

Abstract
Contact hypersensitivity (CHS) is a CD8 T cell-mediated response to hapten sensitization and challenge of the skin. Effector CD8 T cell recruitment into the skin parenchyma to elicit the response to hapten challenge requires prior CXCL1/KC-directed neutrophil infiltration within 3–6 h after challenge and is dependent on IFN-γ and IL-17 produced by the hapten-primed CD8 T cells. Mechanisms directing hapten-primed CD8 T cell localization and activation in the antigen challenge site to induce this early CXCL1 production were investigated. Both TNFα and IL-17, but not IFN-γ, mRNA was detectable in the hapten challenge site within 1 h of challenge of sensitized mice and increased thereafter. Expression of ICAM-1 was observed by 1 h after challenge of sensitized and non-sensitized mice and was dependent on TNFα. The induction of IL-17, IFN-γ and CXCL1 in the challenge site was not observed when ICAM-1 was absent or neutralized by specific antibody. During the elicitation of the CHS response, endothelial cells expressed ICAM-1 and produced CXCL1 suggesting this as the site of CD8 T cell localization and activation. Endothelial cells isolated from challenged skin of naïve and sensitized mice had acquired the hapten and the ability to activate hapten-primed CD8 T cell cytokine production. These results indicate that hapten application to the skin of sensitized animals initiates an inflammatory response promoting hapten-primed CD8 T cell localization to the challenge site through TNFα induced ICAM-1 expression and CD8 T cell activation to produce IFN-γ and IL-17 through endothelial cell presentation of hapten.
Keywords: Skin, CD8 T cells, neutrophils, TNFα, ICAM-1, IL-17, IFN-γ
The inflammation induced in response to tissue injury is initiated by acute phase cytokines produced by the vascular endothelium, resident macrophages, and tissue parenchymal cells. Cytokines, such as TNFα, stimulate the expression of adhesion molecules on vascular endothelium and the production of chemoattractants that synergize to direct leukocyte recruitment to the injury site (1–3). During many inflammatory responses TNFα directly induces the production of the neutrophil chemoattractants IL-8 and CXCL1/KC as well as chemoattractants for monocytes/macrophages and dendritic cells (1–3). Neutrophils are typically the first leukocytes recruited to tissue inflammation sites in response to the production of these chemoattractants (4–6), implicating TNFα as a critical mediator of this infiltration. During some T cell-mediated responses, however, production of these neutrophil chemoattractants requires the initial activation of antigen-specific T cells within the inflammatory site (7). The mechanisms directing these T cells to the site prior to neutrophil infiltration and the mechanisms activating the T cells to stimulate neutrophil chemoattractant production in such responses remain poorly defined.
Contact hypersensitivity (CHS) is a T cell mediated inflammatory response to epidermal sensitization and subsequent challenge with a hapten. Hapten application to the skin triggers antigen acquisition by epidermal and dermal dendritic cells, including Langerhans cells and their migration to the skin draining lymph nodes where they prime effector T cells (8–10). Following hapten challenge of sensitized individuals, the hapten-primed T cells infiltrate the challenge site and are activated to produce cytokines that mediate the characteristic tissue edema that peaks 18–48 hours after challenge and then resolves. CD8 T cells are the primary effector cells mediating CHS responses to many haptens including dinitrofluorobenzene (DNFB), oxazolone (Ox) and urushiol, the reactive hapten in poison ivy and are low-to-absent in mice without CD8 T cells whereas the absence of CD4 T cells typically results in elevated responses (11–13). In support of these roles, sensitization with DNFB and other haptens was shown to prime hapten-specific CD8 T cells producing IFN-γ whereas hapten-primed CD4 T cells produced IL-4, IL-5 and IL-10 (13). More recent studies have documented hapten priming of separate populations of hapten-specific CD8 T cells producing IFN-γ and IL-17 and that the elicitation of CHS to DNFB and Ox requires activation of both the IL-17- and the IFN-γproducing CD8 T cell populations within the challenge site (7, 14, 15).
A critical event during the elicitation of CHS is hapten-primed CD8 T cell recruitment to the challenge site. Studies from this laboratory indicated that this recruitment requires prior CXCL1/KC-mediated neutrophil infiltration into the site (16, 17). This neutrophil infiltration into the challenge site skin parenchyma can be observed within 4–6 hours after hapten challenge of sensitized animals (7). Surprisingly, CD8 T cell infiltration into the skin parenchyma is not observed at this time and does not occur until 8–12 hours later. The production of CXCL1 and CXCL2 in the challenge site that directs this early neutrophil infiltration into the hapten challenged skin, however, requires the activation of both the IL-17- and the IFN-γ-producing CD8 T cell populations within the challenge site. Overall, these results indicate that the elicitation of the CHS response is initiated by the recruitment and activation of these hapten-primed CD8 T cell populations to induce the production of neutrophil chemoattractants that direct the initial infiltration of neutrophils into the skin.
The factors that direct the hapten-primed CD8 T cell populations to the challenge site shortly after challenge and activate them to produce the IL-17 and IFN-γ that induce the neutrophil chemoattractants remain undefined. The goal of the current study was to identify mechanisms mediating this initial CD8 T cell localization and activation in the challenge site. The results implicate hapten-induced production of TNFα within 1 hr. following hapten challenge and TNFα-induced ICAM-1 expression for the recruitment of the hapten-primed CD8 T cell populations to the challenge site. The results also indicate the acquisition and presentation of hapten by endothelial cells in the challenge site that activates the CD8 T cells to produce the IL-17 and IFN-γ that, in turn, stimulate the endothelial cells to produce the neutrophil chemoattractants. Collectively, these studies indicate that hapten application to the skin of immune animals initiates an inflammatory response that promotes both the localization and activation of hapten-primed CD8 T cells to produce the cytokines initiating the innate immune response required for subsequent elicitation of the CD8 T cell mediated response in the skin.
Materials and Methods
Mice
BALB/c (H-2d) and C57/BL6 (H-2b) mice were obtained through Dr. Clarence Reeder (National Cancer Institute, Frederick, MD). C3H (H-2k) mice were obtained from Taconic (Hudson, NY). ICAM-1−/− mice on a C57BL/6 background were purchased from Jackson Laboratory (Bar Harbor, ME). Female mice, 8–10 weeks of age, were used throughout these studies.
Hapten sensitization and elicitation of CHS
Mice were sensitized to 2,4-dinitrofluorobenzene (DNFB) or oxazolone (Ox) by painting the shaved abdomen with 25 μl 0.25% DNFB (Sigma Aldrich, St. Louis, MO) or 25 μl 1% Ox (Sigma Aldrich) and 10 μl to each paw on days 0 and +1 (7, 13). On day +5 hapten sensitized and control, non-sensitized mice were challenged on each side of each ear with 10 μl DNFB or Ox to elicit the CHS response. Ear thickness was measured using an engineer’s micrometer (Mitutoyo, Elk Grove Village, IL) and expressed in units of 10−4 in. The ear swelling response is given as the mean increase of each group of 4 individual animals ± SEM.
Antibodies and cytokines
Purified anti-CD4 mAb YTS 191.1.2 and GK1.5; anti-CD8 mAb YTS 169 and TIB-105; anti-Gr-1 mAb RB6.8C5; anti-mouse IFN-γ mAb XMG1.2; anti-mouse LFA-1 mAb FD441-8; and, anti-mouse ICAM-1 mAb YN1-1.7.4 used for in vivo treatment were purchased from BioXCell (West Lebanon, NH). Anti-mouse IL-17 mAb was purchased from Southern Biotech (Birmingham, AL). Culture supernatant of the anti-mouse anti-TNFα mAb producing hybridoma XT3 was used to purify IgG by protein G chromatography. Recombinant CXCL1/KC, IL-17 and IFN-γ were purchased from R&D Systems, Inc. (Minneapolis, MN).
For in vivo depletion of CD4 T cells, mice were injected with 100 μg of each anti-CD4 mAb, YTS 191 and GK1.5, i.p. on three consecutive days before hapten sensitization on days 0 and +1 as previously described (13, 18). CD8 T cells were depleted by injecting mice with 100 μg of each anti-CD8 mAb, YTS 169 and TIB-150. In each experiment, treated sentinel mice were used to evaluate the efficiency of CD4 or CD8 T cell depletion by antibody staining and flow cytometry analysis of spleen and lymph node cells (LNC) and was always >95% when compared to cells from control, rat IgG treated mice.
In vivo neutralization of TNFα was performed by injecting mice with 250 μg anti-mouse TNFα i.v. at the time of hapten challenge. In vivo antagonism of ICAM-1 and/or LFA-1 was performed by injecting mice with 300 μg anti-mouse ICAM-1 mAb, 300 μg anti-mouse LFA-1 mAb, or 150 μg of each on the day of hapten-challenge.
Quantitation of CXCL1/KC production by immunoassay
Hapten challenged or normal abdominal skin was excised and homogenized in 500 μl proteinase inhibitor cocktail (Sigma Aldrich) with gentle shaking for 30 min. Following centrifugation at 12000 × g for 10 min. the supernatants were collected and the total protein concentration quantified using a Coomasie Plus Protein Assay Reagent Kit (Pierce, Rockford, IL). All samples were diluted to an equivalent total protein concentration and tested for concentrations of CXCL1 by ELISA as previously reported. Supernatants from endothelial cell line cultures were also tested for concentrations of CXCL1 using the ELISA.
Analysis of gene expression by quantitative RT-PCR
Hapten challenged or normal abdominal skin was excised and homogenized in TRIZOL (Invitrogen Life Technologies, Carlsbad, CA) with subsequent chloroform extraction to isolate the whole cell RNA. cDNA was synthesized from 2 μg RNA using the TaqMan Reverse Transcription Reagent Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. PCR was performed using custom primers and FAM dye-labeled probes (Applied Biosystems) for mouse IFN-γ, IL-17, CXCL1, TNFα, ICAM-1, and Mrpl 32 (gene assay ID#: Mm00801778_m1, Mm00439619_m1, Mm00433859_m1, Mm00443258_m1, Mm00516023_m1 and, Mm00777741_sH respectively).
The comparative CT method for relative quantitation of cytokine gene expression was used where log measurements for each sample are made during amplification and the expression level of the Mrpl 32 housekeeping gene is subtracted from the expression level for each test cytokine gene. For each test cytokine, the expression level of a single RNA sample prepared from the unchallenged skin of non-sensitized wild-type mice was used as the calibrator and was arbitrarily set at 1.0 and the expression levels of all other samples were then normalized to the calibrator. Duplicate runs of each individual RNA sample prepared from a single mouse of 3–4 mice per group were tested and the data from 3–4 RNA samples for each group are expressed as mean test cytokine expression level ± SEM.
T cell transfer
C57BL/6 mice were depleted of CD4+ T cells using specific mAb prior to sensitization to DNFB. On day +4 following sensitization, lymph node cell (LNC) suspensions were prepared from the sensitized mice and aliquots of 4 × 106 LNC were transferred i.v. into naïve C57BL/6 or ICAM-1−/− recipients that were immediately challenged on the shaved abdomen with 25 ml of 0.2% DNFB.
Sensitization with TRITC and analysis of hapten-expressing cells
Mice were sensitized to TRITC hapten by application of 10 μl of 6.67 mg/ml TRITC (Sigma Aldrich) to each side of each ear on days 0 and +1. On day +5 TRITC sensitized mice were challenged on the shaved abdomen with 25 μl TRITC. After six hours, the challenged skin, as well as skin from non-sensitized, non-challenged mice, was excised and incubated in 0.5% dispase (Invitrogen) for 18 hours at 4°C. The next day, the epidermis was separated from the dermis and incubated in 0.5% trypsin (Sigma Aldrich) for 60 min. at 37° C, 5% CO2. The epidermis was pressed through renal dialysis tubing to isolate individual cells. The cells were washed twice in HBSS, incubated in 0.2% DNase (Roche, Indianapolis, IN) for 10 min. at room temperature and washed again. Aliquots of 1 × 106 cells were washed in staining buffer (Dulbecco’s PBS with 2% FCS/0.2% NaN3) and incubated in Fc block (BD Pharmingen, San Jose, CA) diluted 125:10,000 in the staining buffer for 20 min on ice. The cells were washed and stained with APC-labeled anti-mouse anti-CD31 mAb MEC13.3 (BD Pharmingen). After 30 min., the cells were washed, resuspended in staining buffer and analyzed by two-color flow cytometry using a FACSCalibur and CellQuest software (Becton-Dickinson, San Jose, CA). The cells were gated to exclude residual tissue debris and non-viable cells and sample data were collected on 20,000 cells.
To obtain hapten-labeled endothelial cells from the skin, cell suspensions were prepared from TRITC sensitized and challenged skin as previously described. TRITC-labeled populations for cell sorting were stained with Pacific Blue-labeled anti-CD31 mAb M-20 (Invitrogen) and sorted from the remaining cells using a FACSAria (Becton-Dickinson). The positively selected hapten-labeled endothelial cells were subsequently cultured with naïve CD8 or purified hapten-immune CD4 or CD8 T cells from TRITC-sensitized mice on day +4.
Analysis of tissue infiltrating cells by flow cytometry
On day +5 the shaved trunk skin of sensitized and non-sensitized mice was challenged with DNFB and 6 hrs. later the challenged skin was removed and digested to prepare cell suspensions as previously described. The isolated cells were washed, stained with fluorochrome-labeled FITC-labeled rat anti-mouse CD45 mAb 30-F11 (BD Pharmingen) and PE-labeled rat anti-mouse Gr-1 mAb, RB6.8C5 (eBioScience, San Diego, CA) and analyzed by two-color flow cytometry.
Histological analyses
Hapten challenged skin was excised from naïve and sensitized mice 6 h after challenge and fixed with acid methanol (60% methanol, 10% acetic acid). Paraffin embedded sections (8 μm) were cut on edge and mounted onto slides. The slides were deparaffinized, rehydrated and boiled in antigen retrieval solution (Biogenex, San Ramon, CA). Overnight staining was done with 5μg/ml polyclonal goat-anti-mouse CXLC1 antibody (R&D Systems Inc.) diluted in PBS/1% BSA solution at 4°C. Control slides were incubated with normal goat serum as the primary antibody (Vector Laboratories, Burlingame, CA). Primary antibody binding was detected using biotinylated rabbit anti-goat IgG followed by streptavidin horse radish peroxidase and developed using the substrate chromagen 3,3′-diaminobenzidine (DAB).
For immunohistochemistry to detect ICAM-1, antigen retrieval was performed on fixed sections by immersion of slides in two changes of Trilogy-EDTA, pH 8.0 (Cell Marque, Hot Springs, AR) in a steamer for 1 h. Endogenous peroxidase activity was blocked by incubation with 0.3% H2O2 in methanol. Non-specific protein activity was blocked by incubation with a serum-free protein block (DAKO Corp, Carpinteria, CA). Staining was performed with an 1:20 dilution of purified polyclonal goat anti-mouse ICAM-1 IgG (R & D Systems, Inc.) for 60 min. at room temperature. Primary antibody was detected using biotinylated anti-goat antibody. Staining was performed with a VECTASTAIN ABC Elite kit (Vector) and developed using DAB.
For immunofluorescent analyses, excised skin was fixed in Histochoice and frozen sections (8 μm) were cut on edge and mounted onto slides. The slides were stained with 5 μg/ml polyclonal goat-anti-mouse CXLC1 antibody (R&D Systems Inc., Minneapolis, MN) and 4 μg/ml rat anti-CD31 mAb M-20 (Santa Cruz Biotech, Santa Cruz, CA) diluted in PBS/1% BSA overnight at 4°C. The slides were washed and stained sequentially for 1 h at room temperature with 2 μg/ml rabbit anti-goat IgG Alexa Fluor 488 and then 2 μg/ml goat anti-rat IgG Alexa Fluor 568 (Molecular Probes, Eugene, OR) diluted in HBSS. After washing in HBSS, slides were mounted with Vectashield/DAPI (Vector Laboratories), were viewed at 488 and 568 nm, and images captured using Image ProPlus 5.0.
Statistical analysis
Statistical analysis to assess differences between experimental groups was performed using Students’ t test. Differences were considered significant when P < 0.05.
Results
TNFα is expressed in the skin challenge site as early as one hour following challenge of sensitized mice
To begin to identify factors that direct the recruitment of the hapten-primed CD8 T cells producing IL-17 and IFN-γ to the skin challenge site to initiate the CHS response, the temporal expression of inflammatory mediators was tested in the skin of naïve and sensitized mice within the first 4 h after challenge with hapten. The prediction was that candidate factors would be expressed equivalently at early times in response to hapten application in both naïve and immune animals but may increase further in the immune animals as the antigen specific CD8 T cell response is initiated and progresses. DNFB challenged skin from naïve and sensitized mice was excised 1, 2, 3 and 4 h after the challenge and whole cell RNA was isolated and tested for expression levels of TNF-α, IL-17, IFN-γ, and ICAM-1 by qRT/PCR. Expression of the test genes was not detected in skin that had not been challenged with hapten (Figure 1). TNFα expression was detected within 1 h after hapten application to the skin of naïve and hapten-sensitized mice although the expression levels were 2-fold higher in hapten challenged skin from sensitized vs. naive mice as early as 1 h after challenge and increased thereafter to the 4 h time-point. ICAM-1 expression was also detected as early as 1 h following hapten challenge but was expressed at similar levels in the challenged skin of both naïve and sensitized mice at 1 and 3 h after challenge. At 4 ho post-challenge ICAM-1 expression increased in the challenged skin of sensitized but not naive mice. Similar to the expression of TNFα, the expression of IL-17A was also evident within an hour of skin challenge of sensitized but not naïve mice and increased with time after challenge. In contrast to the rapid expression of IL-17, the expression of IFN-γ was at low levels in the challenged skin of naïve and sensitized mice until 3 h after challenge and then only increased in the challenged skin of the sensitized mice.
FIGURE 1.

Induction of TNF-α, ICAM-1, IL-17, and IFN-γ expression during elicitation of CHS. Groups of 4 C57BL/6 mice were sensitized with 0.25% DNFB on days 0 and +1. On day +5, the sensitized and groups of control naïve mice were challenged with 0.2% DNFB. Challenged skin was excised at 1, 2, 3, and 4 hrs. post-challenge and snap-frozen. Skin from naïve/non-challenged (NNC) mice was excised as a control. Whole cell RNA was prepared and was used to assess mRNA expression of TNF-α, ICAM-1, IL-17, and IFN-γ in the skin samples by quantitative RT/PCR. The mean expression level for each of 4 samples per group ± SEM is shown. All results are representative of two individual experiments. *p ≤ 0.05.
Early expression of IL-17 and IFN-γ is dependent upon TNFα
The impact of TNFα production on the expression of ICAM-1, IL-17 and IFN-γ in the hapten challenge site was then tested. Groups of naïve and sensitized mice were treated with control rat IgG or with anti-TNFα mAb, challenged with DNFB, and the challenged skin excised 2, 4 and 6 h later to assess expression levels of the target genes (Figure 2A–C). Expression of ICAM-1 observed in challenged skin of both naïve and sensitized mice was significantly decreased by TNFα neutralization. Similarly, expression of IL-17 observed in the challenged skin of sensitized but not naïve mice as early as 1 h after challenge was decreased by neutralization of TNFα and the later expression of IFN-γ in the challenged skin of sensitized mice was also decreased by treatment with the anti-TNFα mAb. The down regulation of these mediators was reflected by the marked inhibition of the CHS response when anti-TNFα mAb was given to sensitized mice at the time of hapten challenge (Figure 2D).
FIGURE 2.

Anti-TNF-α antibody decreases inflammatory mediators during elicitation of CHS. Groups of 4 C57BL/6 mice were sensitized with 0.25% DNFB on days 0 and +1. On day +5, the indicated groups of sensitized and non-sensitized naïve mice were treated with 250 μg anti-TNF-α mAb prior to challenge with 0.2% DNFB. A–C. Challenged skin was excised at 1, 2, 3, and 4 hrs. post-challenge and snap-frozen. Skin from naïve/non-challenged (NNC) mice was excised as a control. Whole cell RNA was prepared and was used to assess mRNA expression of ICAM-1, IL-17, and IFN-γ in the skin samples by quantitative RT/PCR. The mean expression level for each of 4 samples per group ± SEM is shown. D. Ear thickness was monitored pre-challenge and 24 hr. after challenge. The mean increase in ear thickness following hapten challenge is shown in 10-4 in. ± SEM for groups of 4 mice. All results are representative of two individual experiments each. All results are representative of two individual experiments. *p ≤ 0.05.
TNFα, ICAM-1, and LFA-1 are required to induce CXCL1 production and CHS response in vivo
Since neutralization of TNFα down-modulated both the expression of ICAM-1 and CD8 T cell cytokines required for elicitation of CHS, the role of ICAM-1 in the recruitment of the IL-17 and IFN-γ producing CD8 T cells to the challenge site was assessed. This was first approached by testing the expression of the IL-17 and IFN-γ induced CXCL1 in the challenge site when groups of DNFB sensitized wild-type mice were treated with control Ig, anti-ICAM-1 mAb, anti-LFA-1 mAb or with both anti-ICAM-1 plus anti-LFA-1 mAb at the time of challenge. The hapten challenged skin was excised 6 h later and prepared homogenates were tested for the levels of CXCL1 as an indication of IL-17 and IFN-γ production at the site (Figure 3A). Whereas skin from naïve mice challenged with hapten did not produce CXCL1, challenged skin from sensitized mice produced CXCL1 by 6 h after challenge and this production was inhibited by treatment with either anti-ICAM-1 mAb or anti-LFA-1 mAb at the time of DNFB challenge (Figure 3A). Consistent with the decreased CXCL1 production was the attenuated or absent neutrophil infiltration into the skin challenge site at 6 hours post-challenge in mice treated with ICAM-1- and/or LFA-1-specific antibodies (Figure 3B) as well as the absence of CHS responses read at 24 h after the hapten challenge (data not shown).
FIGURE 3.

Neutralization of ICAM-1 or LFA-1 during skin challenge to elicit CHS inhibits CXCL1 production and neutrophil infiltration into the challenge site. Groups of 4 C57B/6 mice were sensitized with 0.25% DNFB on days 0 and +1. On day +5, sensitized mice were treated with either 300 μg anti-ICAM-1 mAb, 300 μg anti-LFA-1 mAb, a mixture of 150 μg of both antibodies, or 300 μg control rat IgG and immediately challenged with 0.2% DNFB. A. Challenged areas of skin were removed at 6 h post-challenge from sensitized treated mice and non-sensitized mice and prepared tissue homogenates were tested for the levels of CXCL1 by ELISA. The mean concentration ± SEM for 4 individual samples per group is shown. All results are representative of two individual experiments each. *p ≤ 0.04. B. The skin was excised 6 h after challenge, digested, and prepared cell suspensions were stained with FITC-labeled anti-CD45 mAb and PE-labeled anti-GR-1 mAb and analyzed by flow cytometry to assess neutrophil infiltration into the challenge site. All results are representative of two individual experiments each.
These studies were extended by testing the expression of CXCL1 in the skin challenge site when sensitized ICAM-1−/− and wild-type mice were challenged with hapten. Whereas high levels of CXCL1 were observed in the challenged skin of sensitized wild-type mice 6 ho after challenge, levels were barely detectable following sensitization and challenge of ICAM-1−/− mice (Figure 4A). However, T cell priming is severely compromised in ICAM-1-deficient mice (19) and is likely to account at least in part for the absence of the CD8 T cell induced CXCL1 at the site. Therefore, we tested the ability of transferred hapten-primed CD8 T cells from sensitized wild-type donors to induce CXCL1 in challenged naïve wild-type and ICAM-1−/− mice. Transfer of hapten-primed wild-type CD8 T cells to naïve wild-type recipients induced high levels of CXCL1 production in response to hapten challenge of the skin. In contrast, transfer of the wild-type CD8 T cells to naïve ICAM-1−/− mice did not induce this production.
FIGURE 4.

Activation of IL-17- and IFN-γ-producing CD8 T cells in the hapten challenge site requires ICAM-1. A. Groups of 4 C57BL/6 mice and B6.ICAM-1−/− mice were sensitized with 0.25% DNFB on days 0 and +1. On day +5, the indicated groups of sensitized and non-sensitized naïve mice were challenged with 0.2% DNFB. In addition, wild-type C57BL/6 mice were sensitized with DNFB and on day +4, lymph node cell suspensions were prepared and aliquots of 4 × 106 T cells were injected i.v. into naïve wild-type or ICAM-1−/− mice immediately prior to challenge with 0.2% DNFB. Challenged skin was excised 6 h after challenge, whole cell RNA was prepared and was used to assess mRNA expression of IL-17, IFN-γ, and CXCL1/KC in the skin samples by quantitative RT/PCR. The mean expression level for each of 4 samples per group ± SEM is shown. B. Total protein was prepared from excised skin samples of each individual in the groups and tested for levels of CXCL1 by ELISA. The mean concentration ± SEM for 4 individual samples per group is shown. All results are representative of two individual experiments each. *p ≤ 0.05.
To directly test the presence or absence of hapten-primed CD8 T cell activation in the skin challenge site, skin was excised 6 h after challenge either of sensitized wild-type and ICAM-1−/− mice or of naïve wild-type and ICAM-1−/− mice that had received hapten-primed wild-type CD8 T cells (Figure 4B). After 6 h the challenged skin from DNFB sensitized wild-type mice expressed high levels of CXCL1, IFN-γ and IL-17. In contrast, the challenged skin from sensitized ICAM-1−/− mice expressed very low to undetectable levels of these genes and this was not corrected by transferring hapten-primed wild-type CD8 T cells to the ICAM-1−/− mice. Thus, ICAM-1 expression is required in the skin challenge site for the activation of the IL-17-and IFN-γ-producing CD8 T cells and the induction of CXCL1 during the initiation of CHS.
Since the expression of ICAM-1 was required for the activation of hapten-primed CD8 T cell expression of IL-17 and IFN-γ within the skin challenge site, the cells expressing ICAM-1 in the site were examined by staining prepared sections of hapten challenged skin from naïve and sensitized mice by immunohistochemistry. ICAM-1 staining was not visible in skin from non-sensitized mice not challenged with hapten (Figure 5A). ICAM-1 staining was also not detected in hapten challenged skin from naïve and sensitized challenged mice at 2 h post-challenge. However, ICAM-1 staining of vascular endothelial cells was observed within 4 h post-challenge in hapten challenged skin from both non-sensitized and sensitized mice. ICAM-1 positive vessels were more abundant in the hapten challenged skin from sensitized vs. naïve mice and this increased in both groups at 6 h post-challenge. Consistent with the mRNA levels detected in the challenge site (Figure 2), treatment of sensitized mice with anti-TNFα mAb at the time of hapten challenge markedly decreased ICAM-1 staining (Figure 5B).
FIGURE 5.

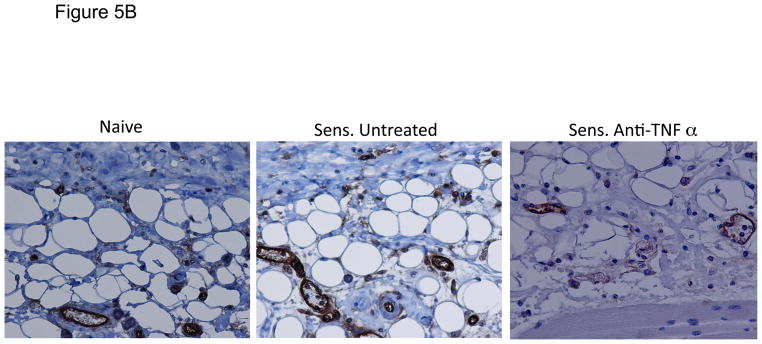
ICAM-1 is induced by hapten application to the skin. C57BL/6 mice were sensitized with 0.25% DNFB on days 0 and +1. On day +5, sensitized and non-sensitized mice were challenged on a shaved square area of trunk skin with 0.2% DNFB. A. Challenged areas of skin were removed at 2, 4, and 6 h post-challenge from challenged and non-challenged mice and fixed in methanol. Paraffin-embedded sections were prepared and stained with anti-ICAM-1 mAb or with control rat IgG. Slides were examined by light microscopy and representative images captured. B. Groups of mice were treated with 250 μg anti-TNFα mAb immediately prior to hapten challenge and skin was prepared as above for analysis of ICAM-1 expression in the skin.
Endothelial cells in the skin challenge site produce CXCL1 during elicitation of CHS
To identify CXCL1-producing cells in the hapten-challenge site of sensitized mice at 6 h post-challenge, immunohistochemical staining was performed. Challenged skin was excised from both DNFB-sensitized and non-sensitized mice at 6 h following DNFB challenge and sections stained with a CXCL1-specific antiserum. In the hapten challenged skin from naïve mice, staining was restricted to low levels in keratinocytes (Figure 7A). In the challenged skin from sensitized mice, sections stained with normal goat serum did not demonstrate any positive staining (data not shown) whereas staining with the CXCL1-specific antiserum indicated many positively staining cells that included the keratinocytes, hair follicles, and cells surrounding dermal vascular structures (Figure 7B).
FIGURE 7.

Acquisition and presentation of challenge hapten by endothelial cells. C57BL/6 mice were sensitized with TRITC hapten on days 0 and +1. On day +5 post-sensitization, sensitized and non-sensitized naïve mice were challenged with TRITC on the shaved abdomen. A. Challenged skin and skin from non-sensitized/non-challenged mice was excised 6 h post-challenge, digested, and cell suspensions were stained with fluorescent antibody to CD31 and analyzed by flow cytometry. B. CD31+ TRITC+ cells from sensitized challenged mice were purified by flow sorting and 2 × 104 cell aliquots were cultured with 1 × 106 naive lymph node CD8 T cells or 1 × 106 hapten-immune CD4 or CD8 T cells from the lymph nodes of TRITC sensitized C57BL/6 mice. Culture supernatants were collected after 6 h and assessed for levels of CXCL1 by ELISA. The mean concentration ± SEM for 4 individual cell culture samples is shown. Results are representative of two individual experiments each. *p ≤ 0.05
To test whether endothelial cells produce CXCL1 in the hapten challenge site of sensitized mice 6 h after challenge, skin sections from sensitized and non-sensitized mice excised at this time-point were simultaneously stained for CXCL1 and for the endothelial cell marker CD31. Endothelial cells were clearly observed in skin from both naïve (Figure 7C) and sensitized skin (Figure 7D) but CXCL1 staining was only observed in the challenged skin from sensitized mice (Figure 7E vs. 7F). Merging the individual images of CXCL1 and CD31 staining clearly indicated endothelial cells staining positive for CXCL1 in skin from sensitized and challenged mice (Figure 7H).
Hapten-expressing endothelial cells isolated from the skin challenge site produce CXCL1 during culture with immune CD8 T cells
Endothelial cells in the skin challenge site were identified as expressing CXCL1. Since CXCL1 is induced by the IFN-γ and IL-17 produced by hapten primed CD8 T cells, the expression of this chemokine by endothelial cells in the challenge site suggested the endothelial cells as the hapten-presenting cells activating the hapten-primed CD8 T cells to initiate the CHS response. Therefore, we examined the relationship between endothelial cells and hapten acquisition and presentation. To identify hapten-expressing endothelial cells in the skin challenge site, challenged skin was removed 6 h after TRITC challenge of sensitized and non-sensitized mice. As a negative control, skin was also excised from non-sensitized/non-challenged mice. The excised skin samples were digested and isolated cell aliquots were stained with fluorescent-labeled anti-CD31 mAb and analyzed by flow cytometry analysis. TRITC-labeled endothelial cells were clearly observed from the skin of both sensitized and naïve mice challenged with the hapten (Figure 8A).
FIGURE 8.

Hapten-immune CD8 T cells induce endothelial cell production of CXCL1 in vitro. A. 2F2B endothelial cells were labeled with DNBS, washed 4 times, and 2 × 104 cells were cultured with 1 × 106 isolated CD4+ or CD8+ T cells from DNFB- or Ox-sensitized C3H mice. Culture supernatants were collected after 6 h and assessed for levels of CXCL1 by ELISA. The mean concentration ± SEM for 4 individual cell culture samples is shown. Results are representative of two individual experiments each. *p ≤ 0.05. B. DNBS-labeled or unlabeled 2F2B cells were cultured with purified CD8 T cells from the lymph nodes of DNFB-sensitized mice and 10 μg mAb to IFN-γ IL-17, TNF-α, ICAM, or LFA-1 was added to each culture as indicated. Culture supernatants were collected after 6 h and tested for CXCL1 by ELISA. The mean concentration ± SEM for 4 individual cell culture samples is shown. *p ≤ 0.05. C. Unlabeled 2F2B cells were cultured with 4 ng aliquots of rIFN-γ, rIL-17, or both recombinant cytokines. Culture supernatants were collected after 6 h and assessed for levels of CXCL1 by ELISA. The mean concentration ± SEM for 4 individual cell culture samples is shown. Results are representative of two individual experiments each. *p ≤ 0.05 vs. 2F2B only. **p ≤ 0.05 vs. cultures with individual cytokines added.
To test the ability of these hapten-expressing endothelial cells to stimulate hapten-primed CD8 T cells, fluorescent-labeled endothelial cells from TRITC-sensitized mice were isolated by flow cell sorting from cell suspensions prepared from digested hapten challenged skin. Aliquots of 2 × 104 sorted cells were cultured alone or with 1 × 106 TRITC-immune CD4 or TRITC-immune or non-immune CD8 T cells. After 6 h culture supernatants were harvested and tested for CXCL1 production by ELISA. CXCL1 was not detected in the culture supernatants from the isolated endothelial cells alone or from endothelial cells cultured with non-immune CD8 T cells (Figure 8B). Whereas TRITC-immune CD4 T cells induced low production of CXCL1 (2.26 ± 0.2 pg/ml), TRITC-immune CD8 T cells induced nearly four-fold higher production (8.53 ± 0.7 pg/ml).
Hapten-presenting endothelial cell production of CXCL1 in vitro
The expression of ICAM-1 on vascular structures and the requirement of ICAM-1 for the expression of the hapten-primed CD8 T cell IL-17 and IFN-γ suggested that the CD8 T cells may interact with the endothelial cells in the challenge site. To investigate these potential interactions in more detail an in vitro culture system was developed using the endothelial cell line 2F2B. Aliquots of DNFB-labeled or unlabeled 2F2B cells were cultured with syngeneic CD4 or CD8 T cells prepared from the lymph nodes of Ox or DNFB-sensitized on day +4 following sensitization. Culture supernatants were removed after 6 h and tested for CXCL1 production by ELISA (Figure 8A). The DNFB-sensitized CD8 T cells induced DNBS-labeled, but not unlabeled, 2F2B cells to produce CXCL1. Culture of the sensitized CD8 T cells alone did not result in CXCL1 production and stimulation of these CD8 T cells with ConA also did not stimulate this production whereas LPS stimulation of unlabeled 2F2B cells did stimulate CXCL1 production (data not shown), indicating that the CXCL1 was produced by the 2F2B cells and not by the hapten-primed CD8 T cells. Culture of DNBS-labeled 2F2B cells with either Ox-sensitized CD8 or DNFB-sensitized CD4 T cells did not stimulate the production of CXCL1 (Figure 6A).
FIGURE 6.

Tissue localization of CXCL1 to endothelial cells during elicitation of CHS. BALB/c mice were sensitized with 0.25% DNFB on days 0 and +1. On day +5 after sensitization, mice were challenged on a shaved square area of trunk skin with 0.2% DNFB. Challenged areas of skin were removed at 6 h post-challenge from DNFB-sensitized or non-sensitized mice and (AB) Paraffin-embedded sections were prepared and stained with CXCL1-specific antiserum. Slides were examined by light microscopy and representative images from (A) naïve and (B) sensitized skin are shown. Magnification, 40×. C–H Frozen sections were prepared and stained with both CXCL1-specific antiserum and anti-CD31 antibody followed by fluorochrome-labeled secondary antibodies (red to react with the CD31 and green to react with the CXCL1-specific antiserum) and were examined by confocal microscopy. Representative images of CD31 (C and D), CXCL1 (E and F), or both CD31 and CXCL1 (G and H) staining in challenged skin from naïve (C, E, and G) and sensitized (D, F, and H) mice are shown. Magnification, 40×.
To directly test the role of the CD8 T cell-derived cytokines IFN-γ and IL-17 in the production of CXCL1 by the hapten-labeled 2F2B cells, neutralizing antibodies to these cytokines were added to cultures of the DNFB-labeled endothelial cells and DNFB-sensitized CD8 T cells (Figure 8B). Culture supernatants were removed after 6 h and tested for CXCL1 production by ELISA. Addition of antibody to either IL-17 or IFN-γ significantly reduced the production of CXCL1 by the 2F2B cells. Addition of antibody to either ICAM-1 or LFA-1 also decreased CXCL1 production to levels observed by neutralization of either anti-IFN-γ or anti-IL-17 indicating the requirement for LFA-1/ICAM-1 interactions for the CD8 T cells to become activated by the hapten-labeled endothelial cells. However, addition of antibody to TNFα did not affect the production of CXCL1 in the immune CD8 T cell-hapten presenting endothelial cell cultures.
Since neutralization of either IL-17 or IFN-γ inhibited hapten-presenting endothelial cell production of CXCL1 during culture with immune CD8 T cells, the ability of these cytokines to directly induce unlabeled 2F2B cells to produce CXCL1 was tested (Figure 8C). Addition of either rIL-17 or rIFN-γ induced low amounts of CXCL1 production whereas addition of equivalent amounts of both rIFN-γ and rIL-17 increased the KC production almost four times higher than addition of either recombinant cytokine alone.
Discussion
An essential event during CD8 T cell mediated immune responses is the recruitment of the antigen-primed CD8 T cells to the tissue site where the response will be elicited. Generally, this recruitment is proposed to involve the synergistic functions of chemokines and adhesion molecules but how the local expression of these chemokines and adhesion molecules is coordinated is not well understood and is likely to be different for specific tissue sites as has been shown for the trafficking of CD4 T cells (20–22). Previous studies from this laboratory have documented that the infiltration of hapten-primed CD8 T cells into the skin to mediate CHS requires prior CXCL1-mediated recruitment and activation of neutrophils (16, 17). Based on work from many laboratories indicating the induction of CXCL1 and other neutrophil and macrophage chemoattractants during the initiation of an inflammatory insult (23–26), we proposed that application of hapten to the skin directly induced cells, such as keratinocytes, in the challenge site to produce the CXCL1 leading to this initial neutrophil infiltration. This was proved wrong when we recently observed that the production of CXCL1/KC and CXCL2/MIP-2 3–6 h after challenge required both IL-17 and IFN-γ produced by two separate populations of hapten-primed CD8 T cells (7). These results suggested that hapten application to the skin of sensitized animals first induces recruitment of the CD8 T cells to the site followed by their activation to produce the IL-17 and IFN-γ that stimulates the CXCL1 and CXCL2. The goal of the current studies was to identify the factors mediating the initial CD8 T cell recruitment to the challenge site to initiate the adaptive-innate-adaptive interactions that result in the tissue edema of the CHS response.
Many studies have identified TNFα as an acute phase cytokine produced early during most incidences of tissue inflammation that induces the participation of additional components to amplify the intensity of inflammation (25, 27, 28). Such TNFα induced down-stream events include the production of chemokines and other pro-inflammatory cytokines and the mobilization of selectins and integrin ligands to the luminal membrane of vascular endothelium (1–3, 29). Inhibition of TNFα through administration of antibody or recombinant TNFα receptors has been shown to attenuate inflammation and antigen-specific immune responses in animal models as well as in patients with psoriasis and inflammatory bowel disease (30–35). Early studies by Piguet and coworkers (36) documented the ability of anti-TNFα antibodies to inhibit the leukocyte infiltration and ear swelling of CHS responses when given to sensitized mice at the time of hapten challenge. CHS responses are also absent following sensitization and challenge of TNFα−/− and TNFα receptor p75−/− mice (37, 38). It is important to note that TNFα is also a critical factor in the activation and mobilization of interstitial dendritic cells including Langerhans cells from the periphery to draining lymphoid organs and TNFα antagonism during sensitization certainly attenuates hapten-specific T cell priming through this mechanism (38, 39). The current studies provide further insights into the role of TNFα specifically during the elicitation of CHS. TNFα expression was observed in the skin as early as 1 h after hapten application to both non-sensitized and sensitized animals but was two fold-higher in the sensitized animals suggesting synergy with a hapten-primed component. The expression of IL-17 (and not IFN-γ) was also evident at this early time post-challenge but as previously reported only in the sensitized mice. It is likely that the IL-17 either directly or in synergy with TNFα amplifies further TNFα expression in the challenge site during the initial elicitation of the response as has been observed in other immune responses (40, 41).
One of the major consequences of this early TNFα production in the site of hapten challenge is the upregulation of ICAM-1 expression. In contrast to TNFα, expression of ICAM-1 was equivalent in the skin of both naïve and sensitized mice challenged with hapten and neutralization of TNFα down-regulated this expression implicating a role for TNFα in ICAM-1 expression following hapten application to the skin of both naïve and sensitized animals. This TNFα induced ICAM-1 expression is required for the localization and activation of the IL-17 and IFN-γ producing CD8 T cell populations in the challenge site. First, induction of IL-17 and IFN-γ were absent in the challenge site of sensitized ICAM-1−/− mice. Since ICAM-1 is also required for optimal antigen priming of T cells and can account for the absence of these cytokines in response to challenge of sensitized ICAM-1-deficient mice (19), we transferred hapten-primed CD8 T cells from sensitized wild-type donors to naïve ICAM-1−/− recipients and observed the same absence of cytokine expression in response to hapten challenge. Second, CXCL1 mRNA expression and protein production required the expression of ICAM-1 in the challenge site and we have previously demonstrated that this production is induced by the IL-17 and IFN-γ produced by the hapten-primed CD8 T cells at the challenge site. Similarly, antagonism of ICAM-1, or of its ligand LFA-1, during challenge of sensitized wild-type animals substantially decreased CXCL1 production and neutrophil infiltration into the skin challenge site.
Immunohistochemistry of hapten challenged skin tissue revealed localized expression of ICAM-1 on endothelial cells. Anti-TNFα antibodies decreased this ICAM-1 expression implicating the effects of TNFα directly on the endothelial cells during the early stages of CHS elicitation. Studies from several laboratories have documented the expression of TNFα receptors on endothelial cells and the induction of inflammatory events in endothelial cells in response to TNFα, including ICAM-1 expression (42, 43). Furthermore, endothelial cells in the hapten challenge site of sensitized animals were the primary source of the CXCL1 observed 6 h after challenge suggesting that the ICAM-1 expressing endothelial cells were stimulated by the hapten-specific CD8 T cell derived IL-17 and IFN-γ to produce the neutrophil chemoattractant where it is accessible to circulating neutrophils.
Vascular endothelial cells are the first cells that antigen-primed T cells in the circulation encounter at an inflammatory site prior to infiltration into peripheral tissues. Although direct perfusion of hapten-protein complexes through the blood into the spleen and through the afferent lymph to the nodes following cutaneous hapten application has been previously documented (44), the presentation of hapten by cutaneous vascular endothelial cells has not. The current experiments show that hapten application to the skin results in endothelial cell acquisition of hapten and presentation to circulating hapten-primed CD8 T cells. To directly test the relationship between endothelial cells and hapten presentation in the skin challenge site, CD31+ endothelial cells expressing TRITC were sorted from cell suspensions prepared from TRITC challenged skin and tested for the ability to activate hapten-specific CD8 T cells. The hapten-presenting endothelial cells were stimulated by syngeneic hapten-primed, but not naïve, CD8+ T cells to produce CXCL1. The consequence of endothelial cell presentation of hapten is the activation of CD8 T cells to produce the IL-17 and IFN-γ that induces the endothelial cells to produce CXCL1. In support of this we have used in vitro models to show that hapten-primed CD8, but not hapten-primed CD4 or naïve CD8, T cells stimulate hapten-presenting endothelial cells to produce CXCL1 within 6 hours of culture initiation. Although the presence of CXCL1 protein was easily detectable in these cultures, IL-17 and IFN-γ protein were below the limit of detection but addition of neutralizing antibodies to the cytokines abrogated endothelial cell production of CXCL1. It is also interesting that in vitro antagonism of ICAM-1 but not TNFα similarly inhibited endothelial cell production of CXCL1. This suggests that in addition to ICAM-1 expression, other TNFα induced events on the endothelium are required to induce CXCL1 production in vivo, under shear force conditions.
Both in vitro and in vivo models have been used to demonstrate that endothelial cell presentation of antigen promotes reactive T cell diapedesis through the endothelial barrier (45–47). In contrast to these models, our previous and current studies indicate that hapten-primed CD8 T cells are activated by endothelial cells in the challenge site to produce IL-17 and IFN-γ which stimulate CXCL1 and CXCL2 but not CD8 T cell traversal through the endothelial barrier into the tissue parenchyma without prior neutrophil infiltration (7). The functions expressed by neutrophils that promote antigen-primed CD8 T cell infiltration into the site are not yet identified. Depletion of neutrophils results in slightly increased levels of CXCL1 and CXCL2 production 6 h after challenge of sensitized animals (results not shown). These increases are likely due to the absence of neutrophil-mediated digestion of the chemokines during transendothelial cell migration (48). Cytokine activation also induces neutrophils to produce T cell chemoattractants such as CXCL9/Mig and CXCL10/IP-10 during transendothelial migration and peripheral tissue infiltration and this is likely to promote the subsequent infiltration of antigen-primed CD8 T cells into the tissue (49–52). Neutrophil dependent leukocyte infiltration into the murine liver during cytomegalovirus infection is associated with neutrophil expression of specific matrix metalloproteinases, suggesting that digestion and possibly structural alteration of extracellular matrix may be required for antigen-primed T cell infiltration into peripheral tissues during certain immune responses (53).
The results of these experiments demonstrate an intricate system of early events initiated by hapten application to the skin of sensitized animals that culminate in the elicitation of the CHS response. The results indicate two immediate consequences of hapten application to the skin. The first is the rapid induction of TNFα that induces the expression of ICAM-1 on endothelial cells and facilitates the localization of hapten-primed CD8 T cells to the challenge site. The second is the acquisition of the hapten by the vascular endothelial cells in the challenge site and their presentation to the hapten-primed T cells resulting in their activation to produce IL-17 and IFN-γ. It is these cytokines that induce the CXCL1 and CXCL2 directing the neutrophils into the site to initiate the innate immune component of the response required for the subsequent infiltration of the CD8 T cells into the skin parenchyma.
Acknowledgments
We thank the staff of the Cleveland Clinic Biological Resources Unit for excellent care of the animals used in this study.
Footnotes
This work was supported by grant RO1 AI45888 from the National Institutes of Allergy and Infectious Diseases.
Disclosures
The authors have no financial conflict of interest to declare.
References
- 1.Alon R, Ley K. Cells on the run: shear-regulated integrin activation in leukocyte rolling and arrest on endothelial cells. Curr Opin Cell Biol. 2008;20:525–532. doi: 10.1016/j.ceb.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petri B, Phillipson M, Kubes P. The physiology of leukocyte recruitment: an in vivo perspective. J Immunol. 2008;180:6439–6446. doi: 10.4049/jimmunol.180.10.6439. [DOI] [PubMed] [Google Scholar]
- 3.Wong CHY, Heit B, Kubes P. Molecular regulators of leukocyte chemotaxis during inflammation. Cardiovasc Res. 2010;86:183–191. doi: 10.1093/cvr/cvq040. [DOI] [PubMed] [Google Scholar]
- 4.Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol. 1997;61:647–653. doi: 10.1002/jlb.61.6.647. [DOI] [PubMed] [Google Scholar]
- 5.El-Sawy T, Belperio JA, Strieter RM, Remick DG, Fairchild RL. Inhibition of polymorphonuclear leukocyte-mediated graft damage synergizes with short-term costimulatory blockade to prevent cardiac allograft rejection. Circulation. 2005;112:320–331. doi: 10.1161/CIRCULATIONAHA.104.516708. [DOI] [PubMed] [Google Scholar]
- 6.Miller AL, Strieter RM, Gruber AD, Ho SB, Lukacs NW. CXCR2 regulates respiratory syncytial virus-induced airway hyperreactivity and mucus overproduction. J Immunol. 2003;170:3348–3356. doi: 10.4049/jimmunol.170.6.3348. [DOI] [PubMed] [Google Scholar]
- 7.Kish DD, Li X, Fairchild RL. CD8 T cells producing IL-17 and IFN-g initiate the innate immune response required for responses to antigen skin challenge. J Immunol. 2009;182:5949–5959. doi: 10.4049/jimmunol.0802830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engeman TM, Gorbachev AV, Gladue RP, Heeger PS, Fairchild RL. Inhibition of functional T cell priming and contact hypersensitivity responses by treatment with anti-secondary lymphoid chemokine antibody during hapten sensitization. J Immunol. 2000;164:5207–5214. doi: 10.4049/jimmunol.164.10.5207. [DOI] [PubMed] [Google Scholar]
- 9.Kripke M, Munn C, Jeevan A, Tang J, Bucana S. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J Immunol. 1990;145:2833–2838. [PubMed] [Google Scholar]
- 10.Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. J Exp Med. 1987;166:1654–1667. doi: 10.1084/jem.166.6.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bour H, Peyron E, Gaucherand M, Garrigue J-L, Desvignes C, Kaiserlian D, Revillard J-P, Nicolas J-F. Major histocompatibility complex class I-restricted CD8+ T cells and class II-restricted CD4+ T cells, respectively, mediate and regulate contact sensitivity to dinitrofluorobenzene. Eur J Immunol. 1995;25:3006–3010. doi: 10.1002/eji.1830251103. [DOI] [PubMed] [Google Scholar]
- 12.Gocinski BL, Tigelaar RE. Roles of CD4+ and CD8+ T cells in murine contact sensitivity revealed by in vivo monoclonal antibody depletion. J Immunol. 1990;144:4121–4128. [PubMed] [Google Scholar]
- 13.Xu H, DiIulio NA, Fairchild RL. T cell populations primed by hapten sensitization in contact sensitivity are distinguished by polarized patterns of cytokine production: interferon-g-produing (Tc1) effector CD8+ T cells and Interleukin (IL) 4/IL-10-producing (Th2) negative regulatory CD4+ T cells. J Exp Med. 1996;183:1001–1012. doi: 10.1084/jem.183.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He D, Wu L, Kim HK, Li H, Elmets CA, Xu H. CD8+ IL-17-producing T cells are important in effector functions for the elicitation of contact hypersensitivity responses. J Immunol. 2006;177:6852–6858. doi: 10.4049/jimmunol.177.10.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antiogen-specific T cell sensitization is impaired in IL-17-deficient mice, cuasing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 16.DiIulio NA, Engeman TM, Armstrong D, Tannenbaum C, Hamilton TA, Fairchild RL. Groa-mediated recruitment of neutrophils is required for elicitation of contact hypersensitivity. Eur J Immunol. 1999;29:3485–3495. doi: 10.1002/(SICI)1521-4141(199911)29:11<3485::AID-IMMU3485>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 17.Engeman TM, Gorbachev AV, Kish DD, Fairchild RL. The intensity of neutrophil infiltration controls the number of antigen-primed CD8 T cells recruited into cutaneous antigen challenge sites. J Leukoc Biol. 2004;76:941–949. doi: 10.1189/jlb.0304193. [DOI] [PubMed] [Google Scholar]
- 18.Xu H, Banerjee A, DiIulio NA, Fairchild RL. Development of effector CD8+ T cells in contact hypersensitivity occurs independently of CD4+ T cells. J Immunol. 1997;158:4721–4728. [PubMed] [Google Scholar]
- 19.Zhang Q-W, Kish DD, Fairchild RL. Absence of allograft ICAM-1 attenuates alloantigen-specific T cell priming, but not primed T cell trafficking into the graft, to mediate acute rejection. J Immunol. 2003;170:5530–5537. doi: 10.4049/jimmunol.170.11.5530. [DOI] [PubMed] [Google Scholar]
- 20.Campbell JJ, Butcher EC. Chemokines in tissue-specific and microenvironment-specific lymphocyte homing. Curr Opin Immunol. 2000;12:336–341. doi: 10.1016/s0952-7915(00)00096-0. [DOI] [PubMed] [Google Scholar]
- 21.Campbell JJ, Haraldsen G, Pan J, Rottman J, Qin S, Ponath P, Andrew DP, Warnke R, Ruffing N, Kassam N, Wu L, Butcher EC. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature. 1999;400:776–780. doi: 10.1038/23495. [DOI] [PubMed] [Google Scholar]
- 22.Morales J, Homey B, Vicari AP, Hudak S, Oldham E, Hendrick J, Orozco R, Copeland NB, Jenkins NA, McEvoy LM, Zlotnik A. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proc Natl Acad Sci USA. 1999;96:14470–14475. doi: 10.1073/pnas.96.25.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miura M, Fu X, Zhang Q-W, Remick DG, Fairchild RL. Neutralizationof Groa and Macrophage inflammatory protein-2 attenuates renal ischemia/reperfusion injury. Am J Pathol. 2001;159:2137–2145. doi: 10.1016/s0002-9440(10)63065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morita K, Miura M, Paolone DR, Engeman TM, Kapoor A, Remick DG, Fairchild RL. Early chemokine cascades in murine cardiac grafts regulate T cell recruitment and progression of acute allograft rejection. J Immunol. 2001;167:2979–2984. doi: 10.4049/jimmunol.167.5.2979. [DOI] [PubMed] [Google Scholar]
- 25.Tracey KJ, Cerami A. Tumor necrosis factor, other cytokines and disease. Annu Rev Cell Biol. 1993;9:317–343. doi: 10.1146/annurev.cb.09.110193.001533. [DOI] [PubMed] [Google Scholar]
- 26.Luster AD. Chemokines-chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 27.Kupper TS. Immunologic targets in psoriasis. N Engl J Med. 2003;349:1987–1990. doi: 10.1056/NEJMp038164. [DOI] [PubMed] [Google Scholar]
- 28.Wajant H, Pfizenmaier K, Scheruich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 29.Rabb H, O’Meara YM, maderna P, Coleman P, Brady HR. Leukocytes, cell adhesion, molecules and ischemic acute renal failure. Kidney Int. 1997;51:1463–1468. doi: 10.1038/ki.1997.200. [DOI] [PubMed] [Google Scholar]
- 30.Chaudhari U, Romano P, Mulcahy LD, Dooley LT, Baker DG, Gottlieb AB. Efficacy and safety of infliximab monotherapy for plaque-type psoriasis: a randomized trial. Lancet. 2001;357:1842–1847. doi: 10.1016/s0140-6736(00)04954-0. [DOI] [PubMed] [Google Scholar]
- 31.Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA. Role of tumor necrosis factor-a in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest. 1990;85:1936–1943. doi: 10.1172/JCI114656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishii D, Schenk AD, Baba S, Fairchild RL. Role of TNFa in early chemokine production and leukcoyte infiltration into heart allografts. Am J Transplant. 2009;10:59–68. doi: 10.1111/j.1600-6143.2009.02921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Present DH, Rutgeerts P, Targan S, Hanauer SB, Mayer L, van Hogezand RA, Podolsky DK, Sands BE, Baakman T, DeWoody KL, Schaible TE, van Deventer SJ. Infliximab for the treatment of fistulas in patients with Crohn’s disease. N Engl J Med. 1999;340:1398–1405. doi: 10.1056/NEJM199905063401804. [DOI] [PubMed] [Google Scholar]
- 34.Schulz R, Aker S, Belosjorow S, Heusch G. TNFalpha in ischemia/reperfusion injury and heart failure. Basic Res Cardiol. 2004;99:8–11. doi: 10.1007/s00395-003-0431-x. [DOI] [PubMed] [Google Scholar]
- 35.Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, Nograles KE, Guttman-Yassky E, Cardinale I, Lowes MA, Kruger JG. Effective treatment of psoriasis with eternacept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;1245:1022–1030. doi: 10.1016/j.jaci.2009.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piguet PF, Grau GE, Hauser C, Vassali P. Tumor necrosis factor is a critical mediator in hapten-induced irritant and contact hypersensitivity reactions. J Exp Med. 1991;173:673–679. doi: 10.1084/jem.173.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibata M, Sueki H, Suzuki H, Watanabe H, Ohtaki H, Shioda S, Nakanishi-Ueda T, Yasuhara H, Sekikawa K, Iijima M. Impaired contact hypersensitivity reaction and reduced production of vascular endothelial growth factor in tumor necrosis factor-alpha gene-deficient mice. J Dermatol. 2005;32:523–533. doi: 10.1111/j.1346-8138.2005.tb00794.x. [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Fujisawa H, Zhuang L, Konod S, Shivji GM, Kim CS, Mak TW, Sauder DN. Depressed Langerhans cell migration and reduced contact sensitivity response in mice lacking TNF receptor p75. J Immunol. 1997;159:6148–6155. [PubMed] [Google Scholar]
- 39.Cumberbatch M, Griffiths CE, Tucker SC, Dearman RJ, Kimber I. Tumor necrosis-factor-a induces Langerhans cell migration in humans. Br J Dermatol. 1999;141:192–200. doi: 10.1046/j.1365-2133.1999.02964.x. [DOI] [PubMed] [Google Scholar]
- 40.Jovanovic DV, DiBattista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines IL-1b and TNF-a by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- 41.Katz Y, Nadiv O, Beer Y. Interleukin-17 enhances tumor necrosis factor a-induced synthesis of interleukins 1, 6 and 8 in skin and synovial fibroblasts: a possible role as a “fine-tuning cytokine” in inflammation processes. Arthr Rheum. 2001;44:2176–2184. doi: 10.1002/1529-0131(200109)44:9<2176::aid-art371>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 42.Al-Lamki RS, Brookes AP, Wang J, Reid MJ, Parameshwar J, Goddard MJ, Tellides G, Wan T, Min W, Pober JS, Bradley JR. TNF receptors differentially signal and are differentially expressed and regulated in the human heart. Am J Transplant. 2009;9:2679–2696. doi: 10.1111/j.1600-6143.2009.02831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slowik MR, De Luca LG, Fiers W, Pober JS. Tumor necrosis factor activates human endothelial cells through the p55 tumor necrosis factor receptor but the p75 receptor contributes to activation at low tumor necrosis factor concentration. Am J Pathol. 1993;143:1724–1730. [PMC free article] [PubMed] [Google Scholar]
- 44.Pior J, Vogl T, Sorg C, Macher E. Free hapten molecules are dispersed by way of the blood stream during contact sensitization to fluorescein isothiocyanate. J Invest Dermatol. 1999;113:888–893. doi: 10.1046/j.1523-1747.1999.00770.x. [DOI] [PubMed] [Google Scholar]
- 45.Marelli-Berg FM, Jams JG, Dangerfield J, Dyson J, Millrain M, Scott D, Simpson E, Nourshaugh S, Lechler RI. Cognate recognition of the endothelium induces HY-specific CD8+ T-lymphocyte transendothelial migration (diapedesis) in vivo. Blood. 2004;103:3111–3116. doi: 10.1182/blood-2003-08-2717. [DOI] [PubMed] [Google Scholar]
- 46.Marelli-Berg FM, Jarmin SJ. Antigen presentation by the endothelium: a green light for antigen-specific T cell trafficking? Immunol Letters. 2004;93:109–113. doi: 10.1016/j.imlet.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 47.Sauvinov AY, Wong FS, Stonebraker AC, Chervonsky AV. Presentation of antigen by endothelial cells and chemoattraction are required for the homing of insulin-specific CD8+ T cells. J Exp Med. 2003;197:643–656. [Google Scholar]
- 48.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 49.Cassatella MA, Gasperini S, Calzetti F, Bertagnin S, Luster AD, McDonald PP. Regulated production of the interferon-gamma-inducilble protein-10 (IP-10) chemokine by human neutrophils. Eur J Immunol. 1997;27:111–115. doi: 10.1002/eji.1830270117. [DOI] [PubMed] [Google Scholar]
- 50.Gasperini S, Marchi M, Calzetti F, Laudanna C, Vicentini L, Olsen H, Murphy M, Liao F, Farber J, Cassatella MA. Gene expression and production of the monokine induced by IFN-g (MIG), IFN-inducible T cell a chemoattractant (I-TAC), and IFN-g inducible protein-109 (IP-10) chemokines by human neutrophils. J Immunol. 1999;162:4928–4937. [PubMed] [Google Scholar]
- 51.Miura M, Morita K, Kobayashi H, Hamilton TA, Burdick MA, Strieter RM, Fairchild RL. Monokine induced by IFN-g is a dominant factor directing T cells into murine cardiac allografts during acute rejection. J Immunol. 2001;167:3494–3504. doi: 10.4049/jimmunol.167.6.3494. [DOI] [PubMed] [Google Scholar]
- 52.Molesworth-Kenyon SJ, Oakes JE, Lausch RN. A novel role for neutrophils as a source of T cell-recruiting chemokines IP-10 and Mig during the DTH response to HSV-1 antigen. J Leukoc Biol. 2005;77:552–559. doi: 10.1189/jlb.0904485. [DOI] [PubMed] [Google Scholar]
- 53.Sitia G, Isogawa M, Iannacone M, Campbell IL, Chisari FV, Duidotti LG. MMPs are required for recruitment of antigen-nonspecific mononuclear cells into the liver by CTLs. J Clin Invest. 2004;113:1158–1167. doi: 10.1172/JCI21087. [DOI] [PMC free article] [PubMed] [Google Scholar]