed]
- Pertz, O., Bozic, D., Koch, A. W., Fauser, C., Brancaccio, A. & Engel, J. (1999). EMBO J. 18, 1738–1747. [DOI] [PMC free article] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed]
- Prakasam, A., Chien, Y.-H., Maruthamuthu, V. & Leckband, D. E. (2006). Biochemistry, 45, 6930–6939. [DOI] [PubMed]
- Rakshit, S., Zhang, Y., Manibog, K., Shafraz, O. & Sivasankar, S. (2012). Proc. Natl Acad. Sci. USA, 109, 18815–18820. [DOI] [PMC free article] [PubMed]
- Reinés, A., Bernier, L.-P., McAdam, R., Belkaid, W., Shan, W., Koch, A. W., Séguéla, P., Colman, D. R. & Dhaunchak, A. S. (2012). J. Neurosci. 32, 6323–6334. [DOI] [PMC free article] [PubMed]
- Schubert, W. D., Urbanke, C., Ziehm, T., Beier, V., Machner, M. P., Domann, E., Wehland, J., Chakraborty, T. & Heinz, D. W. (2002). Cell, 111, 825–836. [DOI] [PubMed]
- Shapiro, L., Fannon, A. M., Kwong, P. D., Thompson, A., Lehmann, M. S., Grübel, G., Legrand, J.-F., Als-Nielsen, J., Colman, D. R. & Hendrickson, W. A. (1995). Nature (London), 374, 327–337. [DOI] [PubMed]
- Shimoyama, Y. & Hirohashi, S. (1991). Cancer Res. 51, 2185–2192. [PubMed]
- Shimoyama, Y., Yoshida, T., Terada, M., Shimosato, Y., Abe, O. & Hirohashi, S. (1989). J. Cell Biol. 109, 1787–1794. [DOI] [PMC free article] [PubMed]
- Sivasankar, S. (2013). J. Investig. Dermatol. 133, 2318–2323. [DOI] [PMC free article] [PubMed]
- Sprecher, E., Bergman, R., Richard, G., Lurie, R., Shalev, S., Petronius, D., Shalata, A., Anbinder, Y., Leibu, R., Perlman, I., Cohen, N. & Szargel, R. (2001). Nature Genet. 29, 134–136. [DOI] [PubMed]
- Taniuchi, K., Nakagawa, H., Hosokawa, M., Nakamura, T., Eguchi, H., Ohigashi, H., Ishikawa, O., Katagiri, T. & Nakamura, Y. (2005). Cancer Res. 65, 3092–3099. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Van Marck, V., Stove, C., Van Den Bossche, K., Stove, V., Paredes, J., Vander Haeghen, Y. & Bracke, M. (2005). Cancer Res. 65, 8774–8783. [DOI] [PubMed]
- Vendome, J., Felsovalyi, K., Song, H., Yang, Z., Jin, X., Brasch, J., Harrison, O. J., Ahlsen, G., Bahna, F., Kaczynska, A., Katsamba, P. S., Edmond, D., Hubbell, W. L., Shapiro, L. & Honig, B. (2014). Proc. Natl Acad. Sci. USA, 111, E4175–E4184. [DOI] [PMC free article] [PubMed]
- Vendome, J., Posy, S., Jin, X., Bahna, F., Ahlsen, G., Shapiro, L. & Honig, B. (2011). Nature Struct. Mol. Biol. 18, 693–700. [DOI] [PMC free article] [PubMed]
- Vunnam, N. & Pedigo, S. (2011). Biochemistry, 50, 2973–2982. [DOI] [PubMed]
- Wakita, H., Shirahama, S. & Furukawa, F. (1998). Microsc. Res. Tech. 43, 218–223. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wu, J. C., Gregory, C. W. & DePhilip, R. M. (1993). Biochem. Biophys. Res. Commun. 195, 1329–1335. [DOI] [PubMed]
- Yoshioka, H. et al. (2012). Cancer Immunol. Immunother. 61, 1211–1220. [DOI] [PMC free article] [PubMed]
- Zhang, C. C. et al. (2010). Clin. Cancer Res. 16, 5177–5188. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human P-cadherin EC1-EC2, 4oy9