

Abstract
Cten is a focal adhesion molecule that is expressed at very low levels in most normal tissues. Nonetheless, its expression has been found to increase dramatically in many types of cancer including colorectal, breast, gastric, and pancreatic cancer, suggesting that cten may play a critical role during tumorigenesis. To study the mechanisms that induce cten expression and the function of up-regulated cten, we examined the effects of several cancer-associated growth factors and cytokines on cten expression. We found that EGF, FGF2, NGF, PDGF, TGF-β, IGF-1, IL-6, and IL-13 were able to induce cten expression in a dose- and time-dependent manner. The Mek-Erk and PI3K-Akt pathways were two main signaling cascades responsible for cten up-regulation, whereas the Jak-Stat pathway could contribute to the increase in some conditions. Since many of these factors are known to promote cell migration, we hypothesized that up-regulated cten might contribute to this process. This hypothesis was investigated in FGF2-mediated cell migration. Silencing of cten not only reduced regular cell motility but also FGF2-mediated cell migration. Overexpression of cten promoted cell migration and FGF2 treatment failed to further enhance cell migration. Our findings that (1) cten is a common downstream molecule of these cancer-associated growth factors and cytokines; and that (2) up-regulated cten modulates cell migration induced by FGF2 and likely other growth factors as well, strongly suggest that cten could be a potential downstream therapeutic target for treating cancers associated with aberrant signaling of these growth factors and cytokines.
Keywords: cten, FGF2, focal adhesion, cell migration
INTRODUCTION
Focal adhesions are the transmembrane links between the extracellular matrix and the cytoskeletal network. They regulate various biological processes such as cell adhesion, migration, proliferation, differentiation, apoptosis, and gene expression.[1–5] Dysregulation of these processes may lead to cell transformation and cancer development. The encoding proteins of the tensin family, including tensin1, tensin2, tensin3, and cten (C-terminal tensin like), localize to the cytoplasmic side of focal adhesions and play roles in signal transduction and organizing cytoskeleton.[6] Among them, cten is the smallest protein in the family and contains the shared C-terminal SH2 and PTB domains but lacks the actin-binding domain that is found in the N-terminus of other tensins, [7] suggesting that cten may not directly interact with actin filaments. The expression of cten is relatively restricted to normal prostate and placenta, and is reduced in advanced prostate cancers.[7] In addition, cten recruits DLC1 (deleted in liver cancer-1) to focal adhesions and this interaction is critical for DLC1s tumor suppressor activity.[2,8] In contrast to its potential role as a tumor suppressor in the prostate, cten expression has been found to be up-regulated in thymoma, gastric, colorectal, breast, lung, and pancreatic cancer, [9–15] suggesting that overexpression of cten in certain tissues may play a critical role in tumorigenesis.
Fibroblast growth factors (FGFs) regulate many developmental pathways. The mammalian FGF family comprises 18 ligands that signal through four conserved tyrosine kinase receptors to activate four key downstream pathways: Ras-Mek-Erk, PI3K-Akt, STAT, and PLC.[16] It is not surprising that dysregulated FGF signaling is found in and may lead to several types of cancer by promoting tumor cell proliferation, survival, migration, and invasion. For example, FGF2 (aka bFGF, FGFβ) is overexpressed in prostate, colon, breast, lung, and bladder cancer. [17–21] Due to the involvement of FGF/FGFR family members in multiple steps of cancer development, several therapeutic approaches are being developed, including small molecule inhibitors, monoclonal antibodies, and ligand traps.[16] These therapeutic strategies have also been applied to many other growth factor or cytokine signaling pathways for treating cancer patients.
Here, we examined several growth factors and cytokines known to be up-regulated in various cancers to determine how each affects cten expression in non-malignant prostate epithelial cells (MLC-SV40 and RWPE-1) and colon adenocarcinomas (SW480). These two cell types were selected because that both express high levels of endogenous cten allowing us to monitor the expression changes, and that one is non-malignant epithelial cells and the other is adenocarcinomas permitting us to compare the potential differential responses between non-malignant cells versus cancer cells. Our results showed that epidermal growth factor (EGF), FGF2, transforming growth factor beta (TGF-β), nerve growth factor (NGF), platelet-derived growth factor (PDGF), insulin-like growth factor (IGF-1), interleukin 6 (IL-6), and interleukin 13 (IL-13) treatments induced dose- and time-dependent cten protein expression in these cells. The up-regulation of cten is mainly dependent on the Mek-Erk and PI3K-Akt pathways in both cell types. On the other hand, the Jak-Stat pathway is more critical in SW480 cells in regulating cten expression. The up-regulation of cten is required for FGF2-mediated cell migration, implicating a possible role of cten in tumor invasion and metastasis.
MATERIALS AND METHODS
Reagents and Cell Culture
EGF was purchased from Invitrogen (Carlsbad, CA); FGF2, PDGF, TFG-β, and IGF-1 were purchased from PeproTech (Rocky Hill, NJ); NGF and IL-6 were purchased from BD (Franklin Lakes, NJ); and IL-13 was purchased from ProSpec (East Brunswick, NJ). MLC-SV40 cells, a gift from J. Rhim (Uniform Services University, Bethesda, MD) were cultured in keratinocyte serum-free medium supplemented with antibiotics, 5 ng/ml human recombinant EGF, and 0.05 mg/ml bovine pituitary extract (Invitrogen). SW480 cells were obtained from American Type Culture Collection (Manassas, VA). SW480 cells were cultured in DMEM with antibiotic and 10% fetal bovine serum. In all cten induction studies, MLC-SV40 cells were cultured in keratinocyte serum-free medium without EGF and bovine pituitary extraction, and SW480 cells were cultured in serum-free DMEM for 24 h prior to growth factor or cytokine treatment. Cells were lysed in radioimmunoprecipitation assay buffer containing protease and phosphastase inhibitors (Roche Applied Science, Indianapolis, IN). The protein expression levels of cten, tensin3, and GAPDH were determined by immunoblotting analysis. Each experiment was repeated at least three times.
RNA Extraction, Reverse Transcription, and Real-Time Quantitative PCR
After growth factor administration for 2 h, total RNAs from MLC-SV40 or SW480 were extracted by using RNeasy mini kit (Qiagen, Valencia, CA), and then reverse transcribed by using SuperScript III (Invitrogen). Real-time PCR was performed by using 7900 HT (Applied Biosystems, Foster City, CA) to monitor the expression of cten and β-actin by using SYBR® green PCR Master Mix (Applied Biosystems). Primers used for cten mRNA detected were 5′-gtggacatacccattgtgctgatcaac-3′ (forward) and 5′-ggt-gggctgcatgtccctggcggg-3′ (reverse). Relative quantification of cten mRNA expression was measured by normalization with β-actin and expressed relative to control by using ΔΔCt method.
Constitutive Active-Mek1/2 Constructs (CA-Mek1/2) and Plasmid Transfection
PCR strategy was applied to generated constitutive active-Mek1 (CA-Mek1) fragment using the CC2 plasmid (Addgene, Cambridge, MA, #21207) as a template. Then the CA-Mek1 fragment was subcloned into BamHI/NotI sites of pCDNA3.1(+) (Invitrogen) plasmid (pCDN3.1-CA-Mek1). For CA-Mek2 expression construct, the CA-Mek2 fragment was isolated directly from M2E1 plasmid (Addgene, #21206) by restriction enzyme digestion then subcloned into BamHI/EcoRI sites of pCDNA3.1(+) (pCDNA3.1-CA-Mek2). Constitutive active-Myr-PIK3CA (#12523) and AKT-T308D/S473D (#14751) plasmids were obtained from Addgene. The plasmids were transfected into SW480 or MLC-SV40 cells by using Lipofetamine 2000® (Invitrogen).
Adenovirus Infection, siRNA Transfection, and Cell Migration
The virus titer of Ad-myc-cten (myc epitope tagged-cten) and Ad-LacZ were determined by using Quick-Titer Adenovirus Titer ELISA kit (Cell Biolabs, Inc., San Diego, CA) according to manufacturer’s instruction. Cells were infected with indicated adenovirus or transfected with control siRNA (Santa Cruz Biotechnology Dallas, Texas, sc-37007) or cten siRNA (5′-GCCCAGUGUCUGAUGUCAGCUAUAU-3′; 20 nM) for 24 h prior to transwell migration assay. Briefly, cells were trypsinized and resuspended in keratinocyte serum-free medium without EGF and bovine pituitary extract, and then transferred (1 × 105 cells in 100 μl medium) to the upper transwell; and 600 μl keratinocyte serum-free medium with or without FGF2 (10 ng/ml) in the absence of EGF and bovine pituitary extract was added into the lower well. After incubation for another 24 h, the cells on the upper surface of the transwell were removed and the cells on the lower surface were fixed, stained, and photographed for counting. Each experiment was repeated at least three times.
RESULTS
Cten Expression Is Induced by Growth Factors and Cytokines
Cten is overexpressed in many types of cancers and may play a critical role during tumorigenesis. To explore possible mechanisms that may regulate cten expression, we have investigated the effects of EGF, FGF2, NGF, TGF-β, PDGF, IGF-1 IL-6, and IL-13 on non-malignant prostate epithelial cells (MLC-SV40) and colon cancer cell line (SW480). EGF was used as a positive control, since we have previously showed that EGF induced cten expression.[12] MLC-SV40 cells cultured in keratinocyte plain medium for 24 h were treated with indicated factors at various concentrations for another 24 h and then cten protein levels were measured by immunoblot analysis. As shown in Figure 1A, EGF (5–60 ng/ml), FGF2 (1–10 ng/ml), NGF (5–50 ng/ml), TGF-β (0.1–10 ng/ml), PDGF (0.3–3 ng/ml), IGF-1 (1–20 ng/ml), IL-6 (0.01–1 ng/ml), and IL-13 (0.01–1 ng/ml) significantly induced cten protein expression in a dose-dependent manner. Next, the effects of EGF (60 ng/ml), FGF2 (10 ng/ml), NGF (20 ng/ml), TGF-β (1 ng/ml), PDGF (3 ng/ml), IGF-1 (5 ng/ml), IL-6 (1 ng/ml), and IL-13 (1 ng/ml) treatments for 1, 3, and 6 h were analyzed. The results showed that EGF, FGF2, NGF, and TGF-β were able to enhance cten expression within 3 h and all factors significantly induce cten protein levels within 6 h (Figure 1B). Meanwhile, the expression of another member of the tensin family, tensin3, was not affected by any of these treatments. Similar results were observed in RWPE-1 cells (data not shown) and SW480 colon cancer cells (Figure 2). Real-time quantitative PCR assays were performed to examine whether cten up-regulation might be mediated through the transcriptional process. The results showed that EGF (60 ng/ml), FGF2 (10 ng/ml), or NGF (50 ng/ml) treatment significantly increased cten mRNA levels in MLC-SV40 and SW480 (Figure 3). Overall, cten expression is induced by these growth factors and cytokines in a dose- and time-dependent fashion in both cell types.
Figure 1.

Cten protein levels are regulated by EGF, FGF2, NGF, TGF-β, PDGF, IGF-1, IL-6, and IL-13 in human prostatic epithelial cells. MLC-SV40 cells cultured in plain medium (without EGF and bovine pituitary extract) for 24 h were treated with indicated concentrations of growth factors or cytokines for another 24 h (A) or treated with indicated concentration for 1, 3, and 6 h (B). Total cell lysates were prepared and protein expression levels were monitored by immunoblotting with indicated antibodies. GAPDH was detected as an equal loading control. The intensity of each cten band was quantified, and expressed as a fold increase using the signal at dose 0 or time 0 as a reference.
Figure 2.

Cten protein levels are regulated by growth factors in human colorectal carcinoma. SW480 cells cultured in serum-free medium for 24 h were treated with indicated concentrations of growth factors or cytokines for another 24 h (A) or treated with indicated concentration for 1, 3, and 6 h (B). Total cell lysates were prepared and protein expression levels were monitored by immunoblotting with indicated antibodies. GAPDH was detected as an equal loading control. The intensity of each cten band was quantified, and expressed as a fold increase using the signal at dose 0 or time 0 as a reference.
Figure 3.
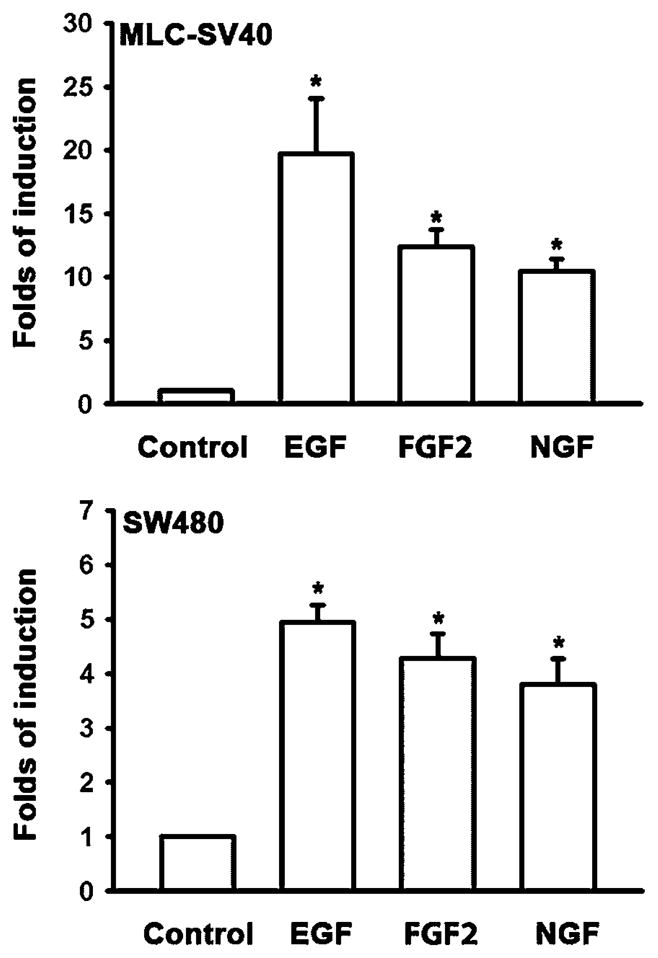
Growth factor treatments up-regulate cten mRNA levels. MLC-SV40 or SW480 cells cultured in plain medium for 24 h were treated with EGF (60 ng/ml), FGF2 (10 ng/ml), or NGF (50 ng/ml) for another 2 h, and then cten mRNA levels were determined by real-time quantitative PCR assays.
Cten Expression Is Regulated by Various Signaling Pathways
Since these factors are able to induce cten expression and they all induce at least two common pathways, namely the Mek-Erk and PI3K-Akt pathways, we have tested whether these signaling cascades are involved in cten up-regulation by using various inhibitors in the presence of these factors. While inhibitors for Mek (U0126 and PD98059) and PI3K (LY294002) significantly blocked cten induction mediated by all seven growth factors and cytokines (except LY294002 in IGF-1mediated induction), the Jak inhibitor (AG490) reduced cten expression initiated by TGF-β, IL-6, or IL-13 in MLC-SV40 cells (Figure 4A). In SW480 cells, inhibitors for Mek, PI3K, and Jak antagonized cten induction by these growth factors with several exceptions (Figure 4A). For example, Jak inhibitor showed no effect on cten induction by IL-6. In addition, blocking the PI3K-Akt pathway appeared to have very limited effect on IGF-1-mediated cten induction. To further demonstrate the positive involvement of Mek1/2 in cten expression, the effects of constitutively activated forms of Mek1 (CA-Mek1) or Mek2 (CA-Mek2) were tested in SW480 (Figure 4B) and MLC-SV40 (not shown) cells. The results showed that both CA-Mek1 and CA-Mek2 up-regulated cten expression. Similarly, expression of either constitutively activated PI3K (myr-PI3KCA) or AKT (T308D/S473D) increased cten protein levels in SW480 (Figure 4B) and MLC-SV40 (not shown) cells. Taken together, these results suggest that Mek-Erk and PI3K-Akt are key main pathways contributing to cten expression.
Figure 4.

Mek-Erk and PI3K-Akt are two main pathways involved in growth factor-mediated cten expression. (A) MLC-SV40 or SW480 cells cultured in plain medium for 24 h were pre-treated with indicated inhibitors for 30 min prior to FGF2, NGF, IL-13, IL-6, IGF-1, PDGF, or TGF-β 24 h incubation. Cten protein levels were determined by immunoblotting. C, control (no inhibitor); AG, AG490 (JAK2 inhibitor, 50 μM); U, U0126 (Mek1/2 inhibitor, 10 μM); LY, LY294002 (PI3K inhibitor, 25 μM); PD, PD98059 (Mek1 inhibitor, 25 μM). (B) Cell lysates prepared from SW480 transfected with control (con), constitutive active Mek1 (CA-Mek1), Mek2 (CA-Mek2), myr-PI3KCA (PI3KCA), Akt-T308D/S473D (Akt) were immunoblotted with indicated antibodies.
Cten Up-Regulation Is Critical for FGF2-Mediated Cell Migration
To understand the functional significance of up-regulated cten, we focused on the analyses of its role in FGF2-mediated cell migration. FGF2 is well known to enhance cell migration in various cell types. In our experimental conditions, FGF2 treatment showed significantly increases of MLC-SV40 cell migration in silence and overexpression controls (Figure 5A, control si and Ad-lacZ). Silencing of cten markedly reduced cell migration and FGF2 treatment had a limited, but not statistically significantly, effect on promoting migration. In parallel experiments, over-expression of cten by adenovirus infection (Ad-cten) greatly induced cell motility and FGF2 treatment failed to further enhanced migration. Same results were observed using SW480 cancer cells (Figure 5B). These results strongly suggest that cten contributes to FGF2-mediated cell migration.
Figure 5.
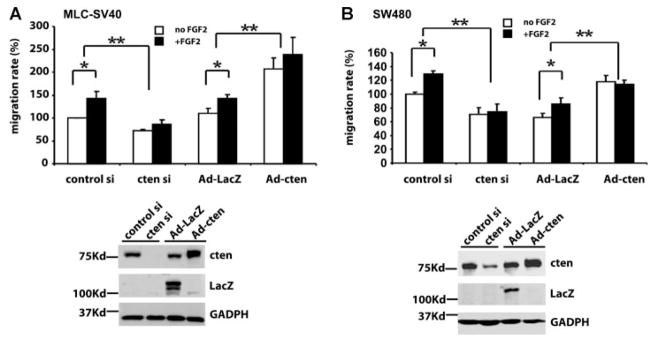
Cten is critical for FGF2-mediated cell migration. MLC-SV40 (A) or SW480 (B) cells transfected with control siRNA or cten siRNA; infected with Ad-Lac or Ad-cten for 24 h were used for cell migration studies in the absence or presence of FGF2 (10 ng/ml). *P < 0.05 and **P < 0.01. Protein levels of indicated molecules were analyzed by immunoblotting.
DISCUSSION
In this study, we reported that cten expression was enhanced by a group of growth factors and cytokines. The effect of EGF on cten expression is consistent with our previous finding in Hela and MCF10A cells.[12] However, we did not detect tensin3 down-regulation by EGF treatment in prostate cell and colon cancer cell systems. Cten expression promoted by IL-6 also echoes a recent report showing that IL-6 mediates cten up-regulation in a Jak-Stat3-dependent manner in MCF10A cells.[22] The positive effects on cten expression of FGF2, NGF, IL-13, PDGF, TGF-β, and IGF-1 have not been reported prior to current study.
The functional relevance of cten up-regulation is illustrated in FGF2-mediated cell migration. Our results have pointed to a critical role of cten in regular cell migration and migration induced by FGF2, since FGF2 treatment shows very limited effects in cten knockdown or overexpression cells. In addition to cten, there are other molecules/pathways involved in FGF2-mediated cell migration, including Rac activation, [23] Mek1/2 activation, [24] enhancing secretion of matrix metalloproteinase-1, plasminogen activator inhibitor-1, and cathepsin L, [25] and inhibition the degradation of hyaluronan by decreasing hyaluroni-dase-2 expression, [26] in a variety of cell types. It will be interesting to examine whether these pathways are cell type specific and whether cten acts synergistically or parallelly with these molecules in the future studies.
Our analyses demonstrate that the Mek-Erk and PI3K-Akt are two critical pathways leading to cten upregulation in both cell types. In agreement with our finding, it has been recently reported that cten is a target of K-Ras, [15] which activates Mek through Raf. On the other hand, the Jak-Stat pathway appears to play a more important role in colon cancer cells (SW480) than in prostate epithelial cells (MLC-SV40). The differences between these two cell lines are the tissue origins and the malignances of the cells. Since the Jak-Stat pathway is involved in both colon and prostate cancer development, [27,28] the discrepancy is likely due to abnormal cellular contexts in SW480 cancer cells. It has been demonstrated that the Jak-Stat pathway is essential for SW480 cell survival.[29] This may explain the more involvement of Jak-Stat pathway in regulating cten expression in SW480 cells.
From our findings, cten appears to be a common downstream molecule of these tumor related growth factors and cytokines. Clinical trials targeting growth factor receptors, including FGFR and EGFR, demonstrated promising results for cancer treatment. However, some patients showed no response due to mutations in their downstream molecules such as Ras or Raf. Being further downstream of these molecules and very low expression levels if any in most of normal tissues, cten might be an excellent alternative therapeutic target for treating these cancer patients.
Acknowledgments
Contract grant sponsor: NIH; Contract grant number: CA102537
This study was supported in part by NIH CA102537 (S.H.L.).
Abbreviations
- Cten
C-terminal tensin like
- SH2
Src homology 2
- PTB
phosphotyrosine binding
- EGF
epidermal growth factor
- FGF2
fibroblast growth factor 2
- NGF
nerve growth factor
- PDGF
platelet-derived growth factor
- TFG-β
transforming growth factor beta
- IGF-1
insulin-like growth factor
- IL-6
interleukin 6
- IL-13
interleukin 13
Footnotes
Conflict of interest: No conflicts of interest, financial or otherwise, relating to this document.
References
- 1.Lo SH. Focal adhesions: what’s new inside. Dev Biol. 2006;294:280–291. doi: 10.1016/j.ydbio.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 2.Liao YC, Si L, Devere White RW, Lo SH. The phosphotyrosine-independent interaction of DLC-1 and the SH2 domain of cten regulates focal adhesion localization and growth suppression activity of DLC-1. J Cell Biol. 2007;176:43–49. doi: 10.1083/jcb.200608015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 4.Lo SH, Janmey PA, Hartwig JH, Chen LB. Interactions of tensin with actin and identification of its three distinct actin-binding domains. J Cell Biol. 1994;125:1067–1075. doi: 10.1083/jcb.125.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- 6.Lo SH. Tensin. Int J Biochem Cell Biol. 2004;36:31–34. doi: 10.1016/s1357-2725(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 7.Lo SH, Lo TB. Cten, a COOH-terminal tensin-like protein with prostate restricted expression, is down-regulated in prostate cancer. Cancer Res. 2002;62:4217–4221. [PubMed] [Google Scholar]
- 8.Liao YC, Lo SH. Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. Int J Biochem Cell Biol. 2008;40:843–847. doi: 10.1016/j.biocel.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao YC, Chen NT, Shih YP, Dong Y, Lo SH. Up-regulation of C-terminal tensin-like molecule promotes the tumorigenicity of colon cancer through beta-catenin. Cancer Res. 2009;69:4563–4566. doi: 10.1158/0008-5472.CAN-09-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasaki H, Yukiue H, Kobayashi Y, Fukai I, Fujii Y. Cten mRNA expression is correlated with tumor progression in thymoma. Tumour Biol. 2003;24:271–274. doi: 10.1159/000076141. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki H, Moriyama S, Mizuno K, et al. Cten mRNA expression was correlated with tumor progression in lung cancers. Lung Cancer. 2003;40:151–155. doi: 10.1016/s0169-5002(03)00037-0. [DOI] [PubMed] [Google Scholar]
- 12.Katz M, Amit I, Citri A, et al. A reciprocal tensin-3-cten switch mediates EGF-driven mammary cell migration. Nat Cell Biol. 2007;9:961–969. doi: 10.1038/ncb1622. [DOI] [PubMed] [Google Scholar]
- 13.Albasri A, Al-Ghamdi S, Fadhil W, et al. Cten signals through integrin-linked kinase (ILK) and may promote metastasis in colorectal cancer. Oncogene. 2011;30:2997–3002. doi: 10.1038/onc.2011.26. [DOI] [PubMed] [Google Scholar]
- 14.Albasri A, Aleskandarany M, Benhasouna A, et al. Cten (C-terminal tensin-like), a novel oncogene overexpressed in invasive breast carcinoma of poor prognosis. Breast Cancer Res Treat. 2011;126:47–54. doi: 10.1007/s10549-010-0890-3. [DOI] [PubMed] [Google Scholar]
- 15.Al-Ghamdi S, Albasri A, Cachat J, et al. Cten is targeted by Kras signalling to regulate cell motility in the colon and pancreas. PLoS ONE. 2011;6:e20919. doi: 10.1371/journal.pone.0020919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437:199–213. doi: 10.1042/BJ20101603. [DOI] [PubMed] [Google Scholar]
- 17.Marzioni D, Lorenzi T, Mazzucchelli R, et al. Expression of basic fibroblast growth factor, its receptors and syndecans in bladder cancer. Int J Immunopathol Pharmacol. 2009;22:627–638. doi: 10.1177/039463200902200308. [DOI] [PubMed] [Google Scholar]
- 18.Galzie Z, Fernig DG, Smith JA, Poston GJ, Kinsella AR. Invasion of human colorectal carcinoma cells is promoted by endogenous basic fibroblast growth factor. Int J Cancer. 1997;71:390–395. doi: 10.1002/(sici)1097-0215(19970502)71:3<390::aid-ijc15>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 19.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res. 1999;5:1063–1071. [PubMed] [Google Scholar]
- 20.Finak G, Bertos N, Pepin F, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 21.Ruotsalainen T, Joensuu H, Mattson K, Salven P. High pretreatment serum concentration of basic fibroblast growth factor is a predictor of poor prognosis in small cell lung cancer. Cancer Epidemiol Biomarkers Prev. 2002;11:1492–1495. [PubMed] [Google Scholar]
- 22.Barbieri I, Pensa S, Pannellini T, et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 2010;70:2558–2567. doi: 10.1158/0008-5472.CAN-09-2840. [DOI] [PubMed] [Google Scholar]
- 23.Sogabe Y, Abe M, Yokoyama Y, Ishikawa O. Basic fibroblast growth factor stimulates human keratinocyte motility by Rac activation. Wound Repair Regen. 2006;14:457–462. doi: 10.1111/j.1743-6109.2006.00143.x. [DOI] [PubMed] [Google Scholar]
- 24.Narong S, Leelawat K. Basic fibroblast growth factor induces cholangiocarcinoma cell migration via activation of the MEK1/2 pathway. Oncol Lett. 2011;2:821–825. doi: 10.3892/ol.2011.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung JH, Im EK, Jin TW, et al. Cathepsin L derived from skeletal muscle cells transfected with bFGF promotes endothelial cell migration. Exp Mol Med. 2011;43:179–188. doi: 10.3858/emm.2011.43.4.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berdiaki A, Nikitovic D, Tsatsakis A, Katonis P, Karamanos NK, Tzanakakis GN. bFGF induces changes in hyaluronan synthase and hyaluronidase isoform expression and modulates the migration capacity of fibrosarcoma cells. Biochim Biophys Acta. 2009;1790:1258–1265. doi: 10.1016/j.bbagen.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Barton BE, Karras JG, Murphy TF, Barton A, Huang HF. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol Cancer Ther. 2004;3:11–20. [PubMed] [Google Scholar]
- 28.Slattery ML, Lundgreen A, Kadlubar SA, Bondurant KL, Wolff RK. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol Carcinog. 2013;52:155–166. doi: 10.1002/mc.21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tadlaoui Hbibi A, Laguillier C, Souissi F, et al. Efficient killing of SW480 colon carcinoma cells by a signal transducer and activator of transcription (STAT) 3 hairpin decoy oligodeoxynucleotide–interference with interferon-gamma-STAT1-mediated killing. FEBS J. 2009;276:2505–2515. doi: 10.1111/j.1742-4658.2009.06975.x. [DOI] [PubMed] [Google Scholar]