

Abstract
Shigellosis is a severe diarrheal disease that affects hundreds of thousands of individuals resulting in significant morbidity and mortality worldwide. Shigellosis is caused by Shigella spp., a gram-negative bacterium that uses a Type 3 Secretion System (T3SS) to deliver effector proteins into the cytosol of infected human cells. Shigella infection triggers multiple signaling programs that result in a robust host transcriptional response that includes the induction of multiple proinflammatory cytokines. PML nuclear bodies (PML-NBs) are dynamic subnuclear structures that coordinate immune signaling programs and have a demonstrated role in controlling viral infection. We show that PML-NB number increases upon Shigella infection. We examined the effects of Shigella infection on SUMOylation and found that upon Shigella infection the localization of SUMOylated proteins is altered and the level of SUMOylated proteins decreases. Although Shigella infection does not alter the abundance of SUMO activating enzymes SAE1 or SAE2, it dramatically decreases the level of the SUMO conjugating enzyme Ubc9. All Shigella-induced alterations to the SUMOylation system are dependent upon a T3SS. Thus, we demonstrate that Shigella uses one or more T3SS effectors to influence both PML-NB number and the SUMOylation machinery in human cells.
Introduction
Shigella spp. (hereafter refered to as Shigella) is the causative pathogen of shigellosis: a severe diarrheal disease that inflicts a major disease burden on the developing world. Shigella damages the colonic epithelium, resulting in massive inflammation and a characteristic bloody diarrhea. Shigella are gram negative bacteria that use a type three secretion system (T3SS) to inject effector proteins into the cytosol of infected human cells (reviewed in [1]). Most virulence factors, including the components of the T3SS, are encoded by a large (220 kb) plasmid that is essential for virulence. The repertoire of effectors for the Shigella T3SS includes many factors that interfere with normal signal transduction in host cells.
T3SS effector proteins are adept at mimicking eukaryotic protein function, even in cases where the signaling components may be completely lacking in bacterial systems [2]. For example, despite the ubiquitin system being absent in prokaryotes Shigella possess many enzymes that act on the ubiquitin pathway. The ubiquitin system is a post-translational modification pathway that controls many eukaryotic processes through alteration of protein stability, localization, and/or activation of enzymatic function [3]. Of the approximately 50 predicted Shigella effectors, at least 14 are dedicated to interfering with or utilizing the ubiquitin system (reviewed in [2, 4]). Shigella effectors that target the Ub system include a class of E3 ubiqutin ligases, the IpaH family [5]. Shigella also use OspG, a protein kinase, that interacts with charged ubiquitin conjugating enzymes to downregulate inflammation [6, 7]. Finally, Shigella also possess OspI; a deamidase that inactivates Ubc13, which is responsible for formation of K63-linked ubiquitin chains, resulting in reduced signaling through the NF-κB cascade[8]. The ability of Shigella to alter ubiquitin-dependent pathways in the host cell is one example of how this intracellular pathogen subverts its host during successful infection. Given that ubiquitin-like modifications often work in concert with the ubiquitin pathway, it is possible that Shigella may possess mechanisms to interfere with these programs [9, 10].
The small ubiquitin-like modifier (SUMO) system is also absent in prokaryotes and is a regulator of diverse processes including genome stability, intracellular transport, immune responses, and transcription [11–13]. Like ubiquitin, SUMOylation is a reversible protein modification where SUMO, a 10kDa protein, is covalently attached to lysine residues of target proteins[14]. SUMOylation, like the ubiquitin system, involves the sequential transfer of SUMO from E1 enzymes (encoded by SAE1 and SAE2) to an E2 conjugating enzyme [15]. In contrast to the ubiquitin system, which may utilize dozens of E2 enzymes, the SUMO system relies on a single enzyme, Ubc9. In addition, there are three isoforms of SUMO (SUMO1, SUMO2 and SUMO3) expressed in human cells, and target proteins may be modified by one or a combination of these isoforms. The impact of Shigella infection on the SUMO pathways is currently unknown; however, it has been shown that Listeria monocytogenes (hereafter Listeria), a bacterium that shares many similarities with Shigella in infection strategies, impairs the SUMO pathway[16]. Listeria use a secreted pore-forming toxin LLO to impair SUMOylation of eukaryotic proteins and gain an advantage in intracellular replication. Listeria infection results in a global reduction in eukaryotic SUMOylation by destabilizing Ubc9 [16]. Similar strategies have been described for many viruses as well (reviewed in [12]). Very recently, it was shown that a virulence factor for the obligate intracellular pathogen, Ehrlichia chaffeensis, is SUMOylated [17]. In that study, it was shown that SUMO1-conjugated proteins colocalized with bacterial inclusions and that impairing SUMOylation resulted in a decreased ability for Ehrlichia chaffeensis to replicate. Another recent report similarly demonstrated that AmpA, a secreted virulence factor for Anaplasma phagocytophilum was also SUMOylated and that pharmacological inhibition of the SUMO program resulted in a significant decrease in bacterial load of infected cells [18]. Thus, while the role of SUMOylation in immunity is emerging, it is clear that successful pathogens have evolved mechanisms to alter or to hijack SUMOylation.
Mammalian cells sense the presence of pathogens using Pattern Recognition Molecules (PRMs). Engagement of PRMs with their microbial ligands, such as peptidoglycan components or flagellin, result in the activation of signaling cascades that activate gene expression (reviewed in [19]). Successful pathogens produce factors that interfere with immune signaling cascades that govern transcription responses, and in some cases can interfere directly with transcription machinery [20]. Within the nucleus, many immune responsive signaling programs converge on subnuclear structures known as promyelocytic leukemia nuclear bodies (PML-NBs) [21, 22]. PML-NBs are dynamic macromolecular structures that coordinate a variety of important cellular processes including anti-viral activity, DNA damage responses, gene expression and cell fate decisions such as apoptosis and senescence [22, 23]. Although the PML protein is an absolute requirement for the formation of PML-NBs, these bodies are structurally heterogeneous with over 100 different cellular proteins associating with PML-NBs either constitutively (e.g. SP100, DAXX) or transiently in response to various stimuli or stresses (e.g. p53) [24–26]. SUMOylation is important for PML-NB formation and function as many proteins that associate with PML-NBs are either SUMO-modified and/or contain SUMO-interacting motifs, including PML itself [27].
In addition to an association with tumour suppression and DNA repair [28], several lines of evidence support a role for PML-NBs in the immune response to pathogens. First, PML-NBs form part of an intrinsic antiviral response through their ability to silence gene expression from certain DNA virus genomes, such as members of the Herpesviridae family, upon infection [12, 29, 30]. Second, PML-NBs are key players in orchestrating the interferon (IFN) response that is triggered by viral and some other intracellular infections and the number of PML-NBs increases upon IFN treatment [31]. Third, viruses have developed specific countermeasures that disrupt PML organization and function [32–36]. To date, the role of PML-NBs in bacterial infection has not been explored, however PML is required for sensitivity to lippopolysaccharide (LPS)-induced septic shock indicating a role for PML in innate immunity to bacterial pathogens [37].
The SUMOylation program was recently shown to play a protective role against Shigella infection [38]. Quantitative proteomic studies showed that Shigella infection results in the SUMO-2 modification of many transcriptional regulators that coordinate the inflammatory response. Overexpression of components of the SUMO pathway significantly decreased the ability of Shigella to enter cultured epithelial cells. These in vitro results were consistent with results obtained in an infection model of haploinsufficient UBC9 +/- mice. When compared with wild type controls, newborn UBC9 +/- mice Infected with Shigella displayed a dramatic increase in the destruction of the colonic tissue, an increase in gut permeability, and an increase in proinflammatory cytokines. Taken together, these data show that the SUMO pathway restricts Shigella infection at multiple steps [38].
Given the importance of the SUMO and PML pathways in innate immunity to multiple cellular pathogens, we assessed the impact of Shigella infection on both PML-NBs and SUMOylation. Our findings indicate that PML-NB number and SUMOylation of cellular proteins are altered during infection with Shigella, and these cellular changes require T3SS effectors.
Results
Shigella infection increases the number of PML bodies in infected cells
PML-NBs are heterogeneous complexes of nuclear factors that have been implicated in genome integrity, immune function, and stress [39]. We examined PML-NB dynamics in HeLa cells infected with Shigella that contained a plasmid expressing the afimbrial adhesin afaE. Infection with wild-type Shigella (M90T) caused a large increase in PML NBs in many of the infected cells (Fig 1A). We quantified the number of PML-NBs per cell and found that uninfected cells had an average of 3.4 PML-NBs with about 87% of all cells having 0–5 PML-NBs and about 13% having 6–10 PML-NBs (Fig 1B and 1C). Cells treated with a non-invasive avirulent strain (BS176) showed a similar distribution of PML-NBs (Fig 1B and 1C). In contrast, M90T-infected cells had and overall 2-fold (p < 0.01) increase in the average number of PML bodies per cell (Fig 1B). This change is due to a significant (p<0.01) reduction in the percentage of cells with 1–5 PML-NBs and a significant increase in cells containing 11 or more PML NBs (p<0.01, Fig 1C). Since cells infected with BS176 had equal numbers of PML-NBs as uninfected cells, we suggest that post-invasion events of Shigella infection are required for numeric changes in these subnuclear domains. It is not clear why only a subset of M90T infected cells contain increased PML-NBs; however, since the BS176 and M90T strains also express RFP, we were able to confirm that all cells in the BS176 and M90T treated conditions (MOI = 10) were associated with these Shigella strains (S1 Fig). Further, the high PML-containing cells were associated with similar levels of M90T as normal PML-containing cells, suggesting that infection “load” per se does not correlate with PML-NB number. It is known that PML protein expression increases in response to cell cycle [40], interferon and other cellular stresses and signaling events [31], which in turn can result in increased PML-NBs. Alternatively, PML body number can increase without increasing PML protein levels through fission events in response to chromatin reorganization resulting from heat shock or DNA damage, resulting in the appearance of twin PML NBs or “doublets” [28, 41]. Alternatively, PML bodies may form through de novo nucleation from the soluble pool of PML, such as PML NB formation at telomeres [42]and association with herpes simplex virus type 1 (HSV-1) genomes [43]. To determine if the observed increase in PML NB number was due to increased PML expression or stability we analyzed control and infected cell lysates for PML levels (Fig 1D). We did not observe a significant change in overall PML protein levels or significant changes in PML isoform distribution during infection with either the non-invasive (BS176) or wild-type (M90T) Shigella strains. Furthermore, we observed multiple closely spaced PML-NB doublets in the infected cells as compared to controls, consistent with an increase in PML-NB number by body fission (Fig 1A, white arrows). Thus, these data indicate that the increase in PML body number occurs in the absence of increased PML protein expression, and may have occurred by PML NB fission or de novo PML body nucleation from soluble PML.
Fig 1. Shigella infection increases PML-NB number.

HeLa cells were not infected (UI), infected with a non-invasive strain (BS176), or infected with an invasive wild-type strain (M90T) as indicated, both strains contained a plasmid expressing the afaE adhesin (A). Cells were incubated for 3 hours, fixed and immunostained for PML (red). M90T-infected cells with normal and high levels of PML nuclear bodies are shown as indicated. DNA was visualized with DAPI (blue) and white arrow heads indicate PML NB “doublets”. Scale bars = 10μm. The average number of PML bodies per cell for each treatment condition was quantified (B) and the p-value of Student’s t-test was calculated (n.s. = not significant). The percentage of cells containing either 0, 1–5, 6–10 or 11+ PML bodies was also determined (C) and the p-value of Student’s t-test was calculated. Values in B and C represent the mean +/- SE, n = 3. Total PML protein levels for each treatment condition was determined by Western blot analysis (D).
Shigella infection alters SUMO1 localization and abundance
In addition to investigating PML, we assessed Shigella-infected cells for changes in SUMO-1, an important post-translational modification involved in PML-NB function as well as important nuclear functions. We examined the localization of SUMO-1 in uninfected cells, cells infected with a non-invasive and non-virulent strain of Shigella, and a virulent and invasive strain of Shigella. In uninfected HeLa cells, SUMO-1 appeared largely in the nucleus and was present in diffuse foci (Fig 2A). This was also the case in cells infected with the non-virulent S. flexneri (Fig 2A, BS176). In contrast, in cells infected with wild-type Shigella SUMO-1 appeared as more condensed punctae with sharper edges and brighter staining (Fig 2A, M90T, left panels, arrows) in comparison to cells that were uninfected or infected with non-invasive Shigella. We quantified the percentage of cells displaying this SUMO-1 phenotype (Fig 2B) and noted a significant (p < 0.05) increase in condensed SUMO-1 in M90T-infected cells (~26% of cells) and BS176-infected cells (~9% of cells) when compared to uninfected cells in which less than 1% of cells showed the condensed SUMO-1 phenotype. SUMOylation of PML is important for the formation and stability of PML-NBs so we investigated if the increase in PML-NB might be due to an increase in the stabilizing effects of PML SUMOylation. We used immunofluoresence microscopy to determine if PML-NBs colocalized with sites of increased SUMO1 concentration in M90T-infected cells (S2 Fig). Although some SUMO1 could often be found associated with PML-NBs in uninfected and BS176-infected cells, PML was rarely present at areas of SUMO1 accumulation (S2A and S2B Fig). In M90T-infected cells with increased PML-NB number or with increased SUMO1 concentration, there was no obvious increase in PML association with SUMO1 structures or vice versa (S2C and S2D Fig). As well, our analysis did not reveal a correlation between PML body number and the condensed SUMO1 phenotype.
Fig 2. Shigella infection induces condensed localizations of SUMO1.

HeLa cells were not infected (UI, uninfected), infected with a non-invasive strain (BS176), or infected with an invasive wild-type strain (M90T) as indicated, both strains contained a plasmid expressing the afaE adhesin (A). Cells were incubated for 3 hours, fixed and immunostained for SUMO1 (green). M90T-infected cells with condensed SUMO1 stain are indicated by arrows (left panels) while those with small nuclei and reduced SUMO1 staining are indicated with arrowheads (right panels). DNA was visualized with DAPI (blue). Scale bars = 10μm. The percentage of cells displaying either condensed SUMO1 staining (B) or small nuclei with reduced SUMO1 staining (C) for each treatment condition was quantified and the p-value of Student’s t-test was calculated. Values = mean +/- SE (n = 3).
We also observed a second SUMO1 phenotype in M90T present in infected cells but not uninfected cells. Specifically, we observed some infected cells with nuclei that were smaller than the average nuclei in uninfected cells and that also showed reduced or absent SUMO1 signal (Fig 2A, M90T, right panels, arrowheads). We quantified the percentage of cells with this second SUMO1 phenotype (Fig 2C) and determined that ~9.5% of M90T-infected (p < 0,05 vs uninfected control) and ~2% of BS176-infected cells had small nuclei with reduced SUMO1 staining. This phenotype was not observed in any of the uninfected cells (Fig 2C). Furthermore, PML-NBs did not appear to either increase or decrease in these SUMO-depleted cells with small nuclei (S2E Fig).
The SUMO1 localization phenotype associated with Shigella infection prompted us to investigate if, as with L. monocytogenes, Shigella infection resulted in a global impairment of SUMOylated proteins in infected cells. In HeLa cells infected for 3 hours, there was a marked reduction in the level of SUMO-conjugated proteins as evidenced by the loss of signal of large molecular weight proteins reacting with SUMO1–specific antisera (Fig 3A). This was also true for SUMO2/3 conjugated proteins, indicating that the general SUMO conjugating machinery may be affected (Fig 3B). To examine if Shigella factors that may be involved in the impaired SUMOylation, we infected Hela cells with a mxiE mutant and a mxiD mutant. The mxiE mutant is as invasive as wild-type Shigella, but does not express the full complement of Shigella effectors [44]. mxiD encodes an essential component of the T3SS apparatus and, as a result, mxiD mutants are non-invasive [45]. We observed that global SUMOylation was impaired in cells infected with the mxiE mutant (Fig 3C). In contrast, the mxiD mutant did not impair global SUMOylation (Fig 3C). We conclude that a functional T3SS is required for Shigella to impair SUMOylation.
Fig 3. A decrease in SUMO-conjugated proteins accompanies Shigella infection.
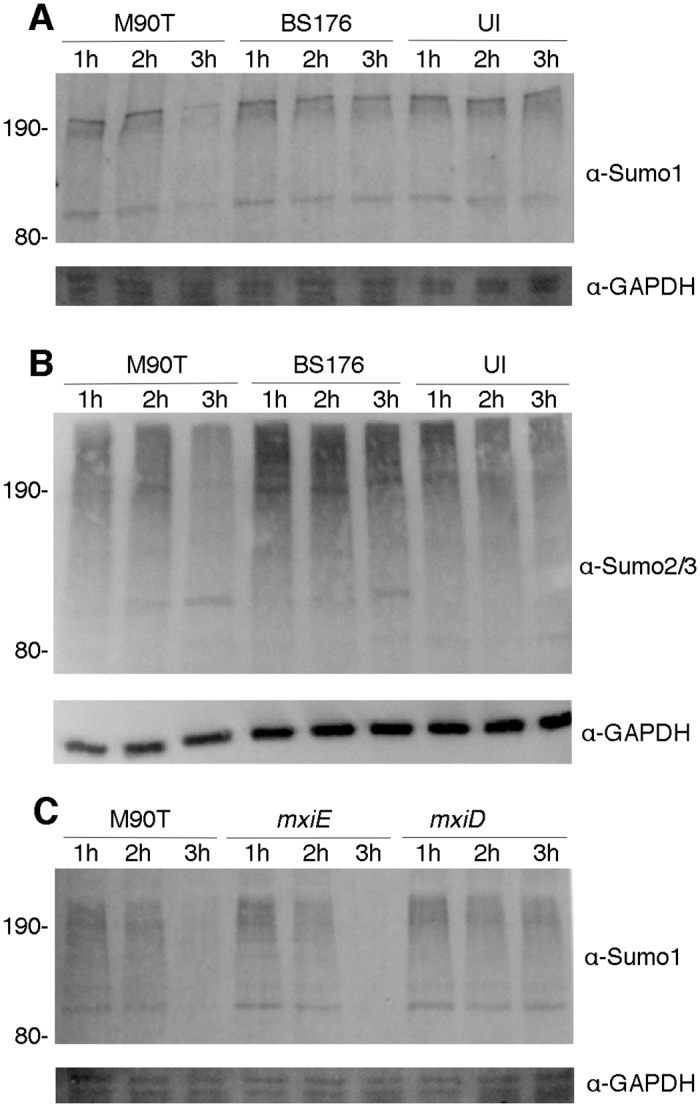
A comparison of SUMO-conjugated proteins from HeLa cells infected with wild type Shigella (M90T), a non-invasive strain (BS176), or uninfected cells for 1, 2, and 3 hours, both strains contained a plasmid expressing the afaE adhesin. Whole cell lysates were prepared from infected cells, separated on 7% polyacrylamide gels and immunoblotted using antibodies specific for SUMO1 (A) or SUMO2/3 (B). In (C) whole cell lysates from HeLa cells infected with wild type Shigella (M90T), a non-invasive mutant defective in the T3SS (mxiD), or an invasive mutant that does not produce a number of effectors (mxiE), was analyzed by immunoblotting using SUMO1-specific antibodies. Immunoblotting using GAPDH served as a loading control.
We examined the effect of Shigella infection on the stability of SUMOylation components. In cells infected with Shigella, the E1 activating enzymes SAE1 and SAE2 were readily detected in equal abundance up to 3 hours after infection (Fig 4A and 4B). In contrast, the SUMO conjugating enzyme Ubc9 decreased in cells infected with wild-type Shigella for 3 hours, while it remained constant in cells that had been infected with a plasmid-cured strain (Fig 4C). The decrease in Ubc9 was less pronounced in cells that had been infected with wild-type Shigella in the presence of the proteasome inhibitor MG132 (Fig 4D). We observed that the E3 ubiquitin conjugating enzyme UbcH5 was readily detected in all experimental conditions, indicating that Ubc9 is specifically destabilized (Fig 4E). We conclude that Shigella causes a decrease in global SUMOylation by destabilizing Ubc9.
Fig 4. Shigella targets SUMO conjugating enzyme Ubc9.

In (A-C) whole cell lysates prepared from HeLa cells infected for 1, 2 or 3 hours with the indicated strains of Shigella bearing a plasmid expressing the afaE adhesin were analyzed by immunoblotting using antibodies specific for SAE1 (A), SAE2 (B), or Ubc9 (C). In (D) HeLa cells were treated with the proteasome inhibitor MG132 prior to infection, or not, as indicated prior to infection and preparation of lysates. Lysates were analyzed for Ubc9 by immunoblotting. In (D) HeLa cells were infected with the indicated strains and lysates were analyzed by immunoblotting for UbcH5.
Because the decrease in Ubc9 levels was inhibited by MG132, we hypothesized that Ubc9 was ubiquitinated in response to Shigella infection, thereby targeting it for destruction by the proteasome. We attempted to immunoprecipitate an epitope-tagged variant of Ubc9 to determine if it was ubiquitinated. However, we were unable to reliably detect Ubc9 from Shigella-infected cells. While our results indicate the depletion of Ubc9 protein levels in Shigella-infected cells is proteasome dependent, we have no evidence to support the modification of Ubc9 directly by either ubiquitin or the SUMO isoforms.
Discussion
We demonstrate that Shigella infection impairs the SUMOylation pathway in infected cells. Shigella infection causes a redistribution of SUMO1 and the destabilization of the E2 conjugating enzyme Ubc9. Shigella infection also causes an increase in the amount of PML-NBs.
Although we cannot associate the observed PML and SUMO phenotypes with a benefit to either the host or the pathogen, this report is the first to demonstrate that bacterial pathogens can alter PML-NB number during infection. The increase in PML-NB number does not appear to be due to an increase in overall PML protein levels (Fig 1D). Therefore we speculate that Shigella infection may induce either a signaling and/or stress event (eg. cytokine signal or DNA damage) that alters PML-NB number, possibly through a fission event or de novo assembly of PML bodies from the soluble pool of PML protein. For example, we have demonstrated previously that PML-NBs respond to DNA damage by increasing in number, and Shigella is known to induce DNA damage signaling in infected cells [46]. In addition, it has been established that interferon signaling is required for restricting Shigella infection [47, 48]. Since PML is an interferon-regulated gene, it is also tempting to speculate that the observed PML-NB changes arise as a result of interferon signaling. Recently it was established that interferon acts through PML-NBs to enhance SUMOylation and inhibit viral replication [49]. We tried to correlate the Shigella-induced phenotypes of increased PML-NB number and decreased SUMO signal in cells but were unable to identify any clear trend to suggest that these phenotypes are connected.
Our results with the proteasome inhibitor MG132 suggest that Ubc9 is targeted by the proteasome, however we do not have direct evidence for ubiquitination of Ubc9. It is possible that MG132 inhibits a second protease, in addition to the proteasome, which is responsible for the destruction of Ubc9. It is also possible that a second protease cleaves Ubc9 initially, and the proteasome provides a mechanism for degrading the resulting peptide fragments, resulting in a method of degradation that is partially proteasome-dependent. Notably, the Listeria-dependent destruction of Ubc9 was shown to be insensitive to MG132 treatment [16]. There have been multiple reports of Ubc9 being targeted for destruction by invading pathogens [50]. Thus, it remains an open question as to whether Ubc9 is directly targeted by multiple pathogens, or if Ubc9 destabilization may be a general mechanism invoked by the host upon infection.
Shigella has efficient countermeasures against cellular programs such as autophagy and the NF-κB signaling that are involved in immune surveillance and bacterial clearance. Our results are in accord with the recently published findings demonstrating that SUMOylation restricts Shigella infection [38]. Inhibition of the SUMOylation pathway by Shigella requires a functional T3SS. This may reflect the need for Shigella to enter cells that may result in membrane damage or activation of intracellular immunity programs. Alternatively, Shigella may possess one or more effectors that are responsible for interfering with SUMOylation. The recent studies by Dunphy and coworkers show that successful pathogens can employ SUMOylation to regulate the fate of their virulence determinants [17]. This is akin to how bacterial pathogens manipulate the ubiqutin conjugation machinery is used to orchestrate effector function within infected cells [51, 52]. We note Shigella uses the effector OspF to effectively interfere with phosphorylation and the deamidase OspI to interfere with ubiquitination [8, 53]. Given that our data now indicates that Shigella targets another pervasive post-translational modification system (i.e. SUMOylation) of eukaryotic cells, it raises the possibility that this bacteria may have specific factors to manipulate the SUMOylation and/or the fate of SUMO-modified proteins to facilitate productive infection.
Materials and Methods
Strains and oligonucleotides
Streptomycin-resistant strain Shigella flexneri serotype 5a (M90T-Sm) was used [54] as a parent strain to create mxiD::tetRA mutants using lambda red recombination as described in [7]. Integration of the knock-out cassette at the desired location was confirmed by PCR using a primer common to the tetRA cassette and one upstream from mxiD. The mxiE::kan mutant was a gift from C. Parsot [44]. Shigella was routinely cultured in or on Trypticase Soy Broth (TSB) plus 0.01% Congo red, with or without 20 mg/mL agar. Tetracycline was used at a concentration of 5 μg/mL to select for the tetracycline resistance cassette (tetRA). Ampicillin, kanamycin and gentamicin were used at 100 μg/mL, 25 μg/mL and 15 μg/mL, respectively, to maintain plasmid selection when applicable.
Cell Culture and infections
Tissue culture cells were treated with 10 μM MG132, or not, one to two hours prior to infection. In all experiments, strains of S. flexneri bearing a plasmid encoding the gene afaE [55] were grown to mid-log phase and used to infected HeLa cells (obtained from ATCC CCL-2) at MOIs of 9.5. To ensure that Shigella entered HeLa cells we performed gentamycin protection assays in parallel to the samples that were used for analysis of proteins according to the methods described in Sidik[56]. We observed that when HeLa cells were treated with 10 μM MG132 for one hour prior to infection, Shigella entered with equal efficiency as those that were untreated. Strains used for immunofluorescence also encoded the gene dsRed on a plasmid [57]. Mammalian cells were incubated at room temperature for fifteen minutes prior to and after infection to synchronize invasion. Infected cells were then incubated at 37°C with 5% CO2 for between one and three hours, as indicated in the figures (Cells used for immunofluorescence were infected for three hours). Cells used for immunoblotting for PML were lysed in one microlitre of lysis buffer per thousand cells (50 mM Tris-Cl pH 6.8, 9M Urea), boiled for 10 minutes and sonicated. The protein concentration for the extracts were determined and equal amounts of protein were separated by SDS-PAGE. For immunoblotting for all other proteins, cells were lysed in 250 μL of SDS sample buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 100 mM DTT, 10% glycerol), boiled for five minutes and separated by SDS-PAGE and blotted for specific antibodies and also GAPDH as a loading control.
Immunofluorescence
Protein localization was visualized by fluorescence microscopy. HeLa cells were plated onto coverslips in a 6-well plate and infected relevant bacterial strains. Three hours post-infection, cells were washed with PBS and fixed in 4% paraformaldehyde. Cells were permeabilized with 0.5% Triton X-100 in PBS, and blocked with 4% BSA in PBS. Cells were then immunolabeled with primary antibodies (1 h, diluted in blocking buffer), washed with PBS and incubated with fluorescently labeled secondary antibodies (45 min, diluted in blocking buffer). Following washes in PBS, cells were incubated with 1 μg/mL of 4',6-diamidino-2-phenylindole (DAPI) stain to visualize DNA. Fluorescent images were captured with Zeiss Axiovert 200M Microscope and Hamamatsu Orca R2 Camera or with Zeiss Cell Observer Microscope under a 63× immersion oil objective lens. Images were processed using only linear adjustments (e.g.brightness/contrast) with Slidebook (Intelligent Imaging Innovations, Boulder, CO) and Adobe Photoshop CS5. Quantification the SUMO1 phenotypes was conducted by coding the image filenames such that treatment condition parameters were concealed during analysis. At least 30 cells for each condition were assessed as either normal, having small nuclei with low SUMO1 expression or having condensed SUMO1. The samples were then decoded, organized by treatment category and the percentage of “low SUMO1” cells and “condensed SUMO1” cells were determined. Quantification of PML nuclear bodies was determined by counting the PML bodies in at least 30 cells per condition from each of three independent experiments. The average number of PML bodies was determined as was the percentage of cells containing either 0, 1–5, 6–10 or 11+ PML bodies. For PML and SUMO1 quantification, the means and standard errors from three independent experiments were calculated. Statistical significance was determined using the student's t-test in Microsoft Excel 2007.
Immunoblotting
Primary antibodies were purchased from the following sources: PML: Santa Cruz Biotechnologies product number sc-377390, Actin: Cell Signaling product number 4967, SUMO-1: Abcam product number ab32058, SUMO-2/3: Invitrogen product number 51–9100, SAE1: Abcam product number ab56957, SAE2: Imgenex product number IMG-5111A, Ubc9: Millipore product number MAB217, UbcH5b: Santa Cruz product number sc-100617, GAPDH: Ambion product number AM4300 and Abcam product number Ab9483. SDS-PAGE gels were transferred to PVDF membranes overnight using a wet-transfer apparatus, then blocked in 5% skim milk powder/TBST (145 mM NaCl, 5 mM Tris-Cl pH 7.5, 0.1% Tween-20, 5% skin milk powder) for one hour. Membranes were incubated in primary antibody for one hour, then washed four times for five minutes in TBST. Membranes were incubated in secondary antibody for one hour, then four additional five-minute washes were performed using TBST. Membranes were then developed using ECL Plus (Pierce) and imaged using a Kodak Electrophoresis Documentation and Analysis System or a Versadoc Imaging System (BioRad).
Supporting Information
Hela cells were not infected (A), infected with the non-invasive strain BS176 (B) or infected with the invasive wild-type strain M90T (C-E). Cells were incubated for 3 hours, fixed and immunostained for PML (red, alexa-649 labelled) or SUMO1 (green, alexa-488 labelled) as indicated. DNA was visualized with DAPI (blue) and RFP-expressing Shigella (BS176 and M90T) are shown in grayscale. Representative images of M90T-infected cells showing high PML (C), small nuclei with reduced SUMO1 (D) and condensed SUMO1 (E) are marked with an asterisk (*). Arrows indicate RFP-labelled bacteria associated with the asterisked cells. Scale bars = 10μm.
(TIF)
HeLa cells were not infected (A), infected with the non-invasive strain BS176 (B) or infected with the invasive wild-type strain M90T (C-E). Cells were incubated for 3 hours, fixed and immunostained for SUMO1 (green) and PML (red) as indicated. Representative images of M90T infected cells with the increased PML (C), condensed SUMO1 (D) and small nuclei with reduced SUMO1 (E) phenotypes are presented. DNA was visualized with DAPI (blue). Scale bars = 10μm.
(TIF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Natural Sciences and Engineering Research Council: RGPIN 386297 Canadian Institutes of Health Research: MOP-102594 and MOP-84260 SS was an Alexander Graham Bell Canada Graduate Scholar, GD is a Senior Scientist of the Beatrice Hunter Cancer Research Institute (BHCRI), JS was supported by a trainee award from the Beatrice Hunter Cancer Research Institute with funds provided the Terry Fox Foundation as part of a Strategic Health Research Training Program in Cancer Research at CIHR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Parsot C. Shigella type III secretion effectors: how, where, when, for what purposes? Curr Opin Microbiol. 2009;12(1):110–6. Epub 2009/01/23. S1369-5274(08)00188-4 [pii] 10.1016/j.mib.2008.12.002 . [DOI] [PubMed] [Google Scholar]
- 2. Ashida H, Kim M, Sasakawa C. Exploitation of the host ubiquitin system by human bacterial pathogens. Nat Rev Microbiol. 2014;12(6):399–413. Epub 2014/05/08. nrmicro3259 [pii] 10.1038/nrmicro3259 . [DOI] [PubMed] [Google Scholar]
- 3. Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. 2011;18(5):520–8. Epub 2011/05/05. nsmb.2066 [pii] 10.1038/nsmb.2066 . [DOI] [PubMed] [Google Scholar]
- 4. Huibregtse J, Rohde JR. Hell's BELs: bacterial E3 ligases that exploit the eukaryotic ubiquitin machinery. PLoS Pathog. 2014;10(8):e1004255 Epub 2014/08/15. doi: 10.1371/journal.ppat.1004255 PPATHOGENS-D-14-01027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, Parsot C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe. 2007;1(1):77–83. Epub 2007/11/17. S1931-3128(07)00006-6 [pii] 10.1016/j.chom.2007.02.002 . [DOI] [PubMed] [Google Scholar]
- 6. Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci U S A. 2005;102(39):14046–51. Epub 2005/09/16. 0504466102 [pii] 10.1073/pnas.0504466102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pruneda JN, Smith FD, Daurie A, Swaney DL, Villen J, Scott JD, et al. E2~Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. EMBO J. 2014;33(5):437–49. Epub 2014/01/22. embj.201386386 [pii] 10.1002/embj.201386386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanada T, Kim M, Mimuro H, Suzuki M, Ogawa M, Oyama A, et al. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature. 2012;483(7391):623–6. Epub 2012/03/13. nature10894 [pii] 10.1038/nature10894 . [DOI] [PubMed] [Google Scholar]
- 9. Perry JJ, Tainer JA, Boddy MN. A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem Sci. 2008;33(5):201–8. Epub 2008/04/12. S0968-0004(08)00061-3 [pii] 10.1016/j.tibs.2008.02.001 . [DOI] [PubMed] [Google Scholar]
- 10. Merlet J, Burger J, Gomes JE, Pintard L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol Life Sci. 2009;66(11–12):1924–38. Epub 2009/02/06. 10.1007/s00018-009-8712-7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pinder JB, Attwood KM, Dellaire G. Reading, writing, and repair: the role of ubiquitin and the ubiquitin-like proteins in DNA damage signaling and repair. Front Genet. 2013;4:45 Epub 2013/04/05. 10.3389/fgene.2013.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Everett RD, Boutell C, Hale BG. Interplay between viruses and host sumoylation pathways. Nat Rev Microbiol. 2013;11(6):400–11. Epub 2013/04/30. nrmicro3015 [pii] 10.1038/nrmicro3015 . [DOI] [PubMed] [Google Scholar]
- 13. Ulrich HD. Two-way communications between ubiquitin-like modifiers and DNA. Nat Struct Mol Biol. 2014;21(4):317–24. Epub 2014/04/05. nsmb.2805 [pii] 10.1038/nsmb.2805 . [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem. 2001;276(16):12654–9. Epub 2001/01/02. 10.1074/jbc.M009476200M009476200 . [DOI] [PubMed] [Google Scholar]
- 15. Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin's mysterious cousin. Nat Rev Mol Cell Biol. 2001;2(3):202–10. Epub 2001/03/27. 10.1038/35056591 . [DOI] [PubMed] [Google Scholar]
- 16. Ribet D, Hamon M, Gouin E, Nahori MA, Impens F, Neyret-Kahn H, et al. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature. 2010;464(7292):1192–5. Epub 2010/04/24. nature08963 [pii] 10.1038/nature08963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dunphy PS, Luo T, McBride JW. Ehrlichia chaffeensis Exploits Host SUMOylation Pathways to Mediate Effector-Host Interactions and Promote Intracellular Survival. Infect Immun. 2014. Epub 2014/07/23. IAI.01984-14 [pii] 10.1128/IAI.01984-14 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beyer AR, Truchan HK, May LJ, Walker NJ, Borjesson DL, Carlyon JA. The Anaplasma phagocytophilum effector AmpA hijacks host cell SUMOylation. Cell Microbiol. 2014. Epub 2014/10/14. 10.1111/cmi.12380 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7(12):1250–7. Epub 2006/11/18. ni1412 [pii] 10.1038/ni1412 . [DOI] [PubMed] [Google Scholar]
- 20. Lebreton A, Lakisic G, Job V, Fritsch L, Tham TN, Camejo A, et al. A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science. 2011;331(6022):1319–21. Epub 2011/01/22. science.1200120 [pii] 10.1126/science.1200120 . [DOI] [PubMed] [Google Scholar]
- 21. Maarifi G, Chelbi-Alix MK, Nisole S. PML control of cytokine signaling. Cytokine Growth Factor Rev. 2014. Epub 2014/05/28. S1359-6101(14)00041-0 [pii] 10.1016/j.cytogfr.2014.04.008 . [DOI] [PubMed] [Google Scholar]
- 22. Bourdeau V, Baudry D, Ferbeyre G. PML links aberrant cytokine signaling and oncogenic stress to cellular senescence. Front Biosci (Landmark Ed). 2009;14:475–85. Epub 2009/03/11. 3256 [pii]. . [DOI] [PubMed] [Google Scholar]
- 23. Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays. 2004;26(9):963–77. Epub 2004/09/08. 10.1002/bies.20089 . [DOI] [PubMed] [Google Scholar]
- 24. Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, et al. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol. 1999;147(2):221–34. Epub 1999/10/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Damme E, Laukens K, Dang TH, Van Ostade X. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci. 2010;6(1):51–67. Epub 2010/01/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Negorev D, Maul GG. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene. 2001;20(49):7234–42. Epub 2001/11/13. 10.1038/sj.onc.1204764 . [DOI] [PubMed] [Google Scholar]
- 27. Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. The mechanisms of PML-nuclear body formation. Mol Cell. 2006;24(3):331–9. Epub 2006/11/04. S1097-2765(06)00661-7 [pii] 10.1016/j.molcel.2006.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dellaire G, Ching RW, Ahmed K, Jalali F, Tse KC, Bristow RG, et al. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J Cell Biol. 2006;175(1):55–66. Epub 2006/10/13. jcb.200604009 [pii] 10.1083/jcb.200604009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol. 2006;80(16):7995–8005. Epub 2006/07/29. 80/16/7995 [pii] 10.1128/JVI.00734-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tavalai N, Stamminger T. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 2011;157(2):128–33. Epub 2010/10/12. S0168-1702(10)00361-8 [pii] 10.1016/j.virusres.2010.10.002 . [DOI] [PubMed] [Google Scholar]
- 31. Bloch DB, Chiche JD, Orth D, de la Monte SM, Rosenzweig A, Bloch KD. Structural and functional heterogeneity of nuclear bodies. Mol Cell Biol. 1999;19(6):4423–30. Epub 1999/05/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adamson AL, Kenney S. Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J Virol. 2001;75(5):2388–99. Epub 2001/02/13. 10.1128/JVI.75.5.2388-2399.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ahn JH, Hayward GS. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology. 2000;274(1):39–55. Epub 2000/08/11. 10.1006/viro.2000.0448S0042-6822(00)90448-3 . [DOI] [PubMed] [Google Scholar]
- 34. Everett RD, Chelbi-Alix MK. PML and PML nuclear bodies: implications in antiviral defence. Biochimie. 2007;89(6–7):819–30. Epub 2007/03/09. S0300-9084(07)00013-2 [pii] 10.1016/j.biochi.2007.01.004 . [DOI] [PubMed] [Google Scholar]
- 35. Sivachandran N, Wang X, Frappier L. Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. J Virol. 2012;86(11):6146–58. Epub 2012/04/12. JVI.00013-12 [pii] 10.1128/JVI.00013-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Salsman J, Zimmerman N, Chen T, Domagala M, Frappier L. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog. 2008;4(7):e1000100 Epub 2008/07/12. 10.1371/journal.ppat.1000100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lunardi A, Gaboli M, Giorgio M, Rivi R, Bygrave A, Antoniou M, et al. A Role for PML in Innate Immunity. Genes Cancer. 2011;2(1):10–9. Epub 2011/07/23. 10.1177/1947601911402682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fritah S, Lhocine N, Golebiowski F, Mounier J, Andrieux A, Jouvion G, et al. Sumoylation controls host anti-bacterial response to the gut invasive pathogen Shigella flexneri. EMBO Rep. 2014;15(9):965–72. Epub 2014/08/07. embr.201338386 [pii] 10.15252/embr.201338386 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eskiw CH, Bazett-Jones DP. The promyelocytic leukemia nuclear body: sites of activity? Biochem Cell Biol. 2002;80(3):301–10. Epub 2002/07/19. . [DOI] [PubMed] [Google Scholar]
- 40. Dellaire G, Eskiw CH, Dehghani H, Ching RW, Bazett-Jones DP. Mitotic accumulations of PML protein contribute to the re-establishment of PML nuclear bodies in G1. J Cell Sci. 2006;119(Pt 6):1034–42. Epub 2006/02/24. jcs.02817 [pii] 10.1242/jcs.02817 . [DOI] [PubMed] [Google Scholar]
- 41. Eskiw CH, Dellaire G, Mymryk JS, Bazett-Jones DP. Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J Cell Sci. 2003;116(Pt 21):4455–66. Epub 2003/09/18. 10.1242/jcs.00758jcs.00758 . [DOI] [PubMed] [Google Scholar]
- 42. Brouwer AK, Schimmel J, Wiegant JC, Vertegaal AC, Tanke HJ, Dirks RW. Telomeric DNA mediates de novo PML body formation. Mol Biol Cell. 2009;20(22):4804–15. Epub 2009/10/02. E09-04-0309 [pii] 10.1091/mbc.E09-04-0309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Everett RD, Murray J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol. 2005;79(8):5078–89. Epub 2005/03/30. 79/8/5078 [pii] 10.1128/JVI.79.8.5078-5089.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mavris M, Page AL, Tournebize R, Demers B, Sansonetti P, Parsot C. Regulation of transcription by the activity of the Shigella flexneri type III secretion apparatus. Mol Microbiol. 2002;43(6):1543–53. Epub 2002/04/25. . [DOI] [PubMed] [Google Scholar]
- 45. Allaoui A, Sansonetti PJ, Parsot C. MxiD, an outer membrane protein necessary for the secretion of the Shigella flexneri lpa invasins. Mol Microbiol. 1993;7(1):59–68. Epub 1993/01/01. . [DOI] [PubMed] [Google Scholar]
- 46. Bergounioux J, Elisee R, Prunier AL, Donnadieu F, Sperandio B, Sansonetti P, et al. Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium's epithelial niche. Cell Host Microbe. 2012;11(3):240–52. Epub 2012/03/20. S1931-3128(12)00055-8 [pii] 10.1016/j.chom.2012.01.013 . [DOI] [PubMed] [Google Scholar]
- 47. Way SS, Borczuk AC, Dominitz R, Goldberg MB. An essential role for gamma interferon in innate resistance to Shigella flexneri infection. Infect Immun. 1998;66(4):1342–8. Epub 1998/04/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jehl SP, Nogueira CV, Zhang X, Starnbach MN. IFNgamma inhibits the cytosolic replication of Shigella flexneri via the cytoplasmic RNA sensor RIG-I. PLoS Pathog. 2012;8(8):e1002809 Epub 2012/08/23. doi: 10.1371/journal.ppat.1002809 PPATHOGENS-D-11-01395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sahin U, Ferhi O, Carnec X, Zamborlini A, Peres L, Jollivet F, et al. Interferon controls SUMO availability via the Lin28 and let-7 axis to impede virus replication. Nat Commun. 2014;5:4187 Epub 2014/06/20. ncomms5187 [pii] 10.1038/ncomms5187 . [DOI] [PubMed] [Google Scholar]
- 50. Gomes R, Guerra-Sa R, Arruda E. Coxsackievirus B5 induced apoptosis of HeLa cells: effects on p53 and SUMO. Virology. 2010;396(2):256–63. Epub 2009/11/13. S0042-6822(09)00614-X [pii] 10.1016/j.virol.2009.10.005 . [DOI] [PubMed] [Google Scholar]
- 51. Patel JC, Hueffer K, Lam TT, Galan JE. Diversification of a Salmonella virulence protein function by ubiquitin-dependent differential localization. Cell. 2009;137(2):283–94. Epub 2009/04/22. S0092-8674(09)00157-3 [pii] 10.1016/j.cell.2009.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kubori T, Shinzawa N, Kanuka H, Nagai H. Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog. 2010;6(12):e1001216 Epub 2010/12/15. 10.1371/journal.ppat.1001216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, et al. The phosphothreonine lyase activity of a bacterial type III effector family. Science. 2007;315(5814):1000–3. Epub 2007/02/17. 315/5814/1000 [pii] 10.1126/science.1138960 . [DOI] [PubMed] [Google Scholar]
- 54. Onodera NT, Ryu J, Durbic T, Nislow C, Archibald JM, Rohde JR. Genome sequence of Shigella flexneri serotype 5a strain M90T Sm. J Bacteriol. 2012;194(11):3022 Epub 2012/05/15. 194/11/3022 [pii] 10.1128/JB.00393-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Labigne-Roussel AF, Lark D, Schoolnik G, Falkow S. Cloning and expression of an afimbrial adhesin (AFA-I) responsible for P blood group-independent, mannose-resistant hemagglutination from a pyelonephritic Escherichia coli strain. Infect Immun. 1984;46(1):251–9. Epub 1984/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sidik S, Kottwitz H, Benjamin J, Ryu J, Jarrar A, Garduno R, et al. A Shigella flexneri Virulence Plasmid Encoded Factor Controls Production of Outer Membrane Vesicles. G3 (Bethesda). 2014;4(12):2493–503. Epub 2014/11/08. g3.114.014381 [pii] 10.1534/g3.114.014381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sorensen M, Lippuner C, Kaiser T, Misslitz A, Aebischer T, Bumann D. Rapidly maturing red fluorescent protein variants with strongly enhanced brightness in bacteria. FEBS Lett. 2003;552(2–3):110–4. Epub 2003/10/07. S0014579303008561 [pii]. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hela cells were not infected (A), infected with the non-invasive strain BS176 (B) or infected with the invasive wild-type strain M90T (C-E). Cells were incubated for 3 hours, fixed and immunostained for PML (red, alexa-649 labelled) or SUMO1 (green, alexa-488 labelled) as indicated. DNA was visualized with DAPI (blue) and RFP-expressing Shigella (BS176 and M90T) are shown in grayscale. Representative images of M90T-infected cells showing high PML (C), small nuclei with reduced SUMO1 (D) and condensed SUMO1 (E) are marked with an asterisk (*). Arrows indicate RFP-labelled bacteria associated with the asterisked cells. Scale bars = 10μm.
(TIF)
HeLa cells were not infected (A), infected with the non-invasive strain BS176 (B) or infected with the invasive wild-type strain M90T (C-E). Cells were incubated for 3 hours, fixed and immunostained for SUMO1 (green) and PML (red) as indicated. Representative images of M90T infected cells with the increased PML (C), condensed SUMO1 (D) and small nuclei with reduced SUMO1 (E) phenotypes are presented. DNA was visualized with DAPI (blue). Scale bars = 10μm.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.