

Abstract
Hemophilia A results from an insufficiency of factor VIII (FVIII). Although replacement therapy with plasma-derived or recombinant FVIII is a life-saving therapy for hemophilia A patients, such therapy is a life-long treatment rather than a cure for the disease. In this review we discuss the possibilities, progress and challenges that remain in the development of a cell-based cure for hemophilia A. The success of cell therapy depends on the type and availability of donor cells, the age of the host and method of transplantation, and the levels of engraftment and production of FVIII by the graft. Early therapy, possibly even prenatal transplantation, may yield the highest levels of engraftment by avoiding immunological rejection of the graft. Potential cell sources of FVIII include a specialized subset of endothelial cells known as liver sinusoidal endothelial cells (LSECs) present in the adult and fetal liver, or patient-specific endothelial cells derived from induced pluripotent stem cells (iPSCs) that have undergone gene editing to produce FVIII. Achieving sufficient engraftment of transplanted LSECs is one of the obstacles to successful cell therapy for hemophilia A. We discuss recent results from transplants performed in animals that show production of functional and clinically relevant levels of FVIII obtained from donor LSECs. Hence, the possibility of treating hemophilia A can be envisioned through persistent production of FVIII from transplanted donor cells derived from a number of potential cell sources or through creation of donor endothelial cells from patient-specific iPSCs.
Introduction
Hemophilia has been a scourge throughout human history (reviewed in [1,2]). It was first described in ancient Egypt, mentioned in the Talmud in the 2nd century A.D., and also described in 10th century by Arabian physician Albucasis. Hemophilia garnered close attention in 19th century when the royal queen of England, Victoria, was found to be a carrier of the hemophilia gene that she passed to the Spanish, German and Russian royal families through her offspring. In Russia, the royal hemophilia gene had dramatic geopolitical consequences. Alexei, son of the last Russian Tsar was born with hemophilia and his parents were so obsessed with Alexei’s health that it contributed to their loss of control over the political situation in Russia, contributing to the Russian Revolution of 1917 [3].
Hemophilia affects 1 in 5000 males or about 400,000 individuals worldwide [4]. Hemophilia A is a genetic disease caused by various mutations in F8 gene located on the X-chromosome resulting in deficient production of factor VIII protein (FVIII). FVIII participates in the intrinsic pathway of blood coagulation and is a cofactor for factor IXa that, in the presence of Ca2+ and phospholipids, converts factor X to the activated form Xa. The FVIII gene, F8, encodes two alternatively spliced transcripts. Transcript variant 1 encodes a large 2351 amino acid single-chain glycoprotein, isoform a, that circulates in plasma associated with von Willebrand factor (VWF) in a noncovalent complex. Transcript variant 2 encodes a putative small protein, isoform b, that consists primarily of the phospholipid binding domain of factor VIIIc. This binding domain is essential for coagulant activity [5]. Deficiency in FVIII leads to spontaneous bleeding and in severe cases internal hemorrhage, especially in the knees, elbows and ankles that can cause disability and lead to death if left untreated. Three forms of Hemophilia A are distinguished based on the levels of FVIII in plasma: severe, less than 1% of normal levels; moderate 1 to 5%; and mild, 6 to 30% [4].
The first successful treatment of hemophilia A with whole blood transfusion was reported in 1840 [6]. Subsequently, treatment with plasma was introduced and then, in 1964, the cryoprecipitate fraction of plasma enriched in FVIII was first utilized [7]. The regular administration of purified FVIII began in 1970s [8,9]. However, despite lifesaving treatment with FVIII this form of therapy has unfortunate and even tragic consequences. In the 1980s, before the availability and wide-spread implementation of donor screening, nearly 90% of hemophilia A patients receiving donor plasma became infected with human immunodeficiency or hepatitis viruses. After 1985, heat inactivation has been used to kill virus in plasma. Safety and treatment was further enhanced by the invention of recombinant FVIII (rFVIII), generated through cloning of FVIII in 1984 [10–12]. rFVIII infusion has improved the life expectancy of patients with mild to moderate hemophilia A, reaching levels comparable to that of the general population. However, an ongoing concern is the development of inhibitory antibodies to plasma-derived FVIII or rFVIII. About 30% of children develop FVIII inhibitors, complicating the treatment of hemophilia A [13]. Additionally, the short half-life of rFVIII necessitates repeated infusions of the protein, in turn making it a very costly long-term treatment option for patients. The requirement for continuous medical monitoring and care makes hemophilia A among the most costly of medical conditions [14].
Gene therapy has been extensively explored as a possible treatment modality that would allow patients to produce their own FVIII instead of receiving FVIII prophylaxis. Various vectors such as adeno-associated virus, retrovirus and lentivirus have been studied for F8 transgene delivery. Different approaches for the delivery of the transgene have been envisioned, including direct injection into the blood to allow widespread dissemination of the vector or ex vivo transduction of transplantable target cells. The choice of target cells (Fig. 1), capable to produce sufficient and lasting levels of FVIII, has a major impact on the success of such therapy. Two main approaches have been suggested: 1) integrating gene-expressing vectors into stem cells or 2) into long-lived quiescent cells, such as hepatocytes or skeletal muscle cells [15]. The effectiveness of transplanting hematopoietic stem cells (HSCs) [16–18] and mesenchymal stromal cells (MSCs) [19] transduced with the human F8 gene in secreting clinically-significant levels of FVIII has been demonstrated in various small and large animal models of hemophilia A (Table 1).
Figure 1.

Potential cell sources for the treatment of hemophilia A. Tissues and cell types are indicated in black lettering. Procedures are written in blue lettering. Descriptions of the potential cell sources are provided in the text with red letters denoting sections of the figure referred to in the text.
Table 1.
Summary of studies in which extra-hepatic cells are transplanted into animals leading to LSEC engraftment and/or FVIII production.
| Donor cells | Animal model | Outcome | Reference |
|---|---|---|---|
| Bone Marrow Cells | |||
| Rat CD133+/CD45+/CD31+ bone marrow cells | Rat, 70% partial hepatectomy | LSEC engraftment, FVIII was not tested | [48] |
| Rat CD133+ bone marrow cells | Rat, monocrotaline | LSEC engraftment, FVIII was not tested | [47] |
| Rat CD133+/CD45+/CD31+ bone marrow cells | Rat, dimethylnitrosamine, partial hepatectomy | LSEC engraftment, FVIII was not tested | [49] |
| Mouse bone marrow cells and mouse Kupffer-Browicz cells | Hemophilia A mice | Hemophilia correction by mononuclear cells, Kupffer-Browicz cells and MSCs | [40] |
| Embryonic Spleen Cells | |||
| Pig embryonic spleen cells | NOD-SCID FVIII knockout mice | F8 mRNA, hemophilia correction | [82] |
| Hematopoietic Stem Cells (HSCs) | |||
| Murine HSCs with human FVIII | Hemophilia A mice | Hemophilia correction | [17] |
| Murine HSCs with human FVIII | Hemophilia A mice | Hemophilia correction | [16] |
| Canine HSCs with human FVIII | Hemophilia A dogs | Platelet expressed FVIII corrected hemophilia >2.5 years | [18] |
| Mesenchymal Stromal Cells (MSCs) | |||
| Murine MSCs with human FVIII transgene | Hemophilia A mice | Human and mouse FVIII in plasma, 32% blood clotting activity | [19] |
| Blood Outgrowth Endothelial Cells (BOECs) | |||
| Mouse hemophilia A BOECs transduced with canine FVIII | Hemophilia A mice | Canine FVIII in mouse plasma 11% blood clotting activity, bleeding time reduced | [46] |
| Human BOECs transfected with human FVIII | NOD/SCID mice | Human FVIII in mouse plasma | [45] |
| Induced Pluripotent Stem Cells (iPSCs) | |||
| Murine iPS | Hemophilia A mice IR | Hemophilia correction | [62] |
Despite these advances, gene therapy approaches face some likely challenges. Even if efficient transduction and lasting expression of FVIII in stem cells is achieved, transplantation of the modified cells may require toxic preconditioning of the patient to foster engraftment. Additionally, ectopic expression of FVIII could cause an immune response against FVIII [20]. Moreover, hepatocytes can present capsid-derived antigens to T cells, which results in inflammation and destruction of transduced hepatocytes. Even though muscle cells are easily modified by gene therapy, they are shown not to be efficient in posttranslational modifications of the transduced proteins [15]. Although there are ongoing clinical trials for hemophilia B, which is caused by the lack of another clotting protein Factor IX, hemophilia A gene therapy is more challenging because of the molecular properties of FVIII. One of the major impediments in the development of successful clinical trials of FVIII gene therapy has been the large size of the F8 gene that exceeds the normal packing capacity of adeno-associated viral vectors and leads to low expression of FVIII. Hence, additional bioengineering, such as deletion of B-domain, needs to be explored to achieve clinically-significant levels of FVIII expression [21,22].
In light of these findings, cell therapy for hemophilia A deserves further consideration to determine if an effective and safe cell-based treatment can be developed. Cell sources could include cells competent for FVIII production, such as liver sinusoidal endothelial cells (LSECs) (Fig. 1A), or stem cells derived from bone marrow (Fig. 1B), blood-outgrowth endothelial cells (BOECs) (Fig. 1C), endothelial progenitor cells derived from the differentiation of induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs) (Fig. 1D). In this review we discuss the therapeutic potential of these cells and the hurdles that must still be overcome to implement a cell based therapy for hemophilia A.
FVIII is produced by LSECs
Although FVIII mRNA is found in different human tissues such as liver, spleen, lymph nodes and kidney [23] transplantation studies in hemophilic animal models and patients have demonstrated liver as the primary source of FVIII [24]. Canine models have played an important role in the study and treatment of hemophilia A and in elucidating the central role played by the liver in this disease [25,26]. The Chapel Hill colony of hemophilia A dogs was founded at the University of North Carolina in 1947 from a male Irish Setter with severe hemophilia A resulting from a gene inversion analogous to one commonly found in humans [27]. Early transplant studies in these dogs showed that liver transplantation could correct the bleeder phenotype, but that transplant of spleen or kidney did not have consistent or lasting positive effects [28]. Beagles also suffer from hemophilia A and have been used to show the efficacy of auxiliary partial orthotopic liver transplantation to treat hemophilia A [29,30]. With the identification of the liver as the major source of FVIII, efforts focused on which cells in this organ were responsible for FVIII production.
Two liver cell types, hepatocytes and liver sinusoidal endothelial cells (LSECs), have each been proposed to be the main sources of FVIII. Stel et al. reported the presence of the FVIII antigen in LSECs, but not in the parenchyma of adult human liver [31]. In contrast, two other reports implicated hepatocytes as the source of FVIII [23,32]. Do et al. observed expression of FVIII by both murine LSECs and hepatocytes at similar levels [33]. Subsequently, Kumaran et al. showed that hemophilia A mice transplanted with unfractionated liver cells, including LSECs, survived a bleeding injury challenge, whereas purified hepatocyte transplantation did not prevent deadly bleeding [34]. More recently, we demonstrated that the only cells producing FVIII mRNA and protein in the fetal human liver are LSECs [35] (Fig. 2) and Shahani et al. came to the same conclusion after an extensive study of adult human liver cells [36]. Moreover, two recent studies elegantly demonstrated that endothelial cells are the source of FVIII in mice, shown using conditional knockout mutations of FVIII. Fahs et al. used promoters to target defects in endothelial cells and hepatocytes and demonstrated a severe hemophilic phenotype was associated with lack of FVIII expression be endothelial cells [37]. Likewise, Everett et al. observed that endothelial cells and not hepatocytes are the source of FVIII [38]. Their observations pointed to LSECs as the main source of FVIII in the liver and also showed that kidney endothelial cells to be a source of FVIII.
Figure 2.
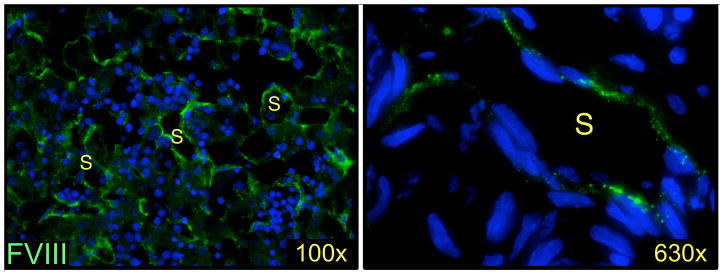
Human fetal LSECs express FVIII. Fetal liver, of 19 weeks’ gestation, stained for FVIII expression (green) using mouse monoclonal antibody 1.B.684 from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Nuclei stained with DAPI (blue), S – sinusoid.
LSECs are semi-permeable barrier cells lining the blood sinusoids in the liver that contribute to the clearance of pathogens, immune responses, and secretion of cytokines and growth factors. Historically, LSECs have been confused with the Kupffer-Browicz cells because of their co-localization, similarity in expressional profile, and some similar functional attributes [39]. Kupffer-Browicz cells are the resident macrophages of the liver sinusoids and express the pan-leukocyte antigen CD45 as well as the lipopolysaccharide receptor CD14. We have shown that LSECs also express CD14 but can be distinguished from Kupffer-Browicz cells by lack of CD45 expression [35]. Interestingly, murine Kupffer-Browicz cells may also produce FVIII according to one recent study [40], although an early study failed to detect mRNA in these macrophages [33]. Expression of FVIII by human Kupffer-Browicz cells has not been confirmed. FVIII expression and various other differences in gene expression differentiate LSECs from endothelial cells found outside the liver, bringing LSECs to the forefront in the design of a cellular therapy for hemophilia A.
Transplantation of murine LSECs demonstrates the potential of cell therapy for hemophilia A
Transplantation of LSECs has been demonstrated in several animal models as a means to treat hemophilia A. Table 2 summarizes a number of reports where liver-originated cells were studied for use in the therapy of hemophilia A. By example, Kumaran et al. transplanted mice, by intraperitoneal injection, with different liver-cell mixtures and found that the presence of endothelial cells could correct clotting dysfunction in mice with hemophilia A [34]. Subsequently, this group transplanted purified mature LSECs into the portal vein and demonstrated liver engraftment and therapeutic correction of hemophilia A with plasma FVIII activity more than 10% of normal plasma levels [41]. The ability to engraft LSECs was significantly improved when mice were treated with a fibronectin-like polymer that can engage extracellular matrix receptors. Additionally, host conditioning with monocrotaline, a pyrrolizidine alkaloid that causes disruption of the sinusoidal endothelial barrier, was also found to aid LSEC engraftment.
Table 2.
Summary of studies in which mice are transplanted with mouse or human LSECs.
| Donor cells | Animal model | Outcome | Reference |
|---|---|---|---|
| Mouse LSECs enriched liver cells | Hemophilia A knockout mice | FVIII in plasma, Phenotypic correction of hemophilia | [34] |
| Purified mouse CD146+CD45− LSECs | NOD/SCID Hemophilia A knockout mice | FVIII in plasma, Phenotypic correction of hemophilia | [41] |
| Rat LSEC enriched liver cells | DPPIV−/− rat external IR (25Gy) + 1/3 partial hepatectomy | CD31+ LSEC engraftment; FVIII was not tested | [77] |
| Mouse LSEC enriched liver cells or CD105+ selected LSECs | RAG2−/− γc−/− mice treated with monocrotaline | Orthotopic LSEC engraftment, inhibition of T cell responses to cognate stimuli; FVIII was not tested | [83] |
| Human fetal liver cells | uPA-NOG mice | Human FVIII in mouse plasma | [35] |
| Human adult hepatocytes enriched fraction containing LSECs | uPA-NOG mice | CD14+CD105+ LSEC engraftment; FVIII was not tested | [35] |
| Human adult cultured CD31+ liver cells | RAG2−/− γc−/− mice treated with monocrotaline | Human CD31+ LSECs engrafted, FVIII was not tested | [42] |
| Cultured human fetal liver CD31+ endothelial cells | RAG2−/− γc−/− mice treated with monocrotaline | Human CD31+ LSECs engrafted, FVIII was not tested | [42] |
Transplantation of adult human LSECs into immunodeficient mice
Successful transplantation of human LSECs isolated from adult liver has recently been demonstrated (Table 2) [35,42]. Filali et al. isolated liver endothelial cells by selection of CD31+ cells and cultured these cells under conditions supportive of endothelial cell growth for up to 1 week [42]. Transplantation into monocrotaline-treated immunodeficient mice yielded engraftment of LSECs identified, in part, by their expression of human lymphatic vessel endothelial receptor-1 (LYVE-1). Although culture conditions caused loss of fenestration of LSECs, normal phenotype was re-acquired after transplantation, pointing to the role of microenvironmental signals in supporting some of the unique features of the LSEC phenotype. Another important observation by Filali and colleagues is that macrovascular endothelial cells obtained from human umbilical vein endothelial cells failed to repopulate liver, and neither did microvascular endothelial cells obtained from adult adipose tissue. These findings indicate irreversible differentiation of these different endothelial cell compartments that cannot be overcome by the signals provided by the liver environment. Accordingly, cellular therapy for hemophilia A may need to rely on LSECs or immature endothelial progenitors as sources of transplantable cells (Fig. 1).
Cell therapy with adult LSECs is limited by access to living donor tissue or cadaverous livers (Fig. 1A). Cryopreservation could be used to bank adult liver cells as LSECs have been shown to engraft uPA-NOG mice after thawing (Fig. 3A) [35]. Ex vivo expansion of LSECs could be used to increase the number of adult LSECs available for transplantation, but the effects of in vitro culture on the viability, proliferation and function of these cells needs to be further evaluated. Due to the restricted proliferative capacity of adult cells, an immortalized cell line was recently developed from adult human LSECs using lentiviral transduction with hTERT [43]. These cells expressed some LSEC specific markers and demonstrated endocytic properties. However, the safety and potential of this cell line to engraft and produce FVIII remains to be tested.
Figure 3.

Transplanted human LSECs engraft mouse liver. A. Frozen adult liver cells engrafted into mouse express panhuman marker β-2 microglobulin (B2M) and the LSEC marker CD14 [35]. B. Fetal liver cells engrafted into mouse express B2M and LSEC-specific markers CD32 and CD32b. Notice clonal character of cell expansion. Human markers are shown in green and mouse cells were stained with H-2Kd (red). Nuclei were stained with DAPI (blue). S – sinusoid.
Other sources of endothelial cells for the treatment of hemophilia A
Alternate sources of cells capable of engrafting the liver and differentiating into LSECs have also been explored by a number of research teams (Table 1). Blood-derived circulating endothelial cells represent a clinically-promising cell type present at a low frequency in adult peripheral blood but which can be expanded in culture for therapy of hemophilia A (Fig. 1C). Most of the freshly isolated blood circulating endothelial cells originate from vascular walls, however 5% are estimated to come from the bone marrow [44]. These bone marrow precursors have a much higher proliferative potential and are responsible for the majority of cell outgrowth in culture and are, thus, referred to as BOECs. After intravenous injection, BOECs primarily engrafted the spleen and bone marrow. Furthermore, BOECs transfected with FVIII can secrete human FVIII into the plasma as shown by transplantation into NOD/SCID mice [45]. FVIII-transduced BOECs have also been grown into sheets and then transplanted subcutaneously resulting in partial correction of the disease phenotype in hemophilia A mice [46].
Other stem cell and progenitor populations have been evaluated for their potential for in vivo FVIII production either by differentiation into LSECs after transplantation or by acting as carrier cells for ectopic FVIII expression as discussed above (Fig. 1B and Table 1). For instance, rat bone marrow-derived CD133+CD45+CD31+ cells have been shown to differentiate into LSECs after transplantation [47–49], but the potential of these cells as a source of FVIII still remains to be evaluated. Though these reports reveal other potential FVIII producing cell sources besides LSECs, further evaluation of the phenotype and lineage relationship of these FVIII-secreting cells found in tissues other than the liver is desired. Given the scarcity of donor liver tissue, cell sources from blood or bone marrow would be a welcome advance, particularly when combined with knowledge of the growth factors required to expand and differentiate these cells towards the LSEC lineage.
Fetal tissues as a source of transplantable LSECs
Fetal LSECs obtained from elective abortions offer another potential source of transplantable cells for the treatment of hemophilia A (Fig. 1A). After determining that LSECs are the source of FVIII in fetal human liver, we sought to determine if fetal LSECs could be transplanted in immunodeficient mice to demonstrate secretion of human FVIII in vivo. As an immunodeficient-hemophilia A mouse model was not available for study, we evaluated transplants in two strains of immunodeficient mice. Without any conditioning prior to transplant, low levels of human LSEC engraftment were observed in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice [50]. However, notable levels of engraftment were achieved in NOD.Cg-Prkdcscid Il2rgtm1Sug Tg(Alb-Plau)11-4/ ShiJic (uPA-NOG) mice [35]. In addition to the immune system defects shared with NSG mice, uPA-NOG mice also overexpress urokinase-type plasminogen activator (uPA) in the liver [51]. These mice, and similar strains uPA transgenic mice, have been widely used to study hepatocytes, as the uPA transgene is toxic to host hepatocytes and, hence, these transgenic mice are more readily engrafted by donor hepatocytes [52,53]. Until recently, it was not appreciated that the uPA transgene could also enabled robust LSEC engraftment leading to circulating human FVIII levels approaching those of normal human plasma (Fig. 3B) [35]. The findings in the uPA-NOG mouse model are very encouraging in in light of the fact that even increasing circulating factor VIII levels 1% above threshold is sufficient to obtain a therapeutic effect in severely affected patients [15].
Clinical transplantation of fetal tissues has been performed in many countries although legal, ethical and safety considerations can pose obstacles and limitations to the use of fetal tissues. In utero transplantation of fetal liver has been performed safely and with clinical success in a number of cases for severe combined immune deficiency, but microbial contamination of the donor tissue has also led to sepsis and death of at least one fetal patient [54]. However, we have shown that the threat of microbial contamination can be significantly reduced and readily managed making fetal liver transplantation a viable treatment consideration [55,56]. Human fetal liver cell infusions have also led to improvement in patients with different liver pathologies. Peritoneal transplantation of human fetal hepatocytes successfully treated acute fatty liver of pregnancy [57]. Treatment of hyperbilirubinemia in biliary atresia and liver cirrhosis of different etiologies was performed by transplantation of fetal hepatic progenitors via the hepatic artery [58,59]. Intrasplenic infusion through the splenic artery was also applied in a patient with end-stage chronic liver failure [60]. Although these cases speak to the feasibility and safety of fetal liver transplantation, no specific cases of fetal LSEC transplantation have been reported to treat hemophilia.
There are some possible advantages in selecting fetal LSECs as a source of donor tissue. The short proliferative history of fetal cells suggests that these cells have an extensive proliferative reserve capacity. Pediatric or in utero transplantation of LSECs would allow the donor cells to grow within the host to provide life-long sustained FVIII production. In the case of in utero transplantation, the immature and tolerogenic host immune system may offer further advantages for allogeneic transplants. Filali et al. observed that the percentage of mouse liver repopulation by human fetal LSECs was at least 3-fold higher compared to adult LSECs [42]. We also observed clonal expansion of engrafted fetal LSEC in uPA-NOG mouse livers that appeared more robust than with adult cell transplants (Fig. 3). These observations suggest fetal LSECs may better engraft patients than adult LSECs and may also be more amenable to ex vivo expansion prior to transplant.
Pluripotent stem cell therapies for hemophilia A
Even though LSECs and stem cells derived from various adult and fetal sources may play a tremendous role in the therapy of hemophilia A, there are potential limitations that may hinder their clinical utility such as: 1) lack of availability of a suitable donor tissue 2) low yield of LSECs from liver homogenates 3) limited expansion potential of stem cells or LSECs due to loss of functional or proliferative capacity in vitro. The extent of these potential limitations is not fully known and requires further study. Nonetheless, to provide sufficient cells to treat hemophilia A, investigation into alternative LSEC sources is prudent as is gaining a greater understanding of the environmental requirements required for the extensive expansion, differentiation and maintenance of the LSEC phenotype in culture.
The discovery of embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) has revolutionized the study of regenerative medicine due to their extensive potential for self-renewal, growth and pluripotency allowing creation of cells belonging to all three primary germ layers and, thus, including LSECs (Fig. 1D). Wang et al. showed that mouse chimeras created by injecting ESCs into FVIII-deficient mouse blastocysts were able to correct the bleeding disorder as FVIII was stably expressed in chimeras [61]. Although this study offers a basic proof of principle that ESCs can differentiate into FVIII-producing cells, much work is required to optimize the culture conditions required to generate transplantable and functional FVIII-producing cells such as LSECs or their precursors from pluripotent stem cells. There are also safety concerns that must be addressed, such as the risk of teratoma formation or genetic mutation of the extensively cultured and selected cells, before pluripotent stem cells can be considered for human transplantation.
Although ESCs are viewed as the gold-standard of pluripotent stem cells, many current studies focus on iPSCs as they offer several advantages over ESCs. Firstly, derivation of iPSCs does not require human embryos and hence circumvents the ethical debate and supply constraints associated with their use in research or the clinic. Generation of disease-specific iPSCs can facilitate research by being used for disease modeling, drug discovery and drug screening. Their potential greatest impact, however, is for clinical use as iPSCs offer the potential for the generation of patient-specific cells, thus reducing the risk of immune rejection by offering an autologous source of cells for transplantation. The genetic abnormalities causing hemophilia A can be corrected using gene-editing technology with subsequent selection, expansion and differentiation of the iPSCs into endothelial cell progenitors or fully-differentiated LSECs. Partial demonstration of such a therapy has recently been reported using a mouse model: transplantable endothelial progenitor cells were derived from iPSC and shown to produce FVIII in hemophilia A mice [62].
There has also been an effort to demonstrate the utility of a combined approach using gene therapy to direct the expression of a human FVIII transgene in iPSC-derived cells [63]. A human artificial chromosome vector containing multiple FVIII expression cassettes under control of megakaryocyte-specific promoter was stably introduced into mouse iPSCs and was shown to produce human FVIII when the stem cells were differentiated into megakaryocytes and platelets. The human artificial chromosome vector is episomal and, thus, there is a low risk of altering the host chromosomes. Nonetheless, the long-term stability and expression from such a system needs further evaluation.
A recent advance came with the creation of a human hemophilia A iPSC line employing transcription activator-like effector nuclease (TALEN) technology to craft one of the most frequently occurring chromosomal inversions affecting the F8 gene that is observed in hemophilic patients [64]. Subsequent TALEN-mediated reversion of the mutated chromosomal segment corrected the disease phenotype in the iPSC line. The study by Park et al. demonstrated that gene-editing technology offers the possibility of repairing the genetic defect in at least one form of hemophilia A as well as providing a method of disease modeling.
Other methods of gene editing have been developed that can be used for correction of genetic abnormalities in iPSCs such as zinc finger nucleases (ZFN) and the RNA-guided clusters of regularly interspaced palindromic repeats (CRISPR)-Cas9 system [65,66]. An approach using TALEN or CRISPR-Cas9, to introduce double-stranded breaks at a specified location in the genome, together with piggyBac transposon technology was recently shown to efficiency repair or induce specific mutations in iPSCs [67,68]. This technology allows for introduction of a selectable marker in the targeting construct followed by seamless excision of the marker gene after its use in the isolation of iPSC clones that have undergone homologous recombination. Such an approach could be used to correct the F8 gene in patient derived iPSCs (Fig. 4). Combining these technologies with recent advances in the derivation of human iPSCs [69–71] and previously described approaches to derive differentiated endothelial progenitor cells from human iPSCs [72,73] offers the possibility of an unlimited source of transplantable endothelial progenitor cells for the treatment of hemophilia A.
Figure 4.

Hemophilia A therapy using endothelial cells created from patient-specific iPSCs that have undergone gene-editing to correct the underlying mutation. The different stages of cell modification are indicated in black lettering and procedures are indicated in blue lettering.
Cell delivery
Delivery method is a critical aspect in the development of a cell therapy of hemophilia A. If engraftment cannot be efficiently achieved, then the therapy will not provide sufficient levels of circulating FVIII. Considering the liver as the likely and natural destination for FVIII-secreting cells, the route used for administration of the cells should optimize hepatic engraftment. Accordingly, the most likely vessels to be used for transplantation are the portal vein, the hepatic artery or the splenic artery (Fig. 1E). Cell-therapy using fetal liver hepatocytes has shown the possibility of such routes of administration [57–60]. Bone marrow cells have also been injected into the hepatic artery or portal vein to treat pediatric malformations and chronic liver failure [74,75], further demonstrating the safety and feasibility of such procedures if applied to the treatment of hemophilia A.
Beyond the localized delivery of cells to the liver, the sinusoids of the liver may need to be treated in a manner to enhance donor cell engraftment. The observation that some LSEC engraftment can be achieved in mice in the absence of cytoablative pretreatment is encouraging [41,50]. Perhaps multiple rounds of cell infusion could result in sufficient engraftment to affect circulating FVIII levels. Nonetheless, animal models have tended to rely on cytotoxic pretreatments to disrupt the sinusoidal endothelial layer to ensure sufficient engraftment using substances such as monocrotaline or cyclophosphamide [41,76]. Additionally, irradiation and partial hepatectomy have been used to engraft both LSECs and parenchymal cells in rats [77]. Clinical transplantation of LSECs would benefit from a relatively non-toxic method of promoting LSEC engraftment.
Understanding the mechanism for the enhanced engraftment of LSECs in uPA-NOG mice may be of great value in devising a safe and effective means of cell engraftment in hemophilia A patients. The mechanism behind the enhanced engraftment of LSECs in uPA-NOG mice is not yet known, but an intriguing observation was that uPA-NOG mice have elevated levels of vascular endothelial growth factor (VEGF) in the liver, which could affect vascular permeability and/or growth of the transplanted LSECs. In addition, uPA activates matrix metalloproteases, which increase the release of VEGF [53]. Increased expression of VEGF after hepatic injury was also demonstrated to be critical for LSEC engraftment in a rat model [49]. Therefore, thinking of clinical possibilities, intrahepatic injections of VEGF could potentially facilitate LSEC engraftment.
With prenatal diagnosis of hemophilia A being feasible, the potential of performing in utero transplantation can also be considered (Fig. 1E). The advantages of in utero transplantation include a reduced immune response to foreign cells that also reduces the risk of developing inhibitory antibodies to FVIII; the small size of the patient reduces the need for a large graft; and the tremendous growth and remodeling that occurs as the liver transitions from a predominantly hematopoietic tissue to an organ composed primarily of hepatocytes may offer greater engraftment potential. Early treatment can also prevent mortality and morbidity before and after birth. Clinical progress has been reported for a number of diseases treated with in utero cell therapy, such as severe immunodeficiency and osteogenesis imperfecta [54,78,79]. Human endothelial colony-forming cells, derived from umbilical cord blood, have been transplanted into fetal sheep resulting in engraftment in the perivascular region of the liver where donor cells comprised 0.11–0.16% of the liver [80]. However, FVIII production by these cells was not evaluated and it is known that endothelial cells isolated from human umbilical vein are not capable of producing FVIII [81]. Nonetheless, the transplant findings demonstrate that some endothelial cell engraftment can be achieved through in utero transplantation, which encourages further study of this method of cell delivery. Consideration of in utero transplantation as a therapy for hemophilia A highlights the possibilities, challenges and overall importance of the developmental stage of the patient as well as the routes and method of cell delivery in determining the success of cell therapy for hemophilia A.
Future perspectives
Transplantation and engraftment of LSECs capable of producing FVIII provides the most natural pathway to a cure for hemophilia A. Such a cell based therapy for hemophilia A offers the potential of a life-long cure if a number of hurdles can be passed. The choice of allogeneic or iPSC-derived syngeneic donor cells each offer advantages and challenges that need further exploration to ascertain their true potential. A deeper understanding of the embryonic development of LSECs and the signals provided by the liver environment that support their growth and differentiation is needed to develop methods for their in vitro development from stem cells or their ex vivo expansion from donor tissue. Attention must also be placed on the methods of transplantation to achieve the greatest possible levels of engraftment. Ultimately, the success of a cell therapy for hemophilia A will depend on the optimization of each of these parameters.
Acknowledgments
The authors wish to thank the administrative staff at Blood Systems Research Institute for their superb assistance in supporting our research endeavors. We also thank the staff and faculty at San Francisco General Hospital Women’s Options Center for assistance in the collection of human fetal tissues.
This work was supported by a gift from the Riva Foundation and the National Institutes of Health: P01DK088760 and P30DK026743 from the National Institute Of Diabetes And Digestive And Kidney Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute Of Diabetes And Digestive And Kidney Diseases or the National Institutes of Health.
Footnotes
Addendum
Drs. M. Fomin, P. P. Togarrati and M. Muench each contributed to the design of the manuscript, literature review, and drafting of the manuscript and figures. All authors read and approved the submission of final manuscript.
References
- 1.Rosendaal FR, Smit C, Briët E. Hemophilia treatment in historical perspective: A review of medical and social developments. Ann Hematol. 1991;62:5–15. doi: 10.1007/BF01714977. [DOI] [PubMed] [Google Scholar]
- 2.Franchini M, Mannucci PM. The history of hemophilia. Semin Thromb Hemost. 2014;40:571–6. doi: 10.1055/s-0034-1381232. [DOI] [PubMed] [Google Scholar]
- 3.Ingram I. The history of haemophilia. J Clin Pathol. 1976;29:469–79. doi: 10.1136/jcp.29.6.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mannucci PM, Franchini M. Haematology clinic: Haemophilia A. Hematology. 2014;19:181–2. doi: 10.1179/1024533214Z.000000000262. [DOI] [PubMed] [Google Scholar]
- 5.Schaub RG. Recent advances in the development of coagulation factors and procoagulants for the treatment of hemophilia. Biochem Pharmacol. 2011;82:91–8. doi: 10.1016/j.bcp.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 6.Lane S. Haemorrhagic diathesis: Successful transfusion of blood. Lancet. 1840;35:185–8. [Google Scholar]
- 7.Pool JG, Gershgold EJ, Pappenhagen AR. High-potency antihaemophilic factor concentrate prepared from cryoglobulin precipitate. Nature. 1964;203:312. doi: 10.1038/203312a0. [DOI] [PubMed] [Google Scholar]
- 8.Webster WP, Roberts HR, Thelin GM, Wagner RH, Brinkhous KM. Clinical use of a new glycine-precipitated antihemophilic fraction. Am J Med Sci. 1965;250:643–51. doi: 10.1097/00000441-196512000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Brinkhous KM, Shanbrom E, Roberts HR, Webster WP, Fekete L, Wagner RH. A new high-potency glycine-precipitated antihemophilic factor (AHF) concentrate. Treatment of classical hemophilia and hemophilia with inhibitors. JAMA. 1968;205:613–7. [PubMed] [Google Scholar]
- 10.Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM. Characterization of the human factor VIII gene. Nature. 1984;312:326–30. doi: 10.1038/312326a0. [DOI] [PubMed] [Google Scholar]
- 11.Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orr EC, Amphlett GW, Foster WB, Coe ML, Knutson GJ, Fass DN, Hewick RM. Molecular cloning of a cdna encoding human antihaemophilic factor. Nature. 1984;312:342–7. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- 12.Wood WI, Capon DJ, Simonsen CC, Eaton DL, Gitschier J, Keyt B, Seeburg PH, Smith DH, Hollingshead P, Wion KL, Delwart E, Tuddenham EG, Vehar GA, Lawn RM. Expression of active human factor VIII from recombinant DNA clones. Nature. 1984;312:330–7. doi: 10.1038/312330a0. [DOI] [PubMed] [Google Scholar]
- 13.Gouw SC, van der Bom JG, Ljung R, Escuriola C, Cid AR, Claeyssens-Donadel S, van Geet C, Kenet G, Mäkipernaa A, Molinari AC, Muntean W, Kobelt R, Rivard G, Santagostino E, Thomas A, van den Berg HM PedNet and RODIN Study Group. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368:231–9. doi: 10.1056/NEJMoa1208024. [DOI] [PubMed] [Google Scholar]
- 14.Fitch K, Pyenson B. Milliman Research Report. New York, NY: Milliman, Inc; 2011. Benefit designs for high cost medical conditions. [Google Scholar]
- 15.Chuah MK, Evens H, VandenDriessche T. Gene therapy for hemophilia. J Thromb Haemost. 2013;11 (Suppl 1):99–110. doi: 10.1111/jth.12215. [DOI] [PubMed] [Google Scholar]
- 16.Moayeri M, Ramezani A, Morgan RA, Hawley TS, Hawley RG. Sustained phenotypic correction of hemophilia a mice following oncoretroviral-mediated expression of a bioengineered human factor VIII gene in long-term hematopoietic repopulating cells. Mol Ther. 2004;10:892–902. doi: 10.1016/j.ymthe.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Moayeri M, Hawley TS, Hawley RG. Correction of murine hemophilia A by hematopoietic stem cell gene therapy. Mol Ther. 2005;12:1034–42. doi: 10.1016/j.ymthe.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du LM, Nurden P, Nurden AT, Nichols TC, Bellinger DA, Jensen ES, Haberichter SL, Merricks E, Raymer RA, Fang J, Koukouritaki SB, Jacobi PM, Hawkins TB, Cornetta K, Shi Q, Wilcox DA. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. doi: 10.1038/ncomms3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Gong X, Gong Z, Ren X, Ren Z, Huang S, Zeng Y. The mesenchymal stem cells derived from transgenic mice carrying human coagulation factor VIII can correct phenotype in hemophilia A mice. J Genet Genomics. 2013;40:617–28. doi: 10.1016/j.jgg.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Wells KE, Maule J, Kingston R, Foster K, McMahon J, Damien E, Poole A, Wells DJ. Immune responses, not promoter inactivation, are responsible for decreased long-term expression following plasmid gene transfer into skeletal muscle. FEBS Lett. 1997;407:164–8. doi: 10.1016/s0014-5793(97)00329-3. [DOI] [PubMed] [Google Scholar]
- 21.High KH, Nathwani A, Spencer T, Lillicrap D. Current status of haemophilia gene therapy. Haemophilia. 2014;20 (Suppl 4):43–9. doi: 10.1111/hae.12411. [DOI] [PubMed] [Google Scholar]
- 22.Powell JS, Ragni MV, White GC, Lusher JM, Hillman-Wiseman C, Moon TE, Cole V, Ramanathan-Girish S, Roehl H, Sajjadi N, Jolly DJ, Hurst D. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102:2038–45. doi: 10.1182/blood-2003-01-0167. [DOI] [PubMed] [Google Scholar]
- 23.Wion KL, Kelly D, Summerfield JA, Tuddenham EG, Lawn RM. Distribution of factor VIII mrna and antigen in human liver and other tissues. Nature. 1985;317:726–9. doi: 10.1038/317726a0. [DOI] [PubMed] [Google Scholar]
- 24.Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92:3983–96. [PubMed] [Google Scholar]
- 25.Sabatino DE, Nichols TC, Merricks E, Bellinger DA, Herzog RW, Monahan PE. Animal models of hemophilia. Prog Mol Biol Transl Sci. 2012;105:151–209. doi: 10.1016/B978-0-12-394596-9.00006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christopherson PW, Bacek LM, King KB, Boudreaux MK. Two novel missense mutations associated with hemophilia A in a family of boxers, and a german shepherd dog. Vet Clin Pathol. 2014;43:312–6. doi: 10.1111/vcp.12172. [DOI] [PubMed] [Google Scholar]
- 27.Lozier JN, Dutra A, Pak E, Zhou N, Zheng Z, Nichols TC, Bellinger DA, Read M, Morgan RA. The chapel hill hemophilia A dog colony exhibits a factor VIII gene inversion. Proc Natl Acad Sci U S A. 2002;99:12991–6. doi: 10.1073/pnas.192219599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webster WP, Zukoski CF, Hutchin P, Reddick RL, Mandel SR, Penick GD. Plasma factor VIII synthesis and control as revealed by canine organ transplantation. Am J Physiol. 1971;220:1147–54. doi: 10.1152/ajplegacy.1971.220.5.1147. [DOI] [PubMed] [Google Scholar]
- 29.Ko S, Tanaka I, Kanokogi H, Kanehiro H, Okayama J, Ori J, Shima M, Yoshioka A, Giles A, Nakajima Y. Efficacy of auxiliary partial orthotopic liver transplantation for cure of hemophilia in a canine hemophilia A model. Transplant Proc. 2005;37:1131–3. doi: 10.1016/j.transproceed.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Ko S, Tanaka I, Kanehiro H, Kanokogi H, Ori J, Shima M, Yoshioka A, Giles A, Nakajima Y. Preclinical experiment of auxiliary partial orthotopic liver transplantation as a curative treatment for hemophilia. Liver Transpl. 2005;11:579–84. doi: 10.1002/lt.20390. [DOI] [PubMed] [Google Scholar]
- 31.Stel HV, van der Kwast TH, Veerman EC. Detection of factor VIII/coagulant antigen in human liver tissue. Nature. 1983;303:530–2. doi: 10.1038/303530a0. [DOI] [PubMed] [Google Scholar]
- 32.Zelechowska MG, van Mourik JA, Brodniewicz-Proba T. Ultrastructural localization of factor VIII procoagulant antigen in human liver hepatocytes. Nature. 1985;317:729–30. doi: 10.1038/317729a0. [DOI] [PubMed] [Google Scholar]
- 33.Do H, Healey JF, Waller EK, Lollar P. Expression of factor VIII by murine liver sinusoidal endothelial cells. J Biol Chem. 1999;274:19587–92. doi: 10.1074/jbc.274.28.19587. [DOI] [PubMed] [Google Scholar]
- 34.Kumaran V, Benten D, Follenzi A, Joseph B, Sarkar R, Gupta S. Transplantation of endothelial cells corrects the phenotype in hemophilia A mice. J Thromb Haemost. 2005;3:2022–31. doi: 10.1111/j.1538-7836.2005.01508.x. [DOI] [PubMed] [Google Scholar]
- 35.Fomin ME, Zhou Y, Beyer AI, Publicover J, Baron JL, Muench MO. Production of factor VIII by human liver sinusoidal endothelial cells transplanted in immunodeficient upa mice. PLoS ONE. 2013;8:e77255. doi: 10.1371/journal.pone.0077255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shahani T, Covens K, Lavend’homme R, Jazouli N, Sokal E, Peerlinck K, Jacquemin M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J Thromb Haemost. 2014;12:36–42. doi: 10.1111/jth.12412. [DOI] [PubMed] [Google Scholar]
- 37.Fahs SA, Hille MT, Shi Q, Weiler H, Montgomery RR. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. 2014;123:3706–13. doi: 10.1182/blood-2014-02-555151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Everett LA, Cleuren AC, Khoriaty RN, Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123:3697–705. doi: 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sródka A, Gryglewski RW, Szczepariski W. Browicz or kupffer cells? Pol J Pathol. 2006;57:183–5. [PubMed] [Google Scholar]
- 40.Follenzi A, Raut S, Merlin S, Sarkar R, Gupta S. Role of bone marrow transplantation for correcting hemophilia A in mice. Blood. 2012;119:5532–42. doi: 10.1182/blood-2011-07-367680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Follenzi A, Benten D, Novikoff P, Faulkner L, Raut S, Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest. 2008;118:935–45. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Filali EE, Hiralall J, van Veen HA, Stolz DB, Seppen J. Human liver endothelial cells, but not macrovascular or microvascular endothelial cells engraft in the mouse liver. Cell Transplant. 2012;22:1801–11. doi: 10.3727/096368912X657594. [DOI] [PubMed] [Google Scholar]
- 43.Parent R, Durantel D, Sallé A, Lahlali T, DaCosta D, Plissonnier ML, Lesca G, Zoulim F, Marion MJ, Bartosch B. An immortalized human liver endothelial sinusoidal cell line for the study of the pathobiology of the liver endothelium. Biochem Biophys Res Commun. 2014;450:7–12. doi: 10.1016/j.bbrc.2014.05.038. [DOI] [PubMed] [Google Scholar]
- 44.Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105:71–7. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin Y, Chang L, Solovey A, Healey JF, Lollar P, Hebbel RP. Use of blood outgrowth endothelial cells for gene therapy for hemophilia A. Blood. 2002;99:457–62. doi: 10.1182/blood.v99.2.457. [DOI] [PubMed] [Google Scholar]
- 46.Tatsumi K, Sugimoto M, Lillicrap D, Shima M, Ohashi K, Okano T, Matsui H. A novel cell-sheet technology that achieves durable factor VIII delivery in a mouse model of hemophilia A. PLoS ONE. 2013;8:e83280. doi: 10.1371/journal.pone.0083280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harb R, Xie G, Lutzko C, Guo Y, Wang X, Hill CK, Kanel GC, DeLeve LD. Bone marrow progenitor cells repair rat hepatic sinusoidal endothelial cells after liver injury. Gastroenterology. 2009;137:704–12. doi: 10.1053/j.gastro.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Wang X, Xie G, Wang L, Hill CK, DeLeve LD. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J Clin Invest. 2012;122:1567–73. doi: 10.1172/JCI58789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Wang X, Wang L, Chiu JD, van de Ven G, Gaarde WA, Deleve LD. Hepatic vascular endothelial growth factor regulates recruitment of rat liver sinusoidal endothelial cell progenitor cells. Gastroenterology. 2012;143:1555–1563. e2. doi: 10.1053/j.gastro.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muench MO, Beyer AI, Fomin ME, Thakker R, Mulvaney US, Nakamura M, Suemizu H, Bárcena A. The adult livers of immunodeficient mice support human hematopoiesis: Evidence for a hepatic mast cell population that develops early in human ontogeny. PLoS ONE. 2014;9:e97312. doi: 10.1371/journal.pone.0097312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suemizu H, Hasegawa M, Kawai K, Taniguchi K, Monnai M, Wakui M, Suematsu M, Ito M, Peltz G, Nakamura M. Establishment of a humanized model of liver using NOD/shi-scid il2rgnull mice. Biochem Biophys Res Commun. 2008;377:248–52. doi: 10.1016/j.bbrc.2008.09.124. [DOI] [PubMed] [Google Scholar]
- 52.Sandgren EP, Palmiter RD, Heckel JL, Daugherty CC, Brinster RL, Degen JL. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell. 1991;66:245–56. doi: 10.1016/0092-8674(91)90615-6. [DOI] [PubMed] [Google Scholar]
- 53.Mohammed FF, Khokha R. Thinking outside the cell: Proteases regulate hepatocyte division. Trends Cell Biol. 2005;15:555–63. doi: 10.1016/j.tcb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 54.Muench MO. In utero transplantation: Baby steps towards an effective therapy. Bone Marrow Transplant. 2005;35:537–47. doi: 10.1038/sj.bmt.1704811. [DOI] [PubMed] [Google Scholar]
- 55.Mychaliska GB, Muench MO, Rice HE, Leavitt AD, Cruz J, Harrison MR. The biology and ethics of banking fetal liver hematopoietic stem cells for in utero transplantation. J Pediatr Surg. 1998;33:394–9. doi: 10.1016/s0022-3468(98)90470-5. [DOI] [PubMed] [Google Scholar]
- 56.Golfier F, Bárcena A, Cruz J, Harrison M, Muench M. Mid-trimester fetal livers are a rich source of CD34+/++ cells for transplantation. Bone Marrow Transplant. 1999;24:451–61. doi: 10.1038/sj.bmt.1701940. [DOI] [PubMed] [Google Scholar]
- 57.Khan AA, Habeeb A, Parveen N, Naseem B, Babu RP, Capoor AK, Habibullah CM. Peritoneal transplantation of human fetal hepatocytes for the treatment of acute fatty liver of pregnancy: A case report. Trop Gastroenterol. 2004;25:141–3. [PubMed] [Google Scholar]
- 58.Khan AA, Parveen N, Mahaboob VS, Rajendraprasad A, Ravindraprakash HR, Venkateswarlu J, Rao P, Pande G, Narusu ML, Khaja MN, Pramila R, Habeeb A, Habibullah CM. Management of hyperbilirubinemia in biliary atresia by hepatic progenitor cell transplantation through hepatic artery: A case report. Transplant Proc. 2008;40:1153–5. doi: 10.1016/j.transproceed.2008.03.110. [DOI] [PubMed] [Google Scholar]
- 59.Khan AA, Shaik MV, Parveen N, Rajendraprasad A, Aleem MA, Habeeb MA, Srinivas G, Raj TA, Tiwari SK, Kumaresan K, Venkateswarlu J, Pande G, Habibullah CM. Human fetal liver-derived stem cell transplantation as supportive modality in the management of end-stage decompensated liver cirrhosis. Cell Transplant. 2010;19:409–18. doi: 10.3727/096368910X498241. [DOI] [PubMed] [Google Scholar]
- 60.Gridelli B, Vizzini G, Pietrosi G, Luca A, Spada M, Gruttadauria S, Cintorino D, Amico G, Chinnici C, Miki T, Schmelzer E, Conaldi PG, Triolo F, Gerlach JC. Efficient human fetal liver cell isolation protocol based on vascular perfusion for liver cell-based therapy and case report on cell transplantation. Liver Transpl. 2012;18:226–37. doi: 10.1002/lt.22322. [DOI] [PubMed] [Google Scholar]
- 61.Wang JJ, Kuang Y, Zhang LL, Shen CL, Wang L, Lu SY, Lu XB, Fei J, Gu MM, Wang ZG. Phenotypic correction and stable expression of factor VIII in hemophilia A mice by embryonic stem cell therapy. Genet Mol Res. 2013;12:1511–21. doi: 10.4238/2013.May.13.4. [DOI] [PubMed] [Google Scholar]
- 62.Xu D, Alipio Z, Fink LM, Adcock DM, Yang J, Ward DC, Ma Y. Phenotypic correction of murine hemophilia A using an ips cell-based therapy. Proc Natl Acad Sci U S A. 2009;106:808–13. doi: 10.1073/pnas.0812090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yakura Y, Ishihara C, Kurosaki H, Kazuki Y, Komatsu N, Okada Y, Doi T, Takeya H, Oshimura M. An induced pluripotent stem cell-mediated and integration-free factor VIII expression system. Biochem Biophys Res Commun. 2013;431:336–41. doi: 10.1016/j.bbrc.2012.12.096. [DOI] [PubMed] [Google Scholar]
- 64.Park CY, Kim J, Kweon J, Son JS, Lee JS, Yoo JE, Cho SR, Kim JH, Kim JS, Kim DW. Targeted inversion and reversion of the blood coagulation factor 8 gene in human ips cells using talens. Proc Natl Acad Sci U S A. 2014;111:9253–8. doi: 10.1073/pnas.1323941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–51. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 66.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie F, Ye L, Chang JC, Beyer AI, Wang J, Muench MO, Kan YW. Seamless gene correction of β-thalassemia mutations in patient-specific ipscs using CRISPR/cas9 and piggybac. Genome Res. 2014;24:1526–33. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye L, Wang J, Beyer AI, Teque F, Cradick TJ, Qi Z, Chang JC, Bao G, Muench MO, Yu J, Levy JA, Kan YW. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Δ32 mutation confers resistance to HIV infection. Proc Natl Acad Sci U S A. 2014;111:9591–6. doi: 10.1073/pnas.1407473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ye L, Muench MO, Fusaki N, Beyer AI, Wang J, Qi Z, Yu J, Kan YW. Blood cell-derived induced pluripotent stem cells free of reprogramming factors generated by sendai viral vectors. Stem Cells Transl Med. 2013;2:558–66. doi: 10.5966/sctm.2013-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Merling RK, Sweeney CL, Choi U, De Ravin SS, Myers TG, Otaizo-Carrasquero F, Pan J, Linton G, Chen L, Koontz S, Theobald NL, Malech HL. Transgene-free ipscs generated from small volume peripheral blood nonmobilized CD34+ cells. Blood. 2013;121:e98–107. doi: 10.1182/blood-2012-03-420273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan HK, Toh CX, Ma D, Yang B, Liu TM, Lu J, Wong CW, Tan TK, Li H, Syn C, Tan EL, Lim B, Lim YP, Cook SA, Loh YH. Human finger-prick induced pluripotent stem cells facilitate the development of stem cell banking. Stem Cells Transl Med. 2014;3:586–98. doi: 10.5966/sctm.2013-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoo CH, Na HJ, Lee DS, Heo SC, An Y, Cha J, Choi C, Kim JH, Park JC, Cho YS. Endothelial progenitor cells from human dental pulp-derived ips cells as a therapeutic target for ischemic vascular diseases. Biomaterials. 2013;34:8149–60. doi: 10.1016/j.biomaterials.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 73.Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–40. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 74.Gupta DK, Sharma S, Venugopal P, Kumar L, Mohanty S, Dattagupta S. Stem cells as a therapeutic modality in pediatric malformations. Transplant Proc. 2007;39:700–2. doi: 10.1016/j.transproceed.2007.01.060. [DOI] [PubMed] [Google Scholar]
- 75.Khan AA, Parveen N, Mahaboob VS, Rajendraprasad A, Ravindraprakash HR, Venkateswarlu J, Rao SG, Narusu ML, Khaja MN, Pramila R, Habeeb A, Habibullah CM. Safety and efficacy of autologous bone marrow stem cell transplantation through hepatic artery for the treatment of chronic liver failure: A preliminary study. Transplant Proc. 2008;40:1140–4. doi: 10.1016/j.transproceed.2008.03.111. [DOI] [PubMed] [Google Scholar]
- 76.Malhi H, Annamaneni P, Slehria S, Joseph B, Bhargava KK, Palestro CJ, Novikoff PM, Gupta S. Cyclophosphamide disrupts hepatic sinusoidal endothelium and improves transplanted cell engraftment in rat liver. Hepatology. 2002;36:112–21. doi: 10.1053/jhep.2002.33896. [DOI] [PubMed] [Google Scholar]
- 77.Krause P, Rave-Fränk M, Wolff HA, Becker H, Christiansen H, Koenig S. Liver sinusoidal endothelial and biliary cell repopulation following irradiation and partial hepatectomy. World J Gastroenterol. 2010;16:3928–35. doi: 10.3748/wjg.v16.i31.3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amin MT, Shazly SA. In utero stem cell transplantation for radical treatment of osteogenesis imperfecta: Perspectives and controversies. Am J Perinatol. 2013;31:829–36. doi: 10.1055/s-0033-1363501. [DOI] [PubMed] [Google Scholar]
- 79.Götherström C, Westgren M, Shaw SW, Aström E, Biswas A, Byers PH, Mattar CN, Graham GE, Taslimi J, Ewald U, Fisk NM, Yeoh AE, Lin JL, Cheng PJ, Choolani M, Le Blanc K, Chan JK. Pre- and postnatal transplantation of fetal mesenchymal stem cells in osteogenesis imperfecta: A two-center experience. Stem Cells Transl Med. 2014;3:255–64. doi: 10.5966/sctm.2013-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wood JA, Colletti E, Mead LE, Ingram D, Porada CD, Zanjani ED, Yoder MC, Almeida-Porada G. Distinct contribution of human cord blood-derived endothelial colony forming cells to liver and gut in a fetal sheep model. Hepatology. 2012;56:1086–96. doi: 10.1002/hep.25753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rosenberg B, Greengard S, Montgomery R. Genetic induction of a releasable pool of factor VIII in human endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:2689–95. doi: 10.1161/01.atv.20.12.2689. [DOI] [PubMed] [Google Scholar]
- 82.Aronovich A, Tchorsh D, Katchman H, Eventov-Friedman S, Shezen E, Martinowitz U, Blazar BR, Cohen S, Tal O, Reisner Y. Correction of hemophilia as a proof of concept for treatment of monogenic diseases by fetal spleen transplantation. Proc Natl Acad Sci U S A. 2006;103:19075–80. doi: 10.1073/pnas.0607012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Banshodani M, Onoe T, Shishida M, Tahara H, Hashimoto S, Igarashi Y, Tanaka Y, Ohdan H. Adoptive transfer of allogeneic liver sinusoidal endothelial cells specifically inhibits T cell responses to cognate stimuli. Cell Transplant. 2012;22:1695–708. doi: 10.3727/096368912X657738. [DOI] [PubMed] [Google Scholar]