

A series of high-resolution crystal structures of R. catesbeiana ferritin M loaded with Fe2+ substrate at different metal-ion diffusion times show a sequence of iron-binding sites that allow the visualization of a possible path of substrate diffusion to the catalytic centre.
Keywords: ferritin M, Rana catesbeiana
Abstract
Ferritin superfamily protein cages reversibly synthesize internal biominerals, Fe2O3·H2O. Fe2+ and O2 (or H2O2) substrates bind at oxidoreductase sites in the cage, initiating biomineral synthesis to concentrate iron and prevent potentially toxic reactions products from Fe2+and O2 or H2O2 chemistry. By freezing ferritin crystals of Rana catesbeiana ferritin M (RcMf) at different time intervals after exposure to a ferrous salt, a series of high-resolution anomalous X-ray diffraction data sets were obtained that led to crystal structures that allowed the direct observation of ferrous ions entering, moving along and binding at enzyme sites in the protein cages. The ensemble of crystal structures from both aerobic and anaerobic conditions provides snapshots of the iron substrate bound at different cage locations that vary with time. The observed differential occupation of the two iron sites in the enzyme oxidoreductase centre (with Glu23 and Glu58, and with Glu58, His61 and Glu103 as ligands, respectively) and other iron-binding sites (with Glu53, His54, Glu57, Glu136 and Asp140 as ligands) reflects the approach of the Fe2+ substrate and its progression before the enzymatic cycle 2Fe2+ + O2 → Fe3+—O—O—Fe3+ → Fe3+—O(H)—Fe3+ and turnover. The crystal structures also revealed different Fe2+ coordination compounds bound to the ion channels located at the threefold and fourfold symmetry axes of the cage.
1. Introduction
Ferritins are nanocage proteins that synthesize and concentrate iron as hydrated ferric oxide biominerals (up to 4500 iron ions) inside the large central cavity of the protein cage (Liu & Theil, 2005 ▶). The 24-subunit ferritins are characterized by having 432 (O; octahedral) point symmetry, with three fourfold, four threefold and six twofold axes. The ferritin polypeptide subunit fold is a four-helix bundle motif (helices H1–H4) completed by a short fifth helix at the C-terminus (H5) external to the bundle. Each ferritin subunit has enzymatic (oxidoreductase) activity (H-type), except in animal ferritins, which coproduce a catalytically inactive subunit (L-type) that co-assembles with the active subunits in H:L ratios that are tissue-specific (Arosio et al., 2009 ▶). Amphibians have a second catalytic subunit, called M (or H′), which shares 64% sequence identity with the human H subunit. Homopolymeric M-subunit ferritins are often used as eukaryotic ferritin models.
In ferritin, Fe2+ is the substrate of the reaction that occurs at the oxidoreductase sites and ferric species are the products. The nature of the iron species involved in the catalytic reaction mechanism has been identified by spectroscopy, but the exact nature and number of metal ligands has remained elusive.
The oxidation of iron in eukaryotic ferritins monitored by iron spectroscopy (rapid freeze–quench Mössbauer and XAS) proceeds through at least three different species, as observed in the Rana catesbeiana ferritin M (RcMf) model (Hwang et al., 2000 ▶). In step one, Fe2+ reacts with O2, converting the diferrous high-spin state iron substrate, with an Fe–Fe distance of 3.44 Å, to a peroxodiferric (DFP) intermediate characterized by a very short Fe–Fe distance of 2.53 Å and with a typical UV–Vis absorbance at 650 nm. DFP appears in less than 25 ms and decays (step two) to a diferric μ-oxo/hydroxo species with an Fe–Fe distance of 3.00 Å. It is the latter diferric oxy species that is proposed to translocate from the catalytic sites to the protein biomineralization cavity (step three; Hwang et al., 2000 ▶). Several amino acids in the interior of the ferritin four-helix bundles may act as metal ligands during the catalytic reaction. Initial attempts to identify ferritin protein ligands for iron using as iron proxies redox-inert, non-native, divalent metal cations, which would remain trapped in the active site, were complicated by the different coordination preferences of the various metal ions used (Ha et al., 1999 ▶; Tosha et al., 2010 ▶; Toussaint et al., 2007 ▶). The lifetime of DFP species is too short for conventional X-ray structural characterization. However, kinetic X-ray crystallography can be used to characterize the iron-binding sites in ferritin and their evolution with time.
In the first reported crystal structure of a vertebrate ferritin (RcMf) with bound iron, a binuclear iron cluster was found bound in each ferritin subunit oxidoreductase site (OS). The observed Fe–Fe distance of 3.1 ± 0.1 Å at 2.7 Å resolution is consistent with the presence of a diferric oxo/hydroxo bridge. One of the Fe3+ ions was coordinated to a monodentate Glu23 and a bridging Glu58 and His61, while the second Fe3+ was coordinated to a bidentate Glu103 and a bridging Glu58 (Bertini et al., 2012 ▶). In another ferritin–iron structure obtained after a shorter iron-diffusion time into the crystal an Fe ion was bound to His54, which lies in close proximity to the above residues. Such data suggest that Fe2+ binds at a site with His54 before enzymatic oxidation (Bertini et al., 2012 ▶).
Here, we present a series of high-resolution anomalous X-ray diffraction experiments on single protein crystals of RcMf treated with ferrous ions for different periods of time under aerobic and anaerobic conditions. The results provide detailed information on the identity of the most populated transient iron-binding sites in the pathway from cage entry pores along the protein cage to the OS, as well as direct analysis of Fe2+ binding at the OS itself.
2. Materials and methods
2.1. Mutagenesis
The plasmid pET-3a carrying the RcMf gene was used to introduce the H54Q mutation. The primer for mutation was designed with the web-based PrimerX software (http://bioinformatics.org/primerx/). The site-directed amino-acid substitution in RcMf was generated by PCR with the expression plasmid pET-3a-RcMf DNA as the template using the QuikChange II Site-Directed Mutagenesis kit from Agilent following the protocol provided. Gene sequencing was performed to confirm the mutation (Primm SRL, Milan, Italy).
2.2. Recombinant expression and purification of R. catesbeiana ferritin M and variant
RcMf was expressed and purified using adapted protocols from the literature (Turano et al., 2010 ▶). Escherichia coli BL21(DE3) pLysS cells containing the mutated plasmid DNA were cultured in rich medium (Luria Broth) and grown at 37°C and 180 rev min−1 until the OD600 reached 0.7. Expression started after the addition of IPTG (1 mM final concentration) and the culture was incubated for 4 h. After harvesting by centrifugation, the cells were lysed using lysozyme and sonication. The purification of the ferritin involved the coagulation of temperature-sensitive proteins and two chromatography steps. Firstly, the supernatant of the centrifuged lysate was heated to 65°C for 15 min and the aggregated proteins were sedimented by ultracentrifugation (40 min, 40 000 rev min−1, 4°C). The supernatant was then dialyzed against 20 mM Tris buffer pH 7.5. Finally, the sample was loaded onto a 120 ml SP Sepharose anion-exchange column (GE Healthcare, Sweden) and eluted with a linear NaCl gradient (0–1 M) in 20 mM Tris buffer pH 7.5. Pertinent fractions were determined by SDS–PAGE and those containing ferritin were concentrated using an Amicon ultracentrifugation filter device with a molecular-weight cutoff of 14 kDa (Millipore, USA). The sample was then loaded onto a 120 ml Dextran size-exclusion column and was eluted with 20 mM Tris buffer pH 7.5. Fractions were monitored by UV–Vis spectroscopy and pooled together to obtain a final ferritin sample. Iron and other metal ions were removed by four dialysis steps at room temperature each using 2 l 20 mM Tris pH 7.5, 5 ml 500 mM EDTA pH 5.8, 10 ml ammonium thioglycolate to reduce and chelate the iron, followed by four dialysis steps at room temperature each using 2 l 20 mM Tris pH 7.5.
2.3. Stopped-flow measurements
The kinetics of iron uptake by demetallated wild-type RcMf (wt-RcMf), Mg-loaded wt-RcMf and demetallated RcMf H54Q variant were monitored as the change in absorbance at 650 nm (diferric peroxo) or at 350 nm [(Fe3+O)n species] (Hwang et al., 2000 ▶). Measurements were carried out using a UV–Vis stopped-flow spectrometer (model SX-18 MV, Applied Photophysics, UK). Absorbance curves were measured after mixing equal volumes of 100 µM protein (as subunits) in 200 mM MOPS, 200 mM NaCl pH 7.0 buffer with a freshly prepared solution of 200 µM ferrous sulfate in 1 mM HCl in order to obtain a complex with two Fe atoms per subunit (or 48 Fe atoms per nanocage). To monitor the effect of Mg2+ on protein-dependent Fe2+-oxidation rates under conditions comparable to those used in the crystallization experiments, these experiments were repeated using ferritin solution pre-incubated for 2 h in 0.1 M MgCl2. Each measurement was performed three times. 4000 data points were collected during 10 s. Initial rates of diferric peroxo and (Fe3+O)n species formation were determined from linear fitting of the linear portion of the absorbance curves recorded at 650 and 350 nm. The data are averages from a set of three experiments (see Supporting Information).
2.4. Protein crystallization
Crystals of wt-RcMf and the H54Q variant were grown in an aerobic environment using the hanging-drop vapour-diffusion method. Drops were prepared by mixing equal volumes of demetallated protein solution (7–14 mg ml−1 in 20 mM Tris–HCl pH 7.5) and precipitant solution ranging from 1.6 to 2 M MgCl2·6H2O in 0.1 M bicine buffer pH 9.0. The final pH in the equilibrated drops was about 8.0. The low-pH RcMf crystals were grown at pH 6.5 using the same protein and precipitant solution in 0.1 M bis-tris propane buffer. Drops were equilibrated over 600 µl precipitant solution at 4°C. Octahedral crystals of the wild-type protein and of the variant appeared in 2–5 d and growth was complete in about 10 d. The crystals harvested for all the experiments were carefully chosen to have similar dimensions ranging between 0.12 and 0.15 mm, as can be appreciated from the pictures shown in Supplementary Fig. S3. All crystals belonged to the cubic space group F432 and contained one RcMf subunit per asymmetric unit.
The time-controlled iron-loading study was performed under aerobic conditions by allowing free diffusion of iron from solid ammonium iron(II) sulfate {Mohr’s salt; [(NH4)2Fe(SO4)2]·6H2O} into crystals of wt-RcMf and the H54Q variant. Mohr’s salt crystals were inserted directly into the crystallization drop containing the RcMf crystals. This methodology was devised in view of the fact that our repeated attempts to use conventional soaking experiments of RcMf crystals in conditions analogous to those reported for Pyrococcus furiosus (Tatur et al., 2007 ▶) or pennate diatoms (Marchetti et al., 2009 ▶; Pfaffen et al., 2013 ▶) never led us to detect any iron ions bound to the protein.
After 1, 2, 5, 15, 30 and 60 min for the wt-RcMf and 1, 15 and 60 min for the RcMf H54Q variant, iron loading was stopped by flash-cooling the crystals in liquid nitrogen.
wt-RcMf crystals were also grown under anaerobic conditions (a controlled nitrogen atmosphere in a glove box equipped with a catalyzer for dioxygen reduction) using the sitting-drop vapour-diffusion method. Drops were prepared by mixing equal volumes of demetallated and deoxygenated protein solution (7 mg ml−1 in 20 mM Tris–HCl pH 7.5) and deoxygenated precipitant solution consisting of 1.6 M MgCl2, 0.1 M bicine pH 9.0. The final pH in the equilibrated drops was again about 8.0. Drops were equilibrated over 800 µl reservoir solution at 4°C. The crystals grew in 6–10 d in the same F432 cubic space group as the aerobically grown crystals. Iron loading was performed in the anaerobic environment using the procedure described above. However, the crystals suffered severe cracking after long exposure times to Mohr’s salt, limiting iron diffusion to a maximum of 3 min.
2.5. X-ray data collection, structure solution and refinement
X-ray crystallographic data were collected on beamline XRD-1 at Elettra, Trieste, Italy equipped with a Pilatus 2M detector and on beamlines BM14 and ID29 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France equipped with a MAR Mosaic 225 CCD and a Pilatus 6M-F detector, respectively. Data-collection statistics are reported in Table 1 ▶. Data were integrated using MOSFLM v.7.0.6 (Leslie, 2006 ▶) or XDS (Kabsch, 2010a ▶,b ▶) and were scaled with SCALA (Evans, 2006 ▶) from the CCP4 suite (Winn et al., 2011 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the outer shell.
| RcMf + Fe | |||||||
|---|---|---|---|---|---|---|---|
| RcMf | 1min | 2min | 5min | 15min | |||
| PDB code | 4lqh | 4lpj | 4lqj | 4lyx | 4lyu | ||
| Beamline | Elettra XRD-1 | Elettra XRD-1 | Elettra XRD-1 | Elettra XRD-1 | Elettra XRD-1 | Elettra XRD-1 | Elettra XRD-1 |
| Wavelength () | 1.240 | 1.240 | 1.000 | 1.722 | 1.759 | 1.240 | 1.240 |
| Data-collection temperature (K) | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Space group | F432 | F432 | F432 | F432 | F432 | F432 | F432 |
| Z | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| No. of subunits in asymmetric unit | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| () | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Unit-cell parameter a () | 184.41 | 184.30 | 184.55 | 184.55 | 184.58 | 184.18 | 183.80 |
| Resolution range () | 21.731.16 (1.221.16) | 24.631.28 (1.351.28) | 46.141.20 (1.261.20) | 46.141.58 (1.661.58) | 46.151.61 (1.701.61) | 22.331.25 (1.321.25) | 41.111.70 (1.781.75) |
| Observed reflections | 515592 (11616) | 415932 (27965) | 768858 (105670) | 344923 (8115) | 320427 (7600) | 488572 (47263) | 299848 (17027) |
| Unique reflections | 86036 (6909) | 67104 (8182) | 83387 (11909) | 35036 (3270) | 32969 (3056) | 73758 (10568) | 27391 (1490) |
| Completeness (%) | 92.8 (52.6) | 97.2 (82.9) | 99.6 (99.0) | 93.7 (61.9) | 93.7 (61.8) | 99.8 (99.5) | 100.0 (100.0) |
| Anomalous completeness (%) | 85.8 (24.6) | 91.4 (58.9) | 99.6 (98.9) | 88.7 (34.8) | 88.6 (34.4) | 98.4 (92.6) | 100.0 (100.0) |
| R meas (%) | 6.9 (35.7) | 6.0 (51.5) | 7.0 (46.0) | 5.6 (16.9) | 5.3 (15.1) | 5.9 (48.3) | 8.1 (40.2) |
| I/(I) | 12.7 (1.8) | 14.1 (2.0) | 16.3 (4.7) | 26.5 (5.3) | 27.6 (5.3) | 15.7 (2.8) | 22.3 (6.6) |
| Multiplicity | 6.0 (1.7) | 6.2 (3.4) | 9.2 (8.9) | 9.8 (2.5) | 9.7 (2.5) | 6.6 (4.5) | 10.9 (11.4) |
| Anomalous multiplicity | 3.2 (1.2) | 3.3 (2.0) | 4.8 (4.5) | 5.4 (1.8) | 5.4 (1.8) | 3.4 (2.3) | 5.8 (5.9) |
| RcMf + Fe | RcMf O2-free + Fe | |||||||
|---|---|---|---|---|---|---|---|---|
| 30min | 60min | 3min | RcMfH54Q | |||||
| PDB code | 4lqv | 4lqn | 4my7 | 4mku | ||||
| Beamline | Elettra XRD-1 | Elettra XRD-1 | Elettra XRD-1 | Elettra XRD-1 | ESRF ID29 | ESRF ID29 | ESRF ID29 | ESRF BM14 |
| Wavelength () | 1.000 | 1.000 | 1.722 | 1.759 | 0.976 | 1.722 | 1.759 | 0.886 |
| Data-collection temperature (K) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Space group | F432 | F432 | F432 | F432 | F432 | F432 | F432 | F432 |
| Z | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| No. of subunits in asymmetric unit | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| () | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Unit-cel parameter a () | 184.08 | 184.20 | 184.48 | 184.49 | 183.83 | 183.71 | 183.71 | 184.58 |
| Resolution range () | 42.231.54 (1.621.54) | 19.641.59 (1.681.59) | 42.321.58 (1.661.58) | 42.331.61 (1.701.61) | 45.961.48 (1.561.48) | 45.931.99 (2.101.99) | 45.932.04 (2.152.04) | 21.171.30 (1.371.30) |
| Observed reflections | 335116 (48464) | 210525 (24162) | 337484 (7339) | 309282 (6715) | 433231 (58436) | 129969 (2021) | 121239 (1917) | 946871 (134276) |
| Unique reflections | 39348 (5716) | 36091 (5097) | 35460 (3385) | 33323 (3079) | 44732 (6381) | 17076 (1315) | 160029 (1238) | 66237 (9508) |
| Completeness (%) | 98.8 (100.0) | 99.2 (98.2) | 94.7 (63.8) | 94.3 (61.5) | 100.0 (100.0) | 91.4 (50.2) | 91.6 (50.6) | 100.0 (100.0) |
| Anomalous completeness (%) | 97.9 (99.3) | 95.1 (90.3) | 89.8 (34.5) | 89.4 (32.6) | 100.0 (99.9) | 83.7 (18.1) | 83.7 (18.8) | 100.0 (100.0) |
| R meas (%) | 16.5 (31.3) | 13.0 (34.3) | 9.9 (41.0) | 9.1 (27.2) | 9.6 (47.0) | 9.9 (40.9) | 9.3 (45.0) | 10.3 (42.7) |
| I/(I) | 9.5 (6.1) | 7.2 (3.4) | 13.8 (1.5) | 14.8 (2.5) | 15.4 (4.7) | 15.0 (2.0) | 16.6 (2.6) | 15.2 (5.9) |
| Multiplicity | 8.5 (8.5) | 5.8 (4.7) | 9.5 (2.2) | 9.3 (2.2) | 9.7 (9.2) | 7.6 (1.5) | 7.6 (1.5) | 14.3 (14.1) |
| Anomalous multiplicity | 4.5 (4.3) | 3.1 (2.5) | 5.2 (1.5) | 5.1 (1.5) | 5.0 (4.6) | 4.5 (1.2) | 4.3 (1.2) | 7.5 (7.3) |
| RcMf H54Q + Fe | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1min | 15min | 60min | |||||||
| PDB code | 4ml5 | 4mn9 | 4mjy | ||||||
| Beamline | ESRF BM14 | Elettra XRD-1 | Elettra XRD-1 | ESRF BM14 | Elettra XRD-1 | Elettra XRD-1 | ESRF BM14 | Elettra XRD-1 | Elettra XRD-1 |
| Wavelength () | 0.954 | 1.739 | 1.751 | 0.954 | 1.739 | 1.761 | 0.954 | 1.739 | 1.761 |
| Data-collection temperature (K) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Space group | F432 | F432 | F432 | F432 | F432 | F432 | F432 | F432 | F432 |
| Z | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| No. of subunits in asymmetric unit | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| () | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Unit-cell parameter a () | 184.23 | 184.44 | 184.45 | 183.93 | 184.19 | 184.20 | 183.79 | 183.98 | 184.04 |
| Resolution range () | 22.511.22 (1.291.22) | 46.441.68 (1.771.68) | 46.111.69 (1.781.69) | 22.471.15 (1.211.15) | 46.051.62 (1.711.62) | 46.051.64 (1.731.64) | 30.631.40 (1.481.40) | 45.992.22 (2.692.55) | 46.012.55 (2.692.55) |
| Observed reflections | 1071136 (98528) | 289157 (5951) | 283837 (5651) | 1137420 (61722) | 324072 (7047) | 312708 (6804) | 629154 (36920) | 88474 (12165) | 87151 (11998) |
| Unique reflections | 79449 (11391) | 28296 (2166) | 28496 (2379) | 93348 (12782) | 32369 (2989) | 31379 (2963) | 51854 (6867) | 9193 (1290) | 9196 (1294) |
| Completeness (%) | 100.0 (100.0) | 90.5 (48.7) | 93.0 (55.5) | 99.2 (95.0) | 94.1 (61.2) | 94.6 (63.5) | 98.6 (91.5) | 100.0 (100.0) | 100.0 (100.0) |
| Anomalous completeness (%) | 99.9 (99.2) | 85.9 (29.3) | 90.3 (40.3) | 98.5 (90.3) | 88.5 (33.8) | 90.8 (41.6) | 98.1 (88.8) | 100.0 (100.0) | 100.0 (100.0) |
| R meas (%) | 8.8 (42.4) | 4.4 (9.2) | 4.1 (8.1) | 8.3 (49.6) | 7.1 (23.9) | 5.6 (15.9) | 7.6 (56.4) | 11.2 (41.4) | 9.6 (44.6) |
| I/(I) | 17.9 (5.1) | 40.7 (8.9) | 40.2 (8.0) | 16.7 (3.3) | 24.2 (3.9) | 29.1 (5.7) | 20.1 (3.0) | 17.3 (6.5) | 17.3 (6.9) |
| Multiplicity | 13.5 (8.6) | 10.2 (2.7) | 10.0 (2.4) | 12.2 (4.8) | 10.0 (2.4) | 10.0 (2.3) | 12.1 (5.4) | 9.6 (9.4) | 9.5 (9.3) |
| Anomalous multiplicity | 7.0 (4.4) | 5.6 (1.9) | 5.4 (1.4) | 6.4 (2.5) | 5.4 (1.7) | 5.3 (1.5) | 6.4 (2.8) | 5.3 (5.0) | 5.2 (4.8) |
Initial models were obtained by molecular replacement using MOLREP (Vagin & Teplyakov, 2010 ▶) and using one subunit of wt-RcMf as a search model for the rotation and translation functions (PDB entry 3ka3; Tosha et al., 2010 ▶) after excluding metal ions and water molecules. The positions of the iron ions were determined from the anomalous Fourier difference maps calculated using FFT from the CCP4 suite. The anomalous signals corresponding to iron ions ranged between 4.5 and 50σ in the anomalous difference maps.
In all experiments we collected anomalous data using X-ray wavelengths that were short enough to obtain high-quality data (little absorption) but where the anomalous signal of iron is still strong. When allowed by the synchrotron beamline characteristics, we also collected full three-wavelength anomalous data from the same crystal at remote high energy, the peak of the Fe K edge and immediately below the Fe K edge to obtain unambiguous proof that the observed anomalous signal belonged to iron and not to adventitious metals or other atoms (see Table 1 ▶).
The structures were refined using REFMAC5 (Murshudov et al., 2011 ▶) from the CCP4 suite. The refinement protocol involved a sequence of iterative manual rebuilding of the model and maximum-likelihood refinement. The molecular-graphics software Coot (Emsley et al., 2010 ▶) was used to visualize the structures and for the manual rebuilding and modelling of missing atoms into the electron density. Water molecules were added using ARP/wARP (Langer et al., 2008 ▶). The final models were inspected manually and checked with Coot (Emsley et al., 2010 ▶) and PROCHECK (Laskowski et al., 1993 ▶). Structure-solution and refinement statistics are reported in Table 2 ▶. All figures were generated using CCP4mg (McNicholas et al., 2011 ▶). The occupancies of almost all of the metal sites are fractional. Metal occupancies have been adjusted to obtain atomic displacement parameters comparable with those of the surrounding protein atoms. Water molecules bound to the metal ion refined at partial occupancy have been refined using the same occupancy as the parent metal ion. The crystallographic evidence refers to a spatial average of the single RcMf subunit constituting the crystal asymmetric unit over the 24 subunits composing the holoprotein. Each subunit is further averaged over the whole crystal. The partial occupancies observed for the iron ions and the different conformations of several amino-acid side chains may be the result of either local disorder and/or of the space and time average of the metal-binding events (differential diffusion in different portions of the crystal) occurring in the crystals before cooling.
Table 2. Refinement statistics.
Values in parentheses are for the outer shell.
| RcMf + Fe | |||||||
|---|---|---|---|---|---|---|---|
| RcMf | 1min | 2min | 5min | 15min | 30min | 60min | |
| PDB code | 4lqh | 4lpj | 4lqj | 4lyx | 4lyu | 4lqv | 4lqn |
| Resolution range () | 21.731.16 (1.191.16) | 21.281.27 (1.301.27) | 46.141.20 (1.231.20) | 20.221.23 (1.261.23) | 34.911.90 (1.951.90) | 42.231.54 (1.581.54) | 19.641.59 (1.631.59) |
| R cryst (%) | 16.0 (34.8) | 17.5 (39.8) | 13.6 (18.3) | 16.1 (37.4) | 16.9 (20.5) | 16.8 (18.3) | 19.2 (22.0) |
| R free (%) | 14.7 (32.0) | 15.0 (34.7) | 14.5 (18.6) | 17.5 (40.4) | 20.6 (25.3) | 19.3 (21.0) | 20.6 (21.4) |
| No. of reflections in refinement | 81663 (1769) | 64389 (3361) | 79211 (5771) | 73219 (5164) | 19659 (1421) | 37346 (2781) | 34279 (2432) |
| No. of reflections in R free set | 4309 (92) | 3428 (176) | 4169 (280) | 3869 (269) | 1063 (97) | 1976 (135) | 1798 (133) |
| No. of atoms | |||||||
| Total | 1967 | 1956 | 1927 | 1994 | 1713 | 1834 | 1832 |
| Protein | 1607 | 1570 | 1546 | 1600 | 1429 | 1445 | 1462 |
| Metal ions | 25 | 27 | 25 | 26 | 10 | 14 | 25 |
| Waters | 335 | 359 | 356 | 368 | 274 | 375 | 345 |
| Average B factor (from Wilson plot) (2) | 11.62 | 13.94 | 10.93 | 14.37 | 12.79 | 12.26 | 11.65 |
| R.m.s.d., bond lengths () | 0.004 | 0.006 | 0.005 | 0.008 | 0.007 | 0.005 | 0.005 |
| R.m.s.d., bond angles () | 0.994 | 1.165 | 0.998 | 1.289 | 1.130 | 1.012 | 1.052 |
| R.m.s.d., chiral volumes (3) | 0.058 | 0.067 | 0.059 | 0.078 | 0.070 | 0.069 | 0.072 |
| R.m.s.d., planes () | 0.001 | 0.002 | 0.004 | 0.007 | 0.005 | 0.004 | 0.005 |
| Estimate error on coordinates based on R value () | 0.031 | 0.040 | 0.029 | 0.038 | 0.120 | 0.065 | 0.081 |
| Ramachandran plot (%) | |||||||
| Favoured | 95.1 | 95.1 | 95.7 | 95.7 | 95.0 | 94.3 | 95.0 |
| Allowed | 4.9 | 4.9 | 4.3 | 4.3 | 5.0 | 5.7 | 5.0 |
| RcMf O2-free + Fe | RcMfH54Q + Fe | ||||
|---|---|---|---|---|---|
| 3min | RcMfH54Q | 1min | 15min | 60min | |
| PDB code | 4my7 | 4mku | 4ml5 | 4mn9 | 4mjy |
| Resolution range () | 41.111.48 (1.521.48) | 20.641.30 (1.331.30) | 21.711.22 (1.251.22) | 22.471.15 (1.181.15) | 29.061.40 (1.441.40) |
| R cryst (%) | 17.5 (22.4) | 12.3 (14.9) | 13.9 (16.9) | 14.7 (22.3) | 17.6 (24.6) |
| R free (%) | 19.5 (27.8) | 13.9 (17.4) | 15.4 (17.2) | 15.3 (23.7) | 18.5 (23.7) |
| No. of reflections in refinement | 42464 (3042) | 62754 (4586) | 75437 (5450) | 88648 (5968) | 2601 |
| No. of reflections in R free set | 2255 (171) | 3355 (234) | 3987 (305) | 4670 (305) | 148 |
| No. of atoms | |||||
| Total | 1810 | 1930 | 1932 | 1898 | 1831 |
| Protein | 1446 | 1530 | 1521 | 1521 | 1479 |
| Metal ions | 19 | 24 | 29 | 24 | 16 |
| Waters | 345 | 375 | 382 | 353 | 337 |
| Average B factor (from Wilson plot) (2) | 13.91 | 9.54 | 9.36 | 9.93 | 12.92 |
| R.m.s.d., bond lengths () | 0.005 | 0.004 | 0.004 | 0.004 | 0.005 |
| R.m.s.d., bond angles () | 0.999 | 1.012 | 1.037 | 0.976 | 0.974 |
| R.m.s.d., chiral volumes (3) | 0.074 | 0.054 | 0.058 | 0.054 | 0.065 |
| R.m.s.d., planes () | 0.005 | 0.004 | 0.004 | 0.004 | 0.004 |
| Estimate error on coordinates based on R value () | 0.060 | 0.034 | 0.031 | 0.027 | 0.053 |
| Ramachandran plot (%) | |||||
| Favoured | 94.3 | 95.1 | 95.1 | 95.0 | 95.0 |
| Allowed | 5.7 | 4.9 | 4.9 | 5.0 | 5.0 |
2.6. PDB codes
Final coordinates and structure factors for RcMf obtained under aerobic conditions have been deposited in the Protein Data Bank (PDB) as entries 4lqh for iron-free RcMf and 4lpj, 4lqj, 4lyx, 4lyu, 4lqv and 4lqn for RcMf loaded with iron for 1, 2, 5, 15, 30 and 60 min, respectively. Final coordinates and structure factors for RcMf obtained under anaerobic conditions and loaded with iron maintaining the oxygen-free environment have been deposited in the PDB as entry 4my7. Final coordinates and structure factors of the RcMf variant H54Q have been deposited in the PDB as entries 4mku for the iron-free state and 4ml5, 4mn9 and 4mjy for the iron-loaded states determined at 1, 15 and 60 min, respectively.
3. Results
A series of structures that identified the intermediate Fe2+-binding sites in RcMf, from the entry channels to the OS, were obtained at high resolution. Crystals of wt-RcMf and of its H54Q variant grown at pH 8.0 using MgCl2 as precipitant (see §2) were mixed with crystalline ferrous ammonium sulfate powder and frozen after 1, 2, 5, 15, 30 or 60 min of free diffusion of the Fe2+ ions in the crystallization drop in the presence of the second reaction substrate, dioxygen. The Fe2+ uptake by wt-RcMf was also studied under anaerobic conditions for comparison. The selected reaction conditions were such to sufficiently slow down the kinetics of Fe2+ movement along ferritin protein cages and reaction with dioxygen, thus allowing the observation of intermediate iron-binding sites. Indeed, [Mg(H2O)6]2+ ions inhibit iron mineralization in RcMf by blocking formation of the diferric peroxide catalytic intermediate (Liu & Theil, 2004 ▶), as shown by stopped-flow kinetic measurements (reported in the Supporting Information). The structures of the iron-bound RcMf crystals obtained at different times were compared with the structure of iron-free RcMf. The Fe2+ oxidation state in the aerobic crystals has been attributed on the basis of the observed Fe–Fe distances and by comparison with the structure determined under anaerobic conditions and at different pH values (see below).
A detailed description of the X-ray method used and of all crystal structures is provided as Supporting Information.
3.1. Time zero structure of RcMf
The crystal structure of iron-free RcMf at atomic resolution (time zero structure) was used as the reference structure to appreciate the changes occurring in the protein upon iron binding. In Fig. 1 ▶, time 0 reports a view of the oxidoreductase site of RcMf occupied by three Mg2+ ions bound at the same locations as previously observed in the RcMf structure crystallized under similar conditions (PDB entry 3ka3; Tosha et al., 2010 ▶). Two of these metal ions (Mg1 and Mg2 in Fig. 1 ▶, time 0) are bound with partial occupancy to sites that are close, but not identical, to the iron-binding sites observed in the iron-bound RcMf low-resolution structures (Bertini et al., 2012 ▶; PDB entries 3rbc and 3rgd). Stopped-flow solution measurements of Fe2+ uptake by RcMf in the presence of an Mg2+ ion concentration comparable to that used for crystallization provide experimental evidence of the slowing down of the iron-oxidation kinetics, as reported in the Supporting Information.
Figure 1.

Effect of different exposure times to Mohr’s salt on metal ion binding to wt-RcMf crystals. wt-RcMf crystallized with MgCl2 (time 0 structure) and iron binding at 1, 2, 15 and 30 min exposure. Magnesium and iron ions are represented by light and dark green spheres, respectively (arbitrary radius). The anomalous difference map contoured at 5.0σ, shown as copper wire, is superimposed on the iron ions. To highlight the speciation capability of the measurements, the time 2 panel shows the side chains of Met19 and Met33 superimposed with the two anomalous maps obtained at the Fe K edge (copper wire) and below the Fe K edge (white wire). The Fourier maps corresponding to other atoms have been omitted for clarity. The amino acids involved in iron binding are shown as sticks. Double conformations of the side chains of residues involved in metal ion binding are also shown. Water molecules are shown as small red spheres. Mg2+ coordination bonds are shown as dashed lines. Fe2+ coordination bonds are shown as continuous lines. The last panel displays a scheme summarizing the positions of the five Fe2+-binding sites observed in average RcMf (and the H54Q variant). Protein ligands are represented by sticks of different colours. Fe ions are represented as spheres colour-coded by their protein ligands. Ligands are as follows. Fe site 1 (coral): Glu23, Glu58 (bridging ligand) and His61. Fe site 2 (magenta): Glu58 (bridging ligand) and Glu103. Fe site 3 (green) involves His54 in RcMf or Gln54 in RcMf H54Q and also Glu57 and Glu103 (bonds not shown for clarity). Fe site 4 (blue) is located on the subunit surface facing the ferritin cavity and involves binding to Glu57, Glu136 and Asp140. The position of His54 is structurally coincident with Gln58 of human H ferritin, shown as lilac sticks. Fe site 5 (yellow) is found in the ferritin H54Q variant (see Fig. 3 ▶, time 1): Glu53 and Glu57 assume alternative conformations with respect to wt-RcMf.
3.2. 1 min free-diffusion structure of RcMf
The first Fe2+-binding site to be populated by iron in RcMf is the OS identified by Glu23, Glu58 and His61 and defined as Fe site 1 (Fig. 1 ▶, time 1). Iron is bound to site 1 with an occupancy of 0.30. No other sites appear to be occupied by iron. On binding to site 1, Fe2+ causes only a small rearrangement of the side chains of the three ligands, while the three Mg2+ ions which occupy the OS in the iron-free protein remain in place.
The coordination geometry of Fe2+ is a quite regular square pyramid completed by two water molecules forming the basal plane with Glu23 and Glu58 carboxylate O atoms. This coordination geometry appears to be dictated by the presence of the Val106 side chain at about 4.0 Å from Fe site 1, which hinders the binding of a sixth ligand to Fe1. Val106 is conserved in catalytically active eukaryotic ferritins. Mononuclear Fe2+ enzymes that activate O2 (e.g. intradiol dioxygenases) contain the so-called 2-His-1-carboxylate facial triad active site (Costas et al., 2004 ▶), where the protein ligands are bound to a face of the Fe2+ coordination octahedron while the opposite face is occupied by three water molecules that provide the access site for O2. In RcMf, Fe1 cannot adopt such a coordination as the side chain of Val106 fills this part of the site, hampering O2 access. This is probably the structural explanation of the observed inability of Fe1 in ferritin to react with O2 in the absence of the second iron ion, as demonstrated by the observed formation in ferritin of a dinuclear peroxo intermediate (Pereira et al., 1998 ▶) displaying a UV–Vis absorption spectrum completely different from that of mononuclear Fe3+ oxo/hydroxo complexes (Cho et al., 2011 ▶).
3.3. 2 min free-diffusion structure of RcMf
After 2 min of free diffusion, Fe2+ is now observed bound to the second iron site in the OS (Fe site 2; ligands Glu58 and Glu103; 0.30 occupancy) and the occupancy of site 1 has increased from 0.30 at 1 min to 0.50 at 2 min (Fig. 1 ▶, time 2). When Fe2+ binds at site 2, two of the Mg2+ ions in the OS are displaced, while the third Mg2+ ion (Mg1 in Fig. 1 ▶, times 0 and 1) observed at time 0 in the OS is unaffected (not shown in Fig. 1 ▶, time 2 for clarity). The longer time needed to observe Fe2+ binding to site 2 can be explained by the necessity to displace two Mg2+ ions from this part of the cavity. Once again this correlates well with the slowing down of the reaction rate observed in the presence of Mg2+. The presence of only two protein ligands for Fe2+ in site 2 suggests a lower metal affinity for this site, in analogy with the findings of isothermal titration calorimetry (ITC) on human H ferritin (HuHf; Bou-Abdallah et al., 2002 ▶, 2003 ▶; Honarmand Ebrahimi et al., 2012 ▶).
2 min of Fe2+ free diffusion in the crystal allows completion of the coordination sphere of the dinuclear iron cluster in the OS. The Fe1–Fe2 distance is 3.63 Å. Fe1 displays square-pyramidal geometry, as in the 1 min structure. The Fe2 ion is bound to Glu58 (bridging it to Fe1) and to Glu103 as a symmetric bidentate ligand. Fe2 coordination is completed by a bridging water/hydroxide molecule (Wb) and by a terminal water molecule. The geometry of the Fe2 site is irregular and is somewhere between a square pyramid and a trigonal bipyramid.
3.4. 15 min free-diffusion structure of RcMf
Free diffusion of Fe2+ for 5 and 15 min does not change the binding of Fe2+ in the OS except for an increase in the occupancy at the enzyme centres from 0.5 to ∼0.7 (Fig. 1 ▶, time 15).
Additionally, two new significant maxima close to Fe2 (Fig. 1 ▶, time 15) appear in the anomalous difference map, which are named Fe sites 3 and 4, respectively. The Fe3 ion (Fig. 1 ▶, time 15) is at a very short distance (2.48 Å) from Fe2 and is bound to the Nδ1 atom of His54 (site 3 in Fig. 1 ▶, all sites) with an incomplete coordination sphere. Such observations indicate that Fe2 and Fe3 cannot be simultaneously bound.
The Fe3 and Fe4 sites appear to represent binding sites for an Fe2+ substrate that is approaching the OS. Interestingly, the positions of the Fe1 and Fe2 sites match those of Co2+ and Cu2+ found in recently reported RcMf crystal structures (Bertini et al., 2012 ▶; Tosha et al., 2010 ▶), where they share the same ligands, with the exception of His54, which binds both Co2+ and Cu2+ in site 2 but not Fe2+, which is bound to His54 in site 3. The fact that Co2+ (Tosha et al., 2010 ▶) and Cu2+ (Bertini et al., 2012 ▶) are inhibitors of the catalyzed reactions of Fe2+ with O2 in ferritin might reflect the similarity of Co2+, Cu2+ and Fe2+ binding at the OS, and the conserved inability of ferritin to oxidize Co2+ and Cu2+ in contrast to Fe2+.
3.5. 30 and 60 min free-diffusion structures of RcMf
A clearer view of iron bound at the ferritin OS is provided by the crystal structure of RcMf obtained after 30 min of free diffusion of Fe2+ (Fig. 1 ▶, time 30). Iron sites 1 and 2 are the only ones occupied by Fe2+ and the Fe–Fe distance is 3.54 Å. The coordination spheres of the two Fe ions are better defined, and retain the square-pyramidal and distorted trigonal bipyramidal geometry observed at shorter time intervals, but with much less disorder of the side chains of the residues involved in metal binding. The 60 min structure provides the same view of the OS. The occupancy of Fe sites 1 and 2 is still partial and decreases from 0.7 to about 0.5 for site 1 and 0.3/0.5 for site 2 (Supplementary Table S1). The lower occupancy of Fe in the ferritin OS after a longer exposure time suggests that in the period of time between 15 and 30 min turnover occurs at both the Fe1 and the Fe2 sites and depletes sites 3 and 4, which appear again empty.
3.6. 60 min free-diffusion structure of RcMf at pH 6.5
Crystals of RcMf have been also obtained in the same crystal form at the lower pH of 6.5 (see §2) with respect to the pH of the previous structures (8.0). This experiment allowed us to check the influence of the crystallization pH on the oxidation state of iron bound in the OS, and to have a comparison with the trigonal low-resolution structure obtained from crystals grown in sodium formate at around neutral pH (PDB entry 3rbc; Bertini et al. 2012 ▶). The pH 6.5 structure was determined after 60 min of free diffusion of Fe2+. Under these conditions, two iron ions bind (0.40 occupancy) to the same oxidoreductase sites 1 and 2, maintaining the same coordination geometry observed at pH 8.0 and bridged by a water species (most probably OH−). However, the two iron ions are now closer, with a Fe–Fe distance of 3.15 (5) Å, which is the same as the Fe3+–Fe3+ distance observed in PDB entry 3rbc (Bertini et al., 2012 ▶) and is characteristic of diferric μ-oxo/hydroxo model compounds (Kurtz, 1990 ▶; Tshuva & Lippard, 2004 ▶; Vincent et al., 1990 ▶). Diferric oxo/hydroxo complexes are products of ferritin enzyme activity (Hwang et al., 2000 ▶) and this structure suggests that in crystallo at pH 6.5 Fe2+ has been oxidized by dioxygen at the ferritin enzyme centres and that at this pH the rate-limiting step is the release of the diferric product of the reaction from the OS.
3.7. Fe–RcMf structure under anaerobic conditions
To validate our attribution of the iron oxidation state in previous structures as Fe2+, the structure of the iron adduct of RcMf was obtained using crystals grown under strictly anaerobic conditions at pH 8.0 and treated anaerobically with Mohr’s salt for about 3 min. The diffusion time was the maximum possible before cracking of the crystals occurs (see §2). Such an observation emphasizes the osmotic pressure that builds up from Fe2+ diffusing into the crystal in the absence of enzymatic turnover and mineral growth; Fe2+ accumulation at ferritin metal-binding sites and at less specific iron-binding sites leads to the breakage of native, intermolecular interactions.
The anomalous difference Fourier maps obtained from data collected anaerobically at and below the Fe K edge (7130 eV) clearly show the presence of four iron ions in the OS (Fig. 2 ▶) that are located, within experimental error, at the same positions as observed in the aerobic crystals after 15 min of treatment with Mohr’s salt. However, in the anaerobic crystals the Fe1 site reaches almost full occupancy, while Fe2 is still at 0.50 occupancy. The Fe1–Fe2 distance is 3.64 Å, which is comparable to the analogous distances in crystals formed aerobically at pH 8.0 (Supplementary Table S2), supporting our assignment to Fe2+ as the iron species present in all crystals used for our kinetic crystallography experiments.
Figure 2.

Oxidoreductase site in wt-RcMf crystallized without the second substrate dioxygen after 3 min exposure to the iron substrate (Mohr’s salt). Four Fe2+ ions (represented by green spheres) are clearly seen bound to the enzyme site. The anomalous difference map obtained from data collected at the Fe K edge is superimposed (copper wire). The four sites are coincident with those found after 15 min of iron free diffusion under aerobic conditions (Fig. 1 ▶, time 15).
3.8. Effect of the H54Q substitution near the OS of RcMf
The protein environment near enzyme sites can play an important role in directing the substrate to the active site. In the case of ferritin, metal ions have been observed bound near the active sites, such as to His54 in the RcMf structures presented here and in our previous work (Bertini et al., 2012 ▶). Gln58 in the human H chain adopts the structural position of RcMf His54. In order to investigate the effect of the substitution of His with Gln on iron binding, we have produced the RcMf H54Q variant crystallized under the same conditions as wt-RcMf.
Crystal structures of RcMf H54Q have been determined in the presence of air, in the iron-free state and after 1, 15 and 60 min of iron treatment.
The iron-free RcMf H54Q structure reported in Fig. 3 ▶ (time 0) shows the same Mg2+-binding pattern as observed in HuHf (PDB entry 3ajo; Masuda et al., 2010 ▶) and different from that of wt-RcMf (Fig. 1 ▶, time 0), as described in detail in the Supporting Information. These findings indicate that the H54Q mutation renders the OS of RcMf very similar to that of HuHf.
Figure 3.

Effect of different exposure times to Mohr’s salt on metal ion binding to the RcMf H54Q variant. The four panels show the Mg-bound RcMf H54Q variant (time 0) and the Fe binding sites populated after 1, 15 and 60 min of iron treatment. Mg2+ ions are represented by light green spheres, Fe2+ ions are represented by dark green spheres of arbitrary radius with the superimposed anomalous difference map contoured at 5.0σ shown as copper wire. The Fourier maps corresponding to other atoms have been omitted for clarity. The amino acids involved in iron binding are shown as sticks. Water molecules are shown as small red spheres. The additional Fe5 site is shown in the time 1 and time 15 panels.
The OS of RcMf H54Q treated for 1 min with Mohr’s salt is shown in Fig. 3 ▶, time 1. As in the analogous wt-RcMf, only the Fe1 site of the oxidoreductase is occupied by iron (about 0.70 occupancy) and the Fe2 site is empty. However, the Fe3 and Fe4 sites are now occupied by iron, as observed in the 15 min and the O2-free RcMf structures (see Fig. 1 ▶, time 15, and Fig. 2 ▶ for comparison). The mutated residue Gln54 binds Fe3. At variance with wt-RcMf, a new iron site (Fe5) is found (Fig. 3 ▶, time 1) located further away.
The structures of the RcMf H54Q variant obtained after 15 and 60 min of treatment with Mohr’s salt show a similar pattern of iron binding (Fig. 3 ▶, times 15 and 60). The oxidoreductase cavity is occupied by a cluster of four iron ions bridged by a series of protein and water/hydroxide ligands.
The scenario depicted by this structure is complicated by the partial occupancy of the sites. Indeed, while the Fe1 occupancy refines to about 0.80, those of Fe2, Fe3, Fe4 and Fe5 range from 0.40 to 0.25, as do those of the water molecules bound to them. Furthermore, the side chains of three protein ligands of the cluster (Gln54, Glu57 and Asp140) are observed in double conformations. Therefore, the Fe2–Fe3–Fe4 cluster and the Fe5 ion most probably do not represent a structure that occupies these sites in all RcMf H54Q subunits, but rather indicate a superposition of different states that are owing to a dynamic process of iron interaction with the protein. The multiple conformations adopted by Glu53, Glu57 and Asp140 suggest that these residues pass the metal ions from one site to the next one in the path to the OS.
3.9. Iron sites located on the ferritin symmetry axes
Inspection of the anomalous difference maps at the threefold pores of wt-RcMf and the RcMf H54Q variant obtained after 60 min of treatment with Mohr’s salt shows two anomalous peaks surrounded by six electron-density maxima, indicating the presence of two iron ions having the regular octahedral structure of a Fe2+ hexaaqua ion (Fig. 4 ▶). The Fe2+ aqua ions are located exactly on the ferritin-cage threefold axis at different depths from the protein surface (about 11.0 and 18.0 Å from the surface, respectively). They are engaging two series of six and nine symmetry-related hydrogen bonds, respectively, to Asp127 and Glu130. The presence of iron ions in the threefold pores of RcMf has already been established in our previous structure of the trigonal form of RcMf (Bertini et al., 2012 ▶), but in that case the chemical composition of the iron complex could not be defined. This represents the first structural evidence of the transit of Fe2+ aqua ions through the ferritin threefold pores. The possibility of observing these Fe2+ ions only after long exposure times to iron is probably a consequence of the saturation of the ferritin sites. Indeed, the progressive occupation of the weak binding sites along the path towards the active site may block incoming Fe2+ aqua ions at the pore entrance, in correspondence to the displaced Mg2+ aqua-ion positions observed in the time 0 structures (iron-free) of both wt-RcMf and the RcMf H54Q variant (see Supporting Information).
Figure 4.

Views along (a) and perpendicular to (b) the threefold pore of the 60 min RcMf H54Q structure (the same arrangement is seen in the analogous wt-RcMf structure). The two Fe2+ hexaaqua ions are sitting on the threefold axis. The iron ions are represented as green spheres superimposed on the anomalous difference map obtained from data collected at the Fe K edge (copper wire) and the 2F o − F c Fourier difference map contoured at 1.2σ (blue wire). The coordinating water molecules are represented as red small spheres. Also superimposed are hydrogen bonds involving the nearby residues, which are shown as dashed lines. The open triangles and the continuous line show the position of the crystallographic threefold axis.
The fourfold channels defined by the four symmetry-related His169 residues in the iron-free structures do not contain metal ions but only chloride from the crystallization solution, as indicated by the anomalous signal that is present (see Fig. 5 ▶ a). After 2 min exposure to the iron salt, the anomalous maps of both wt-RcMf and the H54Q variant (data not shown) demonstrate that one iron ion is bound to the four His169 residues that define the entrance to the fourfold channel (see below and Fig. 5 ▶ b). The occurrence of iron binding is also indicated by the conformational change experienced by the side chains of His169, which move to within coordination distance (2.43 Å) of the metal ion. This Fe2+ ion displays elongated tetragonal coordination geometry with one chloride anion at one of the apices, one water molecule/hydroxide at the other, facing the ferritin cavity, and the four His169 residues bound in the basal plane (see Fig. 5 ▶ b). The presence of the chloride anion is demonstrated by the signal present in the anomalous difference map obtained from data collected at an energy below the Fe K edge.
Figure 5.
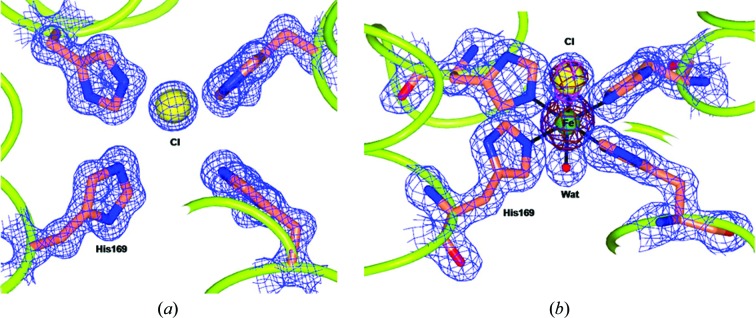
Views of the fourfold pores in wt-RcMf (the same occurs in the H54Q RcMf variant). (a) The fourfold pore in the iron-free structure. Only a chloride anion is present in the pore at a contact distance from the four symmetry-related His169 residues. (b) The fourfold pore as it appears after 2 min or longer of treatment with Mohr’s salt. The species [Fe(His)4Cl(H2O)]+or[Fe(His)4Cl(OH)] is present in the pore as clearly shown by the electron density (2F o − F c difference Fourier map contoured at 1.2σ as a blue wire). The chemical composition of the complex is revealed by the two superimposed anomalous difference maps (the copper wire refers to the anomalous difference map obtained at the Fe K edge; the magenta wire refers to the anomalous difference map obtained below the Fe K edge).
Our structural findings of the Fe2+ aqua ion penetrating the ferritin threefold channels, together with the [Fe(His)4Cl(H2O)]+ {or [Fe(His)4Cl(OH)]} adduct located at the fourfold axes, correlate well with the observed loss of oxidoreductase activity upon mutation of Asp127 and Glu130 to hydrophobic residues (Haldar et al., 2011 ▶; Theil et al., 2014 ▶; Tosha, Behera & Theil, 2012 ▶; Yang et al., 2000 ▶) and with the unaltered catalytic activities of the H169F mutant, which in contrast displays slightly inhibited mineral dissolution (Theil et al., 2014 ▶). Our data provide a structural rationale for the above findings, indicating that the threefold pores are routes for Fe2+ aqua-ion entrance and that the Fe2+ substrate cannot penetrate the cage cavity from the fourfold channels. Conversely, the Fe2+ coordination at the fourfold pores provides a possible model for the interaction that leads to the release of Fe2+ from the ferritin mineral. Iron release from the biomineral ex vivo requires reduction of the iron to Fe2+; this cannot occur under our experimental conditions. Therefore, the Fe2+ at the fourfold pores should come from the Mohr’s salt. When it reaches the pore, the chemical characteristics of the locus (the hydrophobicity and the presence of chloride anion) determine the formation of the observed adduct. In other words, the Fe2+ ions stop at the entrance to the fourfold pore and cannot proceed further, in contrast to what occurs at the threefold pores.
4. Discussion
By allowing the free diffusion of Fe2+ ions in ferritin crystals for different times, followed by flash-cooling, we have been able to obtain a series of time-dependent crystal structures that permit the progressive uptake of Fe2+ ions by RcMf and many of the steps along its pathway to reach the OS to be followed.
The central portion of each subunit, which hosts the OS, is rich in amino acids that can act as metal-ion ligands; each of them can assume multiple conformations, which change depending on the presence of metal ions (Fig. 1 ▶, all times). In the present time-dependent study we have established the structure of the Fe1 and Fe2 sites with the substrate Fe2+ bound. In the time-dependent experiments conducted at pH 8.0 we consistently measured Fe1–Fe2 distances ranging between 3.5 and 3.8 Å, which are the same, within experimental error, as those observed in the Fe–RcMf crystal grown and maintained in the absence of the second substrate O2, strongly supporting the presence of diferrous clusters at the oxidoreductase site in all instances (Supplementary Table S2). At pH 6.5 we instead repeatedly observed (Bertini et al., 2012 ▶ and this work) shorter Fe–Fe distances characteristic of ferric μ-oxo/μ-hydroxo clusters. These observations are consistent with the pH dependence of Fe2+ oxidation kinetics by RcMf and variants reported by Liu & Theil (2004 ▶), which showed an increase of about 100 times in the rate of formation of the diferric product and of the DFP intermediate in going from pH 6.0 to 8.0. The slower release of the diferric product from the RcMf oxidoreductase site in solutions near neutrality allows its direct observation in the crystal (confirming the Mössbauer data on diferrous precursors; Krebs, Bollinger et al., 2002 ▶; Krebs, Edmondson et al., 2002 ▶). In other words, at neutral pH in crystallo the rate-limiting step appears to be the formation and the release of the diferric product from the oxidoreductase sites, and therefore in crystals at pH 6.5 we can observe species with a short intermetal distance characteristic of diferric species. On the other hand, the higher turnover occurring at high pH makes the binding of iron at the Fe1 and Fe2 sites the rate-limiting step of the reaction.
The partial occupancy of all Fe2+-binding sites and the time dependence of this structural parameter indicate their transient character and intrinsically low thermodynamic stability, which is in agreement with the measurements performed by ITC on HuHf and P. furiosus ferritin (Bou-Abdallah et al., 2002 ▶, 2003 ▶; Honarmand Ebrahimi et al., 2012 ▶). Together, these data define the path of Fe2+ ions towards the reaction site, with iron entering inside the bundle and reaching the oxidoreductase site shuttled by wt-RcMf His54 (Gln54 in RcMf H54Q and, most probably, Gln58 in HuHf) and then moving to reach the binuclear reaction centre constituted by the Fe1 and Fe2 sites.
Fe1, with its characteristic square-pyramidal geometry, is the first site to become populated. This finding identifies Fe1 as the site with the highest thermodynamic affinity for Fe2+. Once Fe1 is significantly populated, incoming Fe2+ binds to the second strongest site (Fe2). When both Fe1 and Fe2 are populated, we start observing Fe2+ at the other lower affinity sites. Fe2+, which is continuously diffusing towards the reaction site, accumulates at different positions along its path trapped at sites defined by side chains that can act as transient ligands assisting iron trafficking inside the protein. At times longer than 15 min the occupancy of the Fe1 and Fe2 sites decreases, suggesting depopulation of these two sites owing to reaction turnover. As soon as the diferric oxo/hydroxo product is formed, the diferric cluster leaves the oxidoreductase site, which is then ready for the next catalytic cycle.
Our data provide structural evidence that the natural substrate Fe2+ species that transits through the entry channels at the threefold pores is [Fe(H2O)6]2+, which is observed to interact with the side chains of Asp127 and Glu130. These residues have been proposed to exert an electrostatic attraction on the incoming Fe2+ species. A large body of evidence accumulated over the years using a variety of experimental techniques on different eukaryotic ferritins indicates the key role played by these residues in allowing the Fe2+ ions to enter into the ferritin cage and reach the OS, as reported in Supplementary Table S1 and references therein. In summary, the substitution of these carboxylate residues by hydrophobic residues (Ala or Ile) sensibly reduces the enzymatic activity and the effect has been interpreted in terms of reduced uptake of Fe2+ by the nanocage in their absence. The potential energy profiles of divalent cations in the threefold channels of ferritin have been calculated (Takahashi & Kuyucak, 2003 ▶), providing theoretical support for the results obtained on mutants. From the bottom of the threefold pores, the ferrous aqua ions then proceed inside the inner cage cavity via electrostatic attraction exerted by Glu136–Asp140 (Behera & Theil, 2014 ▶) and the oxidoreductase-site residues, as summarized in Fig. 6 ▶. The present results also support the role of Glu136 and Asp140 in controlling the access of iron to the catalytic site.
Figure 6.

The travel of Fe2+ ions inside the RcMf subunit. The probable pathway of Fe2+ ions of about 50 Å in length from the threefold axis channel to the oxidoreductase site as indicated by the crystallographic structures. The residues involved in Fe2+ binding (green spheres) are shown as sticks. The two Fe2+ ions bound to Fe1 and Fe2 sites are shown as red spheres. The Fe2+ ion trapped at the fourfold axis pore is shown as a green sphere.
Finally, an iron-binding site is identified at the fourfold symmetry pores provided by the four symmetry-related His169 residues. The metal-binding residue His169 is conserved in all catalytically active vertebrate ferritins (H or M subunits). The fourfold pores (and His169 in particular) do not play any role in iron uptake, but are possibly involved in iron release (Theil, 2011 ▶; Theil et al., 2014 ▶). Therefore, the observed Fe at the fourfold pores represents a model of off-path ions with respect to the catalytic process or of the release of iron from the stored iron mineral core, which occurs under the effect of reductants.
The structural characterization of Fe2+ trapped at multiple binding sites along its pathway towards the catalytic centre and of the oxidoreductase reaction intermediates allows us to revisit the available structures of ferritin loaded with metal ions different from the natural metal substrate and those of iron-loaded ferritins from different organisms (Ebrahimi et al., 2012 ▶; Marchetti et al., 2009 ▶). We identified the presence of multiple (Fe3–Fe5) binding sites in the close proximity of the catalytic centre and interpreted them as snapshots of iron moving along its path towards the reaction sites. The analysis of the conformation of side chains acting as iron ligands indicates them as mutually exclusive centres. This finding clearly conflicts with the assumption of previous proposals of the existence of only three iron-binding sites in the OS in eukaryotic, prokaryotic and archaeal ferritins (Honarmand Ebrahimi et al., 2012 ▶). Our structures tell that site 1 always has higher occupancy by Fe2+ and that it corresponds to the high-affinity site identified in other studies (Ha et al., 1999 ▶; Hwang et al., 2000 ▶). On the other hand, an almost full occupancy of site 1 by Fe2+ is reached only in anaerobic crystals, while in the presence of dioxygen it is always significantly lower, suggesting that this site participates in turnover. Our observation that the Fe2+ occupancy of site 2 is always lower than that of site 1 provides an indication that as soon as site 2 is occupied Fe2+ can be oxidized in both sites 1 and 2, leaving a fraction of Fe2+ ions bound to site 1. Our data do not definitely exclude Fe2+ oxidation independently occurring in other sites, such as, for example, sites 3 and 4, but clearly indicate the presence of multiple alternative iron-binding sites rather than the unique ‘third’ site postulated by others (Ebrahimi et al., 2013 ▶; Honarmand Ebrahimi et al., 2012 ▶). The variability of these additional sites in different proteins (and in their functional mutants such as H54Q) does not support the proposal that they can be part of an universal mechanism (Honarmand Ebrahimi et al., 2012 ▶), but rather favours their role as protein-specific modulators of the oxidoreductase reaction kinetics (Bou-Abdallah et al., 2014 ▶).
5. Related literature
The following references are cited in the Supporting Information for this article: Armstrong et al. (1984 ▶), Armstrong & Lippard (1984 ▶), Crichton & Declercq (2010 ▶), Feng et al. (1989 ▶), Harding (1999 ▶), Harding & Hsin (2014 ▶), Levi et al. (1996 ▶), Lv et al. (2014 ▶), Masuda et al. (2010 ▶), Norman, Holz et al. (1990 ▶), Norman, Yan et al. (1990 ▶), Orpen et al. (1989 ▶), Thich et al. (1976 ▶), Tolman et al. (1989 ▶), Tosha, Behara, Ng et al. (2012 ▶) and Yang et al. (2008 ▶).
Supplementary Material
PDB reference: frog ferritin M, native, 4lqh
PDB reference: iron-loaded, 1 min, 4lpj
PDB reference: 2 min, 4lqj
PDB reference: 5 min, 4lyx
PDB reference: 15 min, 4lyu
PDB reference: 30 min, 4lqv
PDB reference: 60 min, 4lqn
PDB reference: iron-loaded under anaerobic environment, 4my7
PDB reference: H54Q mutant, 4mku
PDB reference: H54Q mutant, iron-loaded, 1 min, 4ml5
PDB reference: 15 min, 4mn9
PDB reference: 60 min, 4mjy
Supporting Information.. DOI: 10.1107/S1399004715002333/dz5359sup1.pdf
Acknowledgments
We acknowledge the European Synchrotron Radiation Facility (ESRF), Grenoble, France and Elettra, Trieste, Italy for the provision of synchrotron-radiation facilities and we would like to thank all of the staff for assistance in using the beamlines. We also acknowledge the Italian PRIN projects 2009FAKHZT_001 and 2012SK7ASN_001, the CIRMMP consortium (SM and PT) and NIH DK20251, and the CHORI Partners (ECT) for providing partial financial support to this work.
References
- Armstrong, W. H. & Lippard, S. J. (1984). J. Am. Chem. Soc. 106, 4632–4633.
- Armstrong, W. H., Spool, A., Papaefthymiou, G. C., Frankel, R. B. & Lippard, S. J. (1984). J. Am. Chem. Soc. 106, 3653–3667. [DOI] [PubMed]
- Arosio, P., Ingrassia, R. & Cavadini, P. (2009). Biochim. Biophys. Acta, 1790, 589–599. [DOI] [PubMed]
- Behera, R. K. & Theil, E. C. (2014). Proc. Natl Acad. Sci. USA, 111, 7925–7930. [DOI] [PMC free article] [PubMed]
- Bertini, I., Lalli, D., Mangani, S., Pozzi, C., Rosa, C., Theil, E. C. & Turano, P. (2012). J. Am. Chem. Soc. 134, 6169–6176. [DOI] [PMC free article] [PubMed]
- Bou-Abdallah, F., Arosio, P., Levi, S., Janus-Chandler, C. & Chasteen, N. D. (2003). J. Biol. Inorg. Chem. 8, 489–497. [DOI] [PubMed]
- Bou-Abdallah, F., Arosio, P., Santambrogio, P., Yang, X., Janus-Chandler, C. & Chasteen, N. D. (2002). Biochemistry, 41, 11184–11191. [DOI] [PubMed]
- Bou-Abdallah, F., Yang, H., Awomolo, A., Cooper, B., Woodhall, M. R., Andrews, S. C. & Chasteen, N. D. (2014). Biochemistry, 53, 483–495. [DOI] [PMC free article] [PubMed]
- Cho, J., Jeon, S., Wilson, S. A., Liu, L. V., Kang, E. A., Braymer, J. J., Lim, M. H., Hedman, B., Hodgson, K. O., Valentine, J. S., Solomon, E. I. & Nam, W. (2011). Nature (London), 478, 502–505. [DOI] [PMC free article] [PubMed]
- Costas, M., Mehn, M. P., Jensen, M. P. & Que, L. Jr (2004). Chem. Rev. 104, 939–986. [DOI] [PubMed]
- Crichton, R. R. & Declercq, J.-P. (2010). Biochim. Biophys. Acta, 1800, 706–718. [DOI] [PubMed]
- Ebrahimi, K. H., Hagedoorn, P. L. & Hagen, W. R. (2013). Chembiochem, 14, 1123–1133. [DOI] [PubMed]
- Ebrahimi, K. H., Hagedoorn, P. L., van der Weel, L., Verhaert, P. D. E. M. & Hagen, W. R. (2012). J. Biol. Inorg. Chem. 17, 975–985. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Feng, X., Bott, S. G. & Lippard, S. J. (1989). J. Am. Chem. Soc. 111, 8046–8047.
- Ha, Y., Shi, D., Small, G. W., Theil, E. C. & Allewell, N. M. (1999). J. Biol. Inorg. Chem. 4, 243–256. [DOI] [PubMed]
- Haldar, S., Bevers, L. E., Tosha, T. & Theil, E. C. (2011). J. Biol. Chem. 286, 25620–25627. [DOI] [PMC free article] [PubMed]
- Harding, M. M. (1999). Acta Cryst. D55, 1432–1443. [DOI] [PubMed]
- Harding, M. M. & Hsin, K.-Y. (2014). Methods Mol. Biol. 1091, 333–342. [DOI] [PubMed]
- Honarmand Ebrahimi, K., Bill, E., Hagedoorn, P. L. & Hagen, W. R. (2012). Nature Chem. Biol. 8, 941–948. [DOI] [PubMed]
- Hwang, J., Krebs, C., Huynh, B. H., Edmondson, D. E., Theil, E. C. & Penner-Hahn, J. E. (2000). Science, 287, 122–125. [DOI] [PubMed]
- Kabsch, W. (2010a). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Krebs, C., Bollinger, J. M. Jr, Theil, E. C. & Huynh, B. H. (2002). J. Biol. Inorg. Chem. 7, 863–869. [DOI] [PubMed]
- Krebs, C., Edmondson, D. E. & Huynh, B. H. (2002). Methods Enzymol. 354, 436–454. [DOI] [PubMed]
- Kurtz, D. M. Jr (1990). Chem. Rev. 90, 585–606.
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Levi, S., Santambrogio, P., Corsi, B., Cozzi, A. & Arosio, P. (1996). Biochem. J. 317, 467–473. [DOI] [PMC free article] [PubMed]
- Liu, X. & Theil, E. C. (2004). Proc. Natl Acad. Sci. USA, 101, 8557–8562. [DOI] [PMC free article] [PubMed]
- Liu, X. & Theil, E. C. (2005). Acc. Chem. Res. 38, 167–175. [DOI] [PubMed]
- Lv, C., Zhang, S., Zang, J., Zhao, G. & Xu, C. (2014). Biochemistry, 53, 2232–2241. [DOI] [PubMed]
- Marchetti, A., Parker, M. S., Moccia, L. P., Lin, E. O., Arrieta, A. L., Ribalet, F., Murphy, M. E., Maldonado, M. T. & Armbrust, E. V. (2009). Nature (London), 457, 467–470. [DOI] [PubMed]
- Masuda, T., Goto, F., Yoshihara, T. & Mikami, B. (2010). Biochem. Biophys. Res. Commun. 400, 94–99. [DOI] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Norman, R. E., Holz, R. C., Menage, S., Que, L. Jr, Zhang, J. H. & O’Connor, C. J. (1990). Inorg. Chem. 29, 4629–4637.
- Norman, R. E., Yan, S., Que, L. Jr, Backes, G., Ling, J., Sanders-Loehr, J., Zhang, J. H. & O’Connor, C. J. (1990). J. Am. Chem. Soc. 112, 1554–1562.
- Orpen, A. G., Brammer, L., Allen, F. H., Kennard, O., Watson, D. G. & Taylor, R. (1989). J. Chem. Soc. Dalton Trans., pp. S1–S83.
- Pereira, A. S., Small, W., Krebs, C., Tavares, P., Edmondson, D. E., Theil, E. C. & Huynh, B. H. (1998). Biochemistry, 37, 9871–9876. [DOI] [PubMed]
- Pfaffen, S., Abdulqadir, R., Le Brun, N. E. & Murphy, M. E. P. (2013). J. Biol. Chem. 288, 14917–14925. [DOI] [PMC free article] [PubMed]
- Takahashi, T. & Kuyucak, S. (2003). Biophys. J. 84, 2256–2263. [DOI] [PMC free article] [PubMed]
- Tatur, J., Hagen, W. R. & Matias, P. M. (2007). J. Biol. Inorg. Chem. 12, 615–630. [DOI] [PMC free article] [PubMed]
- Theil, E. C. (2011). Curr. Opin. Chem. Biol. 15, 304–311. [DOI] [PMC free article] [PubMed]
- Theil, E. C., Turano, P., Ghini, V., Allegrozzi, M. & Bernacchioni, C. (2014). J. Biol. Inorg. Chem. 19, 615–622. [DOI] [PMC free article] [PubMed]
- Thich, J. A., Ou, C. C., Powers, D., Vasiliou, B., Mastropaolo, D., Potenza, J. A. & Schugar, H. J. (1976). J. Am. Chem. Soc. 98, 1425–1433.
- Tolman, W. B., Bino, A. & Lippard, S. J. (1989). J. Am. Chem. Soc. 111, 8522–8523.
- Tosha, T., Behera, R. K., Ng, H.-L., Bhattasali, O., Alber, T. & Theil, E. C. (2012). J. Biol. Chem. 287, 13016–13025. [DOI] [PMC free article] [PubMed]
- Tosha, T., Behera, R. K. & Theil, E. C. (2012). Inorg. Chem. 51, 11406–11411. [DOI] [PMC free article] [PubMed]
- Tosha, T., Ng, H.-L., Bhattasali, O., Alber, T. & Theil, E. C. (2010). J. Am. Chem. Soc. 132, 14562–14569. [DOI] [PMC free article] [PubMed]
- Toussaint, L., Bertrand, L., Hue, L., Crichton, R. R. & Declercq, J.-P. (2007). J. Mol. Biol. 365, 440–452. [DOI] [PubMed]
- Tshuva, E. Y. & Lippard, S. J. (2004). Chem. Rev. 104, 987–1012. [DOI] [PubMed]
- Turano, P., Lalli, D., Felli, I. C., Theil, E. C. & Bertini, I. (2010). Proc. Natl Acad. Sci. USA, 107, 545–550. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vincent, J. B., Olivier-Lilley, G. L. & Averill, B. A. (1990). Chem. Rev. 90, 1447–1467.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yang, T.-Y., Dudev, T. & Lim, C. (2008). J. Am. Chem. Soc. 130, 3844–3852. [DOI] [PubMed]
- Yang, X., Arosio, P. & Chasteen, N. D. (2000). Biophys. J. 78, 2049–2059. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: frog ferritin M, native, 4lqh
PDB reference: iron-loaded, 1 min, 4lpj
PDB reference: 2 min, 4lqj
PDB reference: 5 min, 4lyx
PDB reference: 15 min, 4lyu
PDB reference: 30 min, 4lqv
PDB reference: 60 min, 4lqn
PDB reference: iron-loaded under anaerobic environment, 4my7
PDB reference: H54Q mutant, 4mku
PDB reference: H54Q mutant, iron-loaded, 1 min, 4ml5
PDB reference: 15 min, 4mn9
PDB reference: 60 min, 4mjy
Supporting Information.. DOI: 10.1107/S1399004715002333/dz5359sup1.pdf