

Abstract
Intestinal immune regulatory signals govern gut homeostasis. Breakdown of such regulatory mechanisms may result in inflammatory bowel disease (IBD). Lactobacillus acidophilus contains unique surface layer proteins (Slps), including SlpA, SlpB, SlpX, and lipoteichoic acid (LTA), which interact with pattern recognition receptors to mobilize immune responses. Here, to elucidate the role of SlpA in protective immune regulation, the NCK2187 strain, which solely expresses SlpA, was generated. NCK2187 and its purified SlpA bind to the C-type lectin SIGNR3 to exert regulatory signals that result in mitigation of colitis, maintenance of healthy gastrointestinal microbiota, and protected gut mucosal barrier function. However, such protection was not observed in Signr3−/− mice, suggesting that the SlpA/SIGNR3 interaction plays a key regulatory role in colitis. Our work presents critical insights into SlpA/SIGNR3-induced responses that are integral to the potential development of novel biological therapies for autoinflammatory diseases, including IBD.
Keywords: colitis, immune regulation, Lactobacillus acidophilus, SIGNR3, surface layer protein A
See also: G Lugo-Villarino & O Neyrolles (April 2015)
Introduction
The gastrointestinal (GI) microbiota plays a critical role in determining the immunologic outcome of various signaling events in host cells via their gene products, exceeding the human genome by a 100-fold (Ley et al, 2006; Qin et al, 2010). As such, the composition of the GI microbiota and host immunity are mutualistic and continuously influence each other (Maslowski & Mackay, 2011; McDermott & Huffnagle, 2014). Therefore, it is inevitable that intestinal homeostasis is tightly controlled by regulatory immune mechanisms, which are established by the interactions of the trillions of microbes and their gene products with numerous pattern recognition receptors (PRRs), including C-type lectin receptors (CLRs), such as the specific intracellular adhesion molecule-3 grabbing non-integrin homolog-related 3 (SIGNR3) (Konstantinov et al, 2008; Osorio & Reis e Sousa, 2011). Disruption of this delicate balance by inimical signals has devastating consequences that may result in intestinal disorders, including inflammatory bowel disease (IBD). When this occurs, highly activated innate cells trigger intestine-infiltrating pathogenic T-cell subsets (e.g. Th1, Th17), and even regulatory T cells (Tregs) with proinflammatory characteristics (Khazaie et al, 2012; Geremia et al, 2014; Neurath, 2014) that ultimately drive tissue destruction and intestinal disease progression. Innate cells (e.g. dendritic cells, macrophages) are the initial targets of the culpable microbes and their gene products, which subsequently affect the regulation/stimulation of intestinal immunity (Ivanov & Honda, 2012; Atarashi et al, 2013; Yang et al, 2014). Given these entwined relationships, it is not surprising that microbial products have been linked to the pathology of intestinal autoinflammation (Nicholson et al, 2012). The underlying associations between gut microbes and inflammatory diseases (e.g. IBD) have already been well documented; however, the cellular and molecular mechanisms by which intestinal commensal gene product(s) and their molecular receptor(s) impact immune responses remain unclear.
Information regarding the immunobiologic functions of Lactobacillus acidophilus surface layer proteins (Slps) is relatively limited. Slps are paracrystalline (glyco) protein arrays that are abundant on the cell surfaces of few eubacteria and archaea, including L. acidophilus (Johnson et al, 2013). The S-layer of L. acidophilus NCFM is composed of three Slp-encoding genes: slpA (LBA0169), slpB (LBA0175), and slpX (LBA0512) (Goh et al, 2009). Diverse functional roles have been proposed for Slps, including cell shape determinants, molecular sieves, protective layers against viral infection, anchoring sites for surface-associated enzymes, and facilitators of cellular adhesion through PRRs, including C-type lectins (CLECs) (Konstantinov et al, 2008).
CLECs recognize carbohydrate structures on self and nonself antigens (Engering et al, 2002; Osorio & Reis e Sousa, 2011). Twenty-nine CLECs, including DC-specific ICAM-3-grabbing non-integrin (DC-SIGN), have been identified on dendritic cells (DCs) and macrophages (MΦs) thus far (van Kooyk & Geijtenbeek, 2003; Ehlers, 2010; Sancho & Reis e Sousa, 2013). DC-SIGN, which was previously shown to bind L. acidophilus SlpA in vitro (Konstantinov et al, 2008), is a calcium-dependent carbohydrate-binding protein with specificity for the mannose-containing glycans of microbial surface components and fucose-containing Lewis antigens (Ehlers, 2010). Of the eight murine homologs of DC-SIGN, SIGNR3 (CD209d) exhibits the most biochemical similarity to human DC-SIGN (Powlesland et al, 2006). SIGNR3 contains a carbohydrate recognition domain (CRD) and signals through a hemi-immunoreceptor tyrosine-based activation motif (hemi-ITAM) (Tanne et al, 2009). Such signaling potentially downregulates the ubiquitously expressed leukotriene A4 hydrolase (LTA4H) (Tobin et al, 2010) that catalyzes proinflammatory leukotriene B4 (LTB4) synthesis from LTA4 (Snelgrove et al, 2010), which subsequently activates interleukin (IL)-1β production. Here, we identify SlpA as a key effector molecule expressed by L. acidophilus and demonstrate its in vivo protective role in murine colitis models. Moreover, we provide evidence that protection by L. acidophilus SlpA is conferred via signaling through a single CLR, namely SIGNR3.
Results
NCK2187 promotes intestinal immune regulation in steady state
Recently, we showed that transient colonization of the colon with NCK2025 (LTA−) significantly mitigated chemical and T-cell-mediated colitis (Mohamadzadeh et al, 2011). Additionally, NCK2025 significantly abated inflammation-promoting polyposis in our novel Apclox468 × TS4-Cre mouse model, where protection correlated with the regulation of innate and T-cell-induced inflammation (Khazaie et al, 2012). We therefore hypothesized that the controlled inflammation resulted from cross talk between NCK2025 SlpA with intestinal cells. To test this hypothesis, the upp-counterselective gene replacement strategy (Goh et al, 2009) was used to generate in-frame deletions in the slpB and slpX genes of NCK2030. The LTA− derivative was created by a deletion of the phosphoglycerol transferase gene (Mohamadzadeh et al, 2011), resulting in NCK2187, which expresses only SlpA (Fig1A and B). To demonstrate that the newly generated NCK2187 transiently colonizes the gut, the clearance kinetics of both the erythromycin-resistant NCK56 and NCK2187 strains were determined in C57BL/6 (B6) mice that were orally treated once with 109 CFU/mouse. Data show that mice cleared both NCK56 and NCK2187 after 3 days, indicating that the deletion of LTA, SlpB, and SlpX in NCK2187 did not alter its transient passage through the GI tract when compared to its WT parent (Fig1C).
Figure 1.
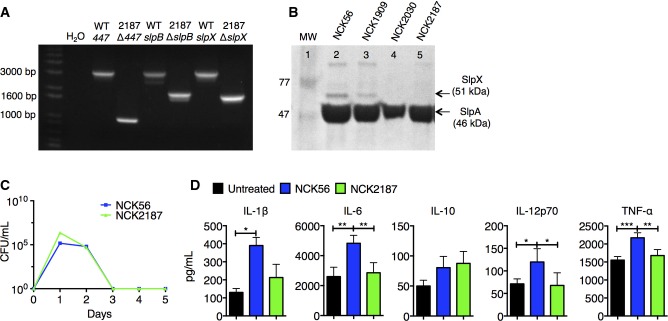
- Agarose gel image illustrating PCR amplicons of ltaS (LBA0447), slpB, and slpX deletions in NCK2187.
- SDS–PAGE gel of 5 M LiCl-purified S-layer fractions from the parental strains, NCK56 and NCK1909, NCK2030 (LTA+, SlpA+, SlpB−, SlpX−), and NCK2187 (LTA−, SlpA+, SlpB−, SlpX−).
- B6 mice were orally gavaged with 109 CFU erythromycin-resistant NCK56 or NCK2187. Fecal pellets were collected daily and tested for the presence of erythromycin-resistant strains. n = 3 mice/group. Data are representative of five independent experiments and are shown as mean ± SEM.
- Colonic LP cells were co-cultured with NCK56 or NCK2187 (1:1), and secreted cytokines were measured in the supernatants. Data are shown as mean ± SEM and are representative of two experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001.
To investigate the activation of colonic DCs when co-cultured with NCK56 or NCK2187, colonic cells were obtained from naïve B6 mice. While such intestinal cell–bacterial co-cultures did not significantly change the expression of DC co-stimulatory molecules (e.g. CD40) (not shown) or IL-10, only NCK56 elevated the levels of IL-1β, IL-6, IL-12, and TNF-α (Fig1D). Next, naïve B6 mice were orally gavaged with NCK56 or NCK2187, and colonic immune responses were analyzed. Treatment with NCK2187 significantly increased the frequency of colonic FoxP3+ Tregs when compared to both untreated (PBS) and NCK56-treated mice (Fig2A). Moreover, IL-17A+ and IFN-γ+ CD4+ T cells were significantly reduced by NCK2187 treatment (Fig2A). NCK2187-treated B6 FoxP3-GFP mice also exhibited higher numbers of colonic IL-10+ TGF-β1+ Tregs than did NCK56-treated and untreated mice (Fig2B and C). Collectively, oral treatment with this novel L. acidophilus strain induced colonic regulatory immune responses.
Figure 2.
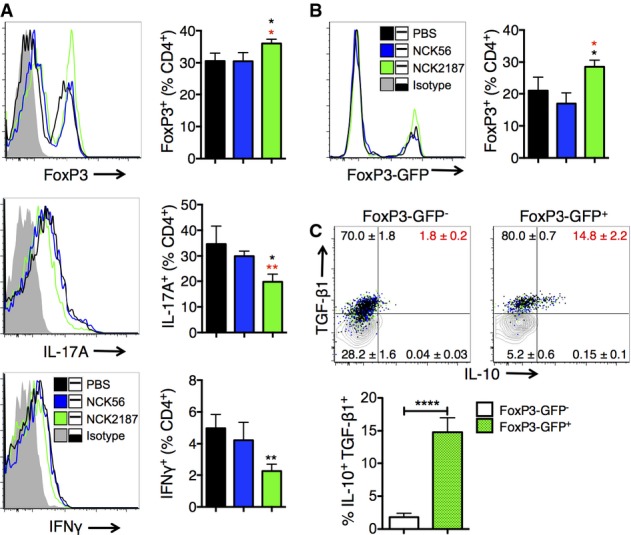
- A B6 mice were orally gavaged with 109 CFU NCK56 (blue) or NCK2187 (green) on days 0, 3, 6, and 9, or left untreated, and immune responses in the colon analyzed at day 14 by flow cytometry.
- B, C B6 FoxP3-GFP mice were treated and evaluated as in (A). Regulatory cytokine production in FoxP3-GFP+ (green-dotted bars) versus FoxP3-GFP− (white bars) cells was measured by intracellular staining and FACS analyses (C).
Protective properties of NCK2187 and its SlpA against intestinal inflammation and dysbiosis
To clarify the consequences of the immunoregulatory responses observed above during inflammation, B6 Rag1−/− mice adoptively transferred with CD45RBhi CD4+ T cells were orally treated with NCK56, NCK2187, its purified SlpA, or PBS (Fig3). Untreated (PBS) and NCK56-treated mice with adoptively transferred T cells developed severe colitis as demonstrated by weight loss, bloody diarrhea, shortening of the colon, and increased damage of the colon (Fig3A–C, Supplementary Fig S1A). Furthermore, the levels of systemically induced proinflammatory IL-1β, IL-6, TNF-α, IFN-γ, G-CSF, and macrophage inflammatory protein (MIP)-1α were significantly enhanced in the sera of these groups of mice (Fig3D). In contrast, similar to the Treg co-transferred mice, NCK2187 and its purified SlpA significantly protected Rag1−/− mice from T-cell-induced colitis (Supplementary Tables S1, S2 and S3). NCK2187- and SlpA-treated mice gained weight throughout the course of the study and did not develop bloody diarrhea in the way that the PBS and NCK56 groups did (Fig3A). Furthermore, cecal and colonic atrophy due to pathogenic inflammation was not observed in these mice, as the tissue destruction and immune cell infiltration associated with T-cell-induced colitis were significantly abrogated in NCK2187- and SlpA-treated groups (Fig3B and C). Accordingly, systemic inflammation was significantly reduced in these groups of mice (Fig3D). Interestingly, the genes encoding the receptors for LTB4, Ltb4r1 and Ltb4r2, were significantly downregulated in the colons of NCK2187- and SlpA-treated mice (Supplementary Fig S1B), which may have contributed to the reduced expression of colonic Il1b (Supplementary Fig S1B).
Figure 3.

- A B6 Rag1−/− mice were injected with 106 CD4+CD45RBhi T cells and then orally gavaged with NCK56 (red), NCK2187 (green), or SlpA (blue), 1 and 3 days after transfer, and subsequently once a week for four consecutive weeks, or left untreated (magenta). A group of mice was co-transferred with CD4+CD25+ T cells as a positive control for protection (Tregs; gray). Colitis severity was determined in part by weight loss, diarrhea scores, and FOB. See Supplementary Tables S1, S2 and S3 for statistical analyses results.
- B, C Colitis scores based on histopathology and gross morphology of the colons of mice treated as in (A) were also used as measures of disease. Scale bar = 50 μm.
- D Circulating levels of proinflammatory cytokines were measured in the sera of the mice transferred and treated as in (A), or sham adoptive transferred (white bars).
T-cell-induced colitis resulted in intestinal epithelial erosions and ulcerations in mice that did not receive NCK2187 or its purified SlpA (Fig3C). Indeed, the colonic expression of tight junction-associated genes was significantly downregulated in PBS- and NCK56-treated Rag1−/− mice (Fig4A). Furthermore, FITC-dextran permeability assays confirmed that these mice were suffering from a dysfunctional intestinal barrier (Fig4B). Accordingly, NCK2187 and SlpA significantly protected barrier integrity and function (Fig4A and B). An immunologically and anatomically weakened intestinal epithelial barrier during intestinal inflammation allows luminal bacteria to interact with the intestinal mucosae and the infiltrating immune cells, initiating inflammatory responses directed against the gut commensals and introducing dysbiosis. To determine the status and composition of the microbiota in the T-cell-induced colitis model (week 7), we analyzed the microbial communities in the colons of the different experimental groups and found that the severity of colitis was associated with significant changes in the microbiota (Fig4C–E, Supplementary Table S4). UniFrac analyses revealed that fecal bacterial diversity in PBS- and NCK56-treated Rag1−/− mice was modified in such a way that these groups were found to cluster separately from each other and from the protected mice (Fig4C). Conversely, SlpA-, NCK2187-, and Treg-treated groups clustered together and showed similar phyla distributions (Fig4C and D). Induced colitis in PBS- and NCK56-treated groups resulted in a significant contraction of members of the Bacteroidetes phyla (Fig4D). Additionally, the normally underrepresented Verrucomicrobia phyla were increased in these colitogenic mice (Fig4D), suggesting a shift in the intestinal milieu and the substrates available in the inflamed colon, which may promote the growth of previously underrepresented microbial communities so that they dominate the population. Alterations in the microbial composition were also manifested at lower taxonomic levels: NCK2187-, SlpA-, and Treg-treated groups once again showed similar relative abundance and distribution of several unclassified genera (Fig4E).
Figure 4.

Lactobacillus acidophilus NCK2187 and its SlpA protect intestinal barrier function and prevent dysbiosis in pathogenic T-cell-induced colitis
- A, B Colonic expression of tight junction-associated genes Cldn3 and Ocln, determined by RT–PCR relative to 18S rRNA (A), as well as passive transepithelial absorption of FITC-dextran (B), were used as measures of epithelial barrier integrity. Sham adoptive transferred B6 Rag1−/− mice (white bars) were used as baseline controls in some cases. n = 5 mice/group. Data represent three individual experiments and are shown as mean ± SEM. *P < 0.05, **P < 0.01. Black asterisks compare NCK2187 to PBS-treated adoptively transferred mice, and red asterisks to NCK56-treated mice.
- C UniFrac analyses were used to calculate distances between the microbial communities of the different samples (week 7), and three-dimensional scatter plots were generated by using principal coordinate analyses (PCoA). Gray dots = CD4+CD45RBhi T cells + PBS; yellow dots = CD4+CD45RBhi T cells + NCK56; green dots = CD4+CD45RBhi T cells + NCK2187; blue dots = CD4+CD45RBhi T cells + SlpA; red dots = CD4+CD45RBhi T cells + Tregs. Each dot represents the fecal microbiota data of an individual mouse.
- D Changes in the relative abundance of different phyla were also analyzed. See Supplementary Table S4 for results of statistical analyses among the different groups. Dark blue = Actinobacteria; green = Bacteroidetes; red = Proteobacteria; yellow = Verrucomicrobia; aqua = Firmicutes; purple = Tenericutes; black = others.
- E Comparison of microbial communities at family or order levels. The heat map depicts the relative value in individual mice. n = 5–6 mice/group.
To further investigate the regulatory role of L. acidophilus SlpA, we then selected an experimentally infectious model using Citrobacter rodentium that results in a breach of the intestinal epithelial barrier, potentially orchestrated by unrestrained proinflammatory immune responses (Lebeis et al, 2009). Accordingly, data demonstrate that treatment with NCK2187 and its purified SlpA significantly accelerated pathogen clearance (Supplementary Fig S2A), resulting in reduced size of the draining lymph nodes (Supplementary Fig S2B), and decreased colonic IL-1β expression (Supplementary Fig S2C). Conversely, this trend was not observed in C. rodentium-infected mice that were treated with NCK56 or PBS, suggesting that L. acidophilus SlpA regulates induced proinflammation (e.g. IL-1β), which results in less colonic damage and bloody diarrhea (Supplementary Fig S2D and E). Histologic analyses of colonic mucosal damage (e.g. goblet cell loss, abnormal crypts) and inflammation with C. rodentium alone or C. rodentium plus NCK56 revealed increased lymphoplasmacytic with lesser but mildly increased neutrophilic infiltrates within the lamina propria and colonic submucosae, which were decreased in L. acidophilus SlpA-treated groups (Supplementary Fig S2D). Future studies are warranted to elucidate the cellular and molecular mechanisms involved in such regulatory protective intestinal responses upon GI pathogen challenge.
Lactobacillus acidophilus SlpA binding to SIGNR3 promotes colonic regulatory immune responses
Symbiotic bacteria and their gene products dictate the nature of innate responses via their sensing receptors (Ivanov & Honda, 2012; Yang et al, 2014); however, such stimulatory signals must be regulated by other receptors to avoid intestinal inflammation. As stated earlier, SIGNR3 exhibits the most biochemical similarity to human DC-SIGN. We screened all known murine SIGNR1–8 and found the Signr1 and Signr3 genes to be differentially activated in the colonic tissue of mice orally treated with NCK2187 (Fig5A), prompting us to evaluate the binding of SlpA to SIGNR1 and SIGNR3. Subsequently, the corresponding extracellular domains of SIGNR1 and SIGNR3 were fused to the Fc portion of human IgG1 (SIGNR1-hFc, SIGNR3-hFc) and then transiently expressed in Chinese hamster ovary (CHO)-S cells (Eriksson et al, 2013) (Supplementary Fig S3). Data demonstrate that while expressed SIGNR3-hFc bound to purified SlpA coated onto charged ferrous beads, SIGNR1-hFc, Ctrl-hFc (control protein tagged with hFc), and the secondary rat anti-human Fc antibody alone did not, suggesting SlpA-binding specificity to SIGNR3 (Fig5B). Additionally, we observed the binding of L. acidophilus SlpA to full-length SIGNR3 but not SIGNR1 expressed on CHO-S cells, indicating once again receptor-binding specificity to this bacterial protein (Fig5C). Moreover, given that SIGNR3 was found to be the mouse ortholog of DC-SIGN, most closely resembling its human homolog in terms of ligand binding (Powlesland et al, 2006), in vitro binding assays confirmed that purified SlpA binds to DC-SIGN-Fc (Fig5D), as well as to CHO-S cells expressing DC-SIGN (Fig5E).
Figure 5.

- A B6 mice were orally gavaged with 109 CFU NCK56 or NCK2187, and the colonic gene expression of C-type lectin receptors were measured by RT–PCR. Each box represents an individual mouse; n = 4. Data represent three individual experiments.
- B Binding of SlpA to various hFc fusion proteins was analyzed by flow cytometry. Gray tinted line = SlpA-coated beads only; orange = SlpA-coated beads + secondary antibody; green = SlpA-coated beads + control fusion protein; blue = SlpA-coated beads + SIGNR1-hFc; red = SlpA-coated beads + SIGNR3-hFc. Binding assay results were confirmed five independent times.
- C Binding of SlpA to SIGNR3, but not SIGNR1 expressed by CHO-S cells. Gray tinted line = untransfected CHO-S cells; blue = untransfected CHO-S cells + labeled SlpA; green = SIGNR1-transfected CHO-S cells + labeled SlpA; red = SIGNR3-transfected CHO-S cells + labeled SlpA. Binding assays in CHO-S cells were performed three individual times.
- D Binding of SlpA to recombinant human DC-SIGN/CD209-Fc chimera was analyzed by flow cytometry. Gray tinted line = SlpA-coated beads only; orange = SlpA-coated beads + secondary antibody; green = SlpA-coated beads + control fusion protein; red = SlpA-coated beads + DC-SIGN-Fc. Binding assays were performed and confirmed three times.
- E Binding of SlpA to DC-SIGN expressed by CHO-S cells. Gray tinted line = untransfected CHO-S cells; blue = untransfected CHO-S cells + labeled SlpA; red = DC-SIGN-transfected CHO-S cells + labeled SlpA. Binding assays in CHO-S cells were performed three individual times.
- F, G IL-1β production by colonic DCs of naïve WT B6 GF or B6 Signr3−/− (KO) mice treated with NCK56 (blue) or NCK2187 (green) four times, or left untreated (black), as determined by flow cytometry. n = 5 mice/group. Data represent four individual experiments and are shown as mean ± SEM. *P < 0.05. Black asterisks compare NCK2187 to untreated (PBS) mice, and red asterisks to NCK56-treated mice.
- H Frequency of colonic FoxP3+ Tregs in KO mice treated with NCK56 or NCK2187 was measured by flow cytometry. n = 5 mice/group. Data represent four individual experiments and are shown as mean ± SEM.
To clarify the role of NCK2187–SlpA/SIGNR3 binding and signaling in vivo, we first orally treated WT B6 and B6 Signr3−/− (KO) mice with our bacterial strains and analyzed the immunologic responses induced in steady state. While NCK2187 treatment led to reduced IL-1β production in both conventional (not shown) and germ-free (GF) B6 mice (Fig5F), no anti-inflammatory effects were observed in NCK2187-treated KO mice (Fig5G). Furthermore, the Treg-promoting properties of NCK2187 (Fig2A and B) were abrogated in KO mice (Fig5H). These data strongly suggest that NCK2187 delivers immunoregulatory signals via its interaction with SIGNR3.
NCK2187 and its SlpA do not prevent dextran sulfate sodium (DSS)-induced colitis in the absence of SIGNR3 signaling
Previous reports have highlighted the role of specific CLRs in experimental colitis. For instance, mice lacking Signr1 expression are less susceptible to induced colitis (Saunders et al, 2010), while mice deficient in Dectin1 and Signr3 exhibit exacerbated disease (Iliev et al, 2012; Eriksson et al, 2013). To further investigate SlpA/SIGNR3 signaling in disease conditions, DSS-treated WT and Signr3−/− mice were orally gavaged with NCK56, NCK2187, or SlpA, and monitored for disease progression. Consistent with our T-cell-induced colitis model, disease progression and severity were significantly reduced in WT mice orally treated with NCK2187 or purified SlpA (Fig6, Supplementary Table S5); however, NCK2187 and SlpA did not confer any protection in KO mice (Fig6). Measurements included weight loss, histopathology-based colitis scores, evaluation of gross mucosal damage, and immune cell recruitment and activation (Figs6 and 7).
Figure 6.

Lactobacillus acidophilus NCK2187 and its SlpA do not protect against DSS-induced colitis in Signr3−/− mice
- A Colitis severity was determined in part by weight loss. n = 5 mice/group.
- B, C Colitis scores based on histopathology and gross morphology of the colons were also used as measures of disease. Scale bar = 50 μm. n = 5 mice/group. Empty bars = WT; lined bars = KO; white bars = untreated; purple bars = DSS; red bars = DSS + NCK56; green bars = DSS + NCK2187; blue bars = DSS + SlpA.
- D Colonoscopies were performed in the different groups with a Multi-Purpose Rigid™ Telescope attached to a TELE PACK X on day 10.
- E Mean relative colonic expression of tight junction-associated genes in WT mice. n = 5 mice/group.
- F Fecal albumin levels in WT mice as a measure of intestinal permeability. n = 5 mice/group.
- G UniFrac analyses were used to calculate distances between the microbial communities of the different samples (day 10), and three-dimensional scatter plots were generated by using PCoA. Light gray: WT + DSS; green: WT + DSS + NCK56; brown: WT + DSS + NCK2187; blue: WT + DSS + SlpA; aqua: KO + DSS; red: KO + DSS + NCK56; yellow: KO + DSS + NCK2187; purple: KO + DSS + SlpA. n = 4–6 mice/group.
- H Species richness and microbial diversity in DSS-treated mice. Top: The Chao richness index was used as a measure of species richness. Bottom: The Shannon diversity index was used to estimate microbial diversity for each group. n = 4–6 mice/group. Color coding is as in (B).
Figure 7.

Lactobacillus acidophilus NCK2187 and its SlpA do not prevent immune infiltration and activation during DSS-induced colitis in Signr3−/− mice
- A Representative plots indicate the frequency of neutrophils in the colons of untreated or DSS-treated WT (left) and KO mice (right). Empty bars = WT; lined bars = KO; white bars = untreated; purple bars = DSS; red bars = DSS + NCK56; green bars = DSS + NCK2187; blue bars = DSS + SlpA.
- B, C Colonic DCs and macrophages (MΦs) were analyzed by flow cytometry for the production of IL-1β (B), and colonic FoxP3+ Tregs were evaluated for co-expression of RORγt+ (C). n = 5 mice/group. Gray tinted line = isotype control; black = untreated; purple = DSS; red = DSS + NCK56; green = DSS + NCK2187; blue = DSS + SlpA.
Disease progression and inflammation were associated with alterations in intestinal barrier integrity and the composition of the gut microbiota (Fig6E–H). Several gut permeability markers were evaluated by RT–PCR. The restoration of claudins (Cldn1, Cldn3, and Cldn5) in WT mice treated with NCK2187 or SlpA indicates that NCK2187 and SlpA were capable of promoting intercellular tight junctions (Fig6E).
Accordingly, only NCK2187 and SlpA treatments of WT mice prevented increased fecal albumin levels after DSS treatment, as is seen with protein-losing enteropathies (Fig6F). On the other hand, no positive effects by NCK2187 or SlpA on barrier integrity were noted in KO mice (data not shown). In terms of microbiota composition, protected WT mice (NCK2187 and SlpA treated) clustered together in UniFrac analyses, while diseased untreated and NCK56-treated WT mice clustered separately (Fig6G). Conversely, the microbial communities of all DSS-treated Signr3−/− mice formed a single cluster, indicating that dysbiosis was uniformly distributed independent of treatment group (Fig6G). Furthermore, richness and diversity, analyzed by the Chao Richness index and Shannon Diversity index, respectively, were maintained in NCK2187- and SlpA-treated WT mice, while no such effects were observed in KO mice (Fig6H).
We then analyzed induced immune responses in the colons of DSS-treated mice to determine the differences, if any, among our treatment groups. Neutrophilic infiltration within the colons of NCK2187- and SlpA-treated SIGNR3-sufficient mice that were given DSS decreased to nearly PBS-treated control mice levels; while, in contrast, an even higher frequency of infiltrating neutrophils was detected in Signr3−/− mice after the induction of colitis, irrespective of the treatment group analyzed (Fig7A, Supplementary Table S6). Similarly, the number of IL-1β-producing DCs and macrophages was significantly decreased with NCK2187 and SlpA treatment in WT mice; however, no changes were observed among the different treatment groups in the absence of SlpA/SIGNR3 signaling (Fig7B). We have previously shown, as have others, that pathogenic inflammation can result in proinflammatory FoxP3+ RORγt+ Tregs (Hovhannisyan et al, 2011; Khazaie et al, 2012). While no major change in the total number of FoxP3+ Tregs was measured among the WT and KO groups, we found that the quality of these Tregs was significantly altered. A large number of FoxP3+ cells co-expressed RORγt after DSS treatment in both WT and KO mice (Fig7C). However, in accordance with the protection observed, NCK2187 and SlpA treatment prevented the generation of FoxP3+ RORγt+ Tregs only in WT mice, but not in KO mice (Fig7C). Correspondingly, the levels of circulating cytokines in the sera (IL-1β, IL-10, IL-17A, IFN-γ, and TNF-α) were rebalanced only in NCK2187- and SlpA-treated WT mice, but not in Signr3−/− mice (Supplementary Fig S4). Collectively, these clinical and immunologic data provide robust evidence in support of an immunoregulatory role for L. acidophilus SlpA that is highly dependent on intact SIGNR3 binding.
Discussion
The human GI tract harbors trillions of microbes, most of which are bacteria (Qin et al, 2010) and are critical determinants of the health of the host (Nicholson et al, 2012; Subramanian et al, 2014). This is especially true in the case of IBD, given the intimate association of the gut microbiota and their gene products with the adjacent colonic tissue (Hold et al, 2014; Huttenhower et al, 2014). Early experiments provided evidence suggesting that susceptibility to pathogenic intestinal inflammation in experimental colitis was dependent upon the presence of enteric antigens (Kuhn et al, 1993), and were later supported by human studies, which demonstrated that an imbalance in the commensal bacterial composition, termed ‘dysbiosis,’ is a defining characteristic of patients suffering from IBD (Sokol et al, 2006; Frank et al, 2007). Accordingly, a major focus in the field has been the identification of effector bacterial strains that influence the immune system (Ahern et al, 2014), and thus may be employed to reprogram undesired immune responses, both locally and systemically.
As reviewed elsewhere (Ahern et al, 2014), it is important that the search for microbes with immunoregulatory properties accounts for differences at the strain level and not merely at the species level. Our goal has been to take this concept one step further and to identify specific bacterial molecule–host receptor interactions that may account for the responses induced by effector bacterial strains. For instance, we previously demonstrated that oral treatment using a L. acidophilus strain lacking the gene responsible for LTA biosynthesis significantly reduced pathogenic inflammation in the GI tract, thereby promoting the mitigation of induced colitis (Mohamadzadeh et al, 2011) and the ablation of colonic polyposis (Khazaie et al, 2012). Yet, the question that remained unaddressed was ‘What molecule(s) of this L. acidophilus strain deficient in LTA significantly diminish the induced pathogenic inflammation that can result in intestinal disorders?’ In addition to LTA, L. acidophilus is composed of various Slps, and several important roles have been attributed to the Slps, including immune effector properties (Konstantinov et al, 2008; Lightfoot & Mohamadzadeh, 2013).
To address this question, we systematically deleted genes using innovative strategies that allow the direct selection of double recombinants, thereby facilitating the construction of novel bacterial strains, such as NCK2187, and the assignment of roles to Slp candidate genes that are responsible for SlpA, SlpB, and SlpX protein expression (Goh et al, 2009). This sophisticated molecular approach to targeting genes in L. acidophilus defined the functional role of SlpA and demonstrated how this bacterial product affects intestinal innate cells and conventional T-cell subset activation, including Tregs, in steady state and murine colitis models. This is the first thorough study that has been carried out to rigorously examine the role of L. acidophilus SlpA in intestinal disorders using immune-mediated, infectious, and irritant-induced colitis models. As seen in Figures3 and 4, NCK2187 and its purified SlpA not only mitigated T-cell-induced colitis by significantly reducing proinflammation, but also protected the composition of the microbiota and intestinal barrier function. Additionally, systemic immune responses were also altered, whereupon the levels of proinflammatory cytokines, including IL-1β, whose detrimental role in IBD was recently demonstrated (Coccia et al, 2012), decreased significantly. These data implicate the IL-1β signaling axis in our model of intestinal protection. Importantly, a similar regulatory trend was also observed in C. rodentium-elicited inflammation, whereupon C. rodentium-infected mice fed NCK2187 or its SlpA exhibited reduced colitis symptoms, including a reduction in diarrhea, colonic atrophy, and inflammation. These data inspire the search to further elucidate molecular mechanisms that could potentially be involved, including the regulation of sensing receptors. Accordingly, gene-screening results, along with SlpA binding to SIGNR3, clearly highlight the involvement of SIGNR3 in the process of tempering highly activated gut immune responses. Additional data regarding SIGNR3 engagement using Signr3−/− mice clarified the role of this signaling molecule in induced immune regulation, as was also documented in the Leishmania infantum murine model (Lefevre et al, 2013). Our data support the notion that the SlpA/SIGNR3 interaction significantly reduces the high-affinity receptors for LTB4 in T-cell-transferred Rag1−/− mice. Downregulation of LTB4 and/or its receptors is critical in preventing inflammasome activation, which otherwise results in increased IL-1β (Lefevre et al, 2013). Interestingly, interrupting the interaction between SlpA and SIGNR3 resulted in hyperactive immunity and the production of IL-1β in KO mice under inflammatory conditions. Such dysregulated immune responses in KO mice promoted neutrophilic infiltration and significantly affected the function of colonic FoxP3+ Tregs, which were converted to proinflammatory FoxP3+ RORγt+ Tregs, all of which significantly contributed to pathologic inflammation, a condition seen in IBD progression. In contrast, balanced immunity was restored in WT mice that were treated with NCK2187 or SlpA. Induced colonic inflammation in WT mice that were treated with NCK56, but not in NCK2187- or SlpA-treated mice, and in Signr3−/− mice, regardless of treatment, resulted in microbial dysbiosis and barrier dysfunction, another hallmark of IBD.
In conclusion, we have shown that the interaction of SlpA with SIGNR3 can impact the status of innate and T-cell polarization in induced colitis. Our findings suggest that effective modulation of these cellular and molecular factors might significantly modify pathogenic inflammation that results in colitis and would therefore restore intestinal homeostasis by rebalancing deteriorated immunity, the composition of the gut microbiota, and mucosal barrier function. More importantly, our data support the translational implications of SlpA/DC-SIGN interaction and the involved regulatory signals, which may advance the development of potential therapeutic approaches for autoinflammatory diseases in the future, including chronic colitis. If hyperactivated innate and T-cell subsets play critical roles in IBD progression, our data provide clues concerning intestinal cellular mechanisms, and the properties of SlpA that may be used for immune therapeutic approaches in future clinical applications.
Materials and Methods
Mice
C57BL/6 (B6) and B6 recombination-activating gene 1-deficient (Rag1−/−) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Germ-free (GF) B6 mice were obtained from the National Gnotobiotic Rodent Resource Center at the University of North Carolina and maintained in the GF facilities at the University of Florida (UF). The mouse strain 031934-UCD, B6 Cd209dtm1.1Cfg/Mmucd (Signr3−/−) was provided by the NIH-sponsored Mutant Mouse Regional Resource Center (MMRRC) National System and was backcrossed at the Max Planck Institute of Colloids and Interfaces, Berlin, Germany. Genotyping of the Signr3 gene in WT and Signr3−/− mice was performed according to a protocol provided by the Consortium for Functional Glycomics. Dr L. Morel (UF) contributed the B6 FoxP3-GFP mice. Mice were bred in-house in the animal facility at the College of Veterinary Medicine, UF. Mice were maintained under specific pathogen-free, Helicobacter-free conditions and sex-matched controls used at 6–8 weeks of age in accordance with the Animal Welfare Act and the Public Health Policy on Humane Care. Mice were randomized into the described treatment groups and disease severity scored blindly. Procedures were approved by UF's Institutional Animal Case and Use Committee (IACUC), protocol number 201406559.
Bacterial strains
The upp-counterselective gene replacement strategy (Goh et al, 2009) was used to generate an in-frame deletion of the phosphoglycerol transferase gene within NCK2030 (LTA+ SlpB− SlpX− SlpA+) to generate NCK2187 (LTA− SlpB− SlpX− SlpA+). Wild-type L. acidophilus NCFM (NCK56) and NCK2187 were propagated anaerobically in MRS broth (Difco, BD, Franklin Lakes, NJ, USA) at 37°C for 15 h. In preparation for oral treatment, bacteria were washed twice with sterile PBS, and the number of colony-forming units (CFU) was estimated by measuring the optical density at 600 nm. The concentration of each L. acidophilus strain was accordingly adjusted to the desired final concentration of 1 × 109 CFU/100 μl. To determine the clearance kinetics of the different L. acidophilus strains, groups of mice (n = 3) were orally gavaged with erythromycin-resistant (Emr) NCK56 or NCK2187 (1 × 109 CFU/100 μl/mouse). Fecal pellets were collected before gavage and every day thereafter for up to 7 days. Each fecal pellet was then resuspended in 10% MRS (0.2 g/2 ml). The homogenized material was serially diluted and plated onto MRS agar containing Em (2 μg/ml). The daily average excreted L. acidophilus was quantified. For the oral gavage of mice, each mouse received either 1 × 109 CFU of NCK56 or NCK2187 in 100 μl of PBS. Mice enrolled in steady-state studies were orally gavaged every 3 days for a total of four times, and immune changes were analyzed at day 14. The gavage schedule was determined based on the clearance kinetics of the bacterial strains in B6 mice, as all mice used in our studies are in the B6 background.
Surface layer protein A isolation
SlpA was purified from NCK2187 with LiCl. Cultures from 18-h-grown NCK2187 were pelleted at 5,000 × g for 10 min. Bacterial pellets were washed with cold PBS and re-pelleted before extraction. Pellets were resuspended in 5 M LiCl (Sigma-Aldrich, St. Louis, MO, USA) and gently stirred for 30 min, and the bacteria were removed by centrifugation (23,000 × g, 15 min). Supernatants were dialyzed against distilled water overnight using a dialysis bag with a cutoff molecular weight of 3 kDa for salt removal. The protein precipitate was dissolved in 1 M LiCl and pelleted at 23,000 × g for 30 min. The SlpA protein preparation was washed with water a minimum of three times before use. SDS–PAGE gels and proteomics analyses were used to confirm SlpA purity. For oral gavage, mice were given 150 μg of SlpA in 300 μl PBS.
Lamina propria leukocyte (LPL) preparation
Freshly isolated colons were cut into 0.5-cm sections, and intraepithelial lymphocytes were removed with a digestion buffer consisting of HBSS (GIBCO, Life Technologies, Grand Island, NY, USA) containing 5 mM EDTA (GIBCO, Life Technologies) and 10 mM HEPES (GIBCO, Life Technologies), for 20 min at 37°C. Remaining colon tissues were digested in DMEM (GIBCO, Life Technologies) supplemented with 0.25 ng/ml Collagenase Type VII (Sigma-Aldrich), 0.125 U/ml Liberase TM Research Grade (Roche Applied Science, Indianapolis, IN), 10 mM HEPES, 0.1 M CaCl2 (Sigma-Aldrich), and 5% FBS (GIBCO, Life Technologies). Three digestions of 10 min each at 37°C were performed. Single-cell suspensions obtained were combined and stained for flow cytometry-based analyses or used for ex vivo studies.
Ex vivo stimulation of colonic LPLs
Isolated colonic LPLs were co-cultured with NCK56 or NCK2187 for 12 h at 37°C. Supernatants were then collected and stored at −80°C for later cytokine analyses using Bio-Plex Pro Mouse Cytokine Immunoassay kits (Bio-Rad, Hercules, CA, USA). Activation phenotypes of DCs were analyzed by flow cytometry using the appropriate antibodies to quantify expression levels of MHC-II molecules and co-stimulatory markers.
Flow cytometry and antibodies
Colonic LPLs were stained with LIVE/DEAD Aqua Dead Cell Stain kit (Molecular Probes, Life Technologies). Washed cells were incubated with Mouse Fc Blocking Reagent (Miltenyi Biotec, Auburn, CA) as per the manufacturer's instructions before staining with combinations of the following antibodies or their corresponding isotype controls: CD45 (30-F11), CD11c (N418), CD11b (M1/70), F4/80 (BM8), GR1 (RB6-8C5), I-A/I-E MHCII (2G9), CD3 (145-2C11), CD4 (RM4-5), CD8 (53-607), Pro-IL-1β (NJTEN3)/Rat IgG1, κ, IFN-γ (XMG1.2)/Rat IgG1, κ, IL-17A (TC11-18H10.1)/Rat IgG1, κ, IL-10 (JES5-16E3)/Rat IgG2b, κ, LAP (TGF-β1, TW7-16B4)/Mouse IgG1, κ, FoxP3 (FJK-16A)/Rat IgG2a, κ, RORγt (AFKJS-9)/Rat IgG2a, κ. For the detection of intracellular cytokines, cells were fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences). Colonic T cells were stimulated with phorbol 12-myristate 13-acetate (PMA) (50 ng/ml) and ionomycin (2.5 μg/ml) in the presence of Brefeldin A (Biolegend) for 2.5 h. The Transcription Factor Fixation/Permeabilization kit from eBioscience was used for FoxP3 staining. After staining, a BD LSRFortessa (BD Biosciences) cell analyzer was used to acquire fixed cells. Data were analyzed with FlowJo software (Tree Star, Ashland, OR, USA). Antibodies and their corresponding isotype controls were purchased from eBioscience (San Diego, CA, USA), Biolegend (San Diego, CA, USA), BD Pharmingen, or R&D Systems (Minneapolis, MN, USA).
T-cell-induced colitis
In preparation for the adoptive transfer of CD45RBhi CD4+ T cells into B6 Rag1−/− mice, spleen and mesenteric lymph node (MLN) single cell suspensions obtained from healthy B6 mice were pooled and incubated in AffiniPure goat anti-mouse IgG (H+L)-coated cell culture plates (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) at 37°C for 1 h. CD4+ T cells were isolated from non-adherent cells using the CD4+ T cell Isolation Kit II (MACS, Miltenyi Biotec, San Diego, CA, USA), and CD25+ CD4+ T cells were then depleted by positive selection (MACS, Miltenyi Biotec). Bound CD25+ CD4+ T cells were collected and injected into the regulatory T cells (Tregs) group. The resulting cell suspensions after negative and positive selection were consistently comprised of > 98% CD25− CD45RBhi CD4+ T cells. Because the T-cell transfer model of colitis is a chronic inflammation model, which requires several weeks for gross inflammation to manifest, B6 Rag1−/− mice were orally gavaged once with NCK56, NCK2187, or SlpA prior to the adoptive transfer of T cells by intraperitoneal injection (i.p.); 1 day later, the mice were orally gavaged once more, and once a week for four consecutive weeks thereafter. Colitis progression was monitored by determining mouse weight loss, diarrhea development, and fecal occult blood (FOB) presence throughout the study. Stool consistency was scored as follows: 0 = normal, 2 = pasty, and 4 = watery with perianal staining.
Murine Citrobacter rodentium infection
Citrobacter rodentium infection in mice causes pathology similar to that seen with enteric infection by the human pathogens, enteropathogenic Escherichia coli and enterohemorrhagic E. coli (Luperchio et al, 2000). These Gram-negative bacteria induce attaching and effacing (AE) lesions on the intestinal epithelium (Kaper et al, 2004), which initiate pathogenic inflammation resulting in intestinal colonization (Mundy et al, 2005; Lupp et al, 2007; Stecher et al, 2007). Briefly, B6 mice were gavaged with either NCK56, NCK2187, SlpA, or PBS at days −3 and −1. Subsequently, these groups of mice were orally infected with C. rodentium (109 CFU/mouse). Mice were then treated with L. acidophilus strains, SlpA, or PBS every other day until day 14 (Supplementary Fig S2). Diarrhea and FOB scores were monitored as measures of disease progression. These groups of mice were then sacrificed on day 14. C. rodentium-induced pathology was evaluated by H&E and real-time PCR. The presence of C. rodentium in the feces that were collected 8 days post-infection were tested by using C. rodentium-specific primers (Luperchio et al, 2000).
DSS-induced colitis
WT and KO mice were treated with 3% DSS in the drinking water for 5 days (made fresh every 2–3 days) to induce colitis. Mice were monitored for disease progression through day 10 after treatment as described above. For prevention studies, mice were orally gavaged with NCK56, NCK2187, or SlpA at days −3 and −1 and then every other day after 3% DSS treatment, for a total of five gavages (two before, and 3 after 3% DSS). Due to the acute nature of this model, and the fact that our bacterial strains are cleared from B6 mice within 3 days, we employed this treatment regimen to ensure the continuous presence of the bacteria during the acute inflammatory window.
Histopathology
Colitis severity in T-cell-, DSS- and C. rodentium infection-induced colitis was determined by histopathology. Tissues were fixed, sectioned, and stained with hematoxylin and eosin (Histology Tech Services, Gainesville, FL, USA). Stained sections were blindly evaluated by a boarded veterinary pathologist. Colitis was graded based on seven parameters (0–17), as previously described (Cheng et al, 2014).
FITC-dextran intestinal permeability assay
Passive transepithelial absorption of FITC-labeled dextran (Sigma-Aldrich) in vivo was used to determine intestinal barrier function as previously described (Napolitano et al, 1996). Mice were gavaged with FITC-dextran, MW 4,000 (60 mg/100 g body weight). Blood was collected retro-orbitally after proper anesthetization; mice were sacrificed after blood collection. Fluorescence intensity in the serum was measured with a fluorimeter (485 nm excitation, 519 nm emission). FITC-dextran concentrations in the mouse sera were determined from standard curves generated by serial dilution of FITC-dextran using blank subtraction in the test samples using sera from mice that were not gavaged with the permeability tracer.
Colonoscopy of DSS- and T-cell-induced colitis mice
Macroscopic damage in the colons of B6 Rag1−/−, WT, and KO mice was visualized with a Multi-Purpose Rigid Telescope attached to a TELE PACK X (Karl Storz–Endoscope, Germany). Mice were fasted for 2–4 h, and subsequently, the colons of the living subjects were imaged under appropriate anesthetic conditions.
Real-time PCR and 16S ribosomal DNA sequencing
Colonic tissues from B6 Rag1−/−, WT, and KO mice were isolated and processed for changes in gene expression as previously described (Lightfoot et al, 2014). Microbiota analyses were performed on the Illumina Miseq (Illumina, Inc., San Diego) as outlined previously (Lightfoot et al, 2014). Primers used, as well as their sequences, are listed in Supplementary Table S7.
SIGNR1, SIGNR3, and DC-SIGN binding assays
C-type lectin receptors, SIGNR1 and SIGNR3, were fused to the Fc part of human IgG1 (SIGNR1-hFc and SIGNR3-hFc) as previously described (Eriksson et al, 2013). Briefly, the extracellular regions of murine SIGNR1 and SIGNR3 were amplified and ligated into the expression vector pFUSE-hIgG1-Fc2 (Invivogen, Toulouse, France) for expression in mycoplasma-free CHO-S cells (ATCC). Expression in CHO-S cells was driven by an hEF1-HTLV promoter, and secretion into the culture supernatant was mediated by an external IL2 signal sequence (IL2ss) (Eriksson et al, 2013). Fusion proteins were incubated with SlpA-coated beads and subsequently with anti-hFc antibodies for detection of biding to beads. Additionally, CHO-S cells were transfected with SIGNR1-, SIGNR3-, or DC-SIGN-expressing commercial vectors (Supplementary Fig S3) and 48 h post-transfection, cells were incubated with beads coated with labeled SlpA for 30 min. The recombinant human DC-SIGN/CD209-Fc chimera was purchased from R&D Systems. Binding of SlpA-coated beads (Dynabeads MyOne Carboxylic Acid, Life Technologies) to fusion proteins or CHO-S cells was analyzed by flow cytometry.
Statistical analyses
Representative data indicate mean ± SEM. Significance was determined by two-tailed unpaired t-tests for two-group comparisons (GraphPad Prism v6.0d for Mac OS X, La Jolla, CA, USA). Statistical significance for differences in weight loss, diarrhea score, and FOB score was calculated using multiple unpaired t-tests correcting for multiple comparisons with the Holm–Sidak method in Prism v6.0d.
Acknowledgments
This work was supported by NIH R01 AI093370, the Department of Defense CA111002, the NIH/NCRR Clinical and Translational Science Award, the Ocala Royal Dames, Gatorade Foundation, the Florida Breast Cancer Foundation to the University of Florida (UL1 RR029890), NIDDK NIH F32 DK101167 (YLL), and the North Carolina Agricultural Foundation. BL acknowledges funding from the German Federal Ministry of Education and Research (Fkz. 0315446) and the Collaborative Research Center (SFB) 765. The authors wish to acknowledge the NIH-sponsored Mutant Mouse Regional Resource Center (MMRRC) National System as the source of genetically altered materials for use in this study, strain 031934-UCD. The materials were produced and deposited to the MMRRC by the Consortium for Functional Glycomics supported by the National Institute of General Medical Sciences (GM62116). We thank Ms. Ashley Mila (UF), Dr. Tridib Ganguly (UF), and Mr. Brant Johnson (NCSU) for technical support.
Author contributions
All authors contributed extensively to the work presented here and discussed data at all stages of the manuscript, which contributed to the writing of the manuscript. All authors read and approved the final manuscript. YLL designed and performed experiments, analyzed data, and wrote the paper; TY performed microbiota and RT–PCR experiments and analyzed data; YJG and KS constructed the NCK2187 strain; KS purified L. acidophilus SlpA; BS purified L. acidophilus SlpA, performed experiments, and analyzed data; MZ designed and performed experiments; JLO evaluated and scored all colonic tissue sections; NC designed and performed experiments; EL performed all biostatistical analyses for interpretation of microbiota data; TJ and BL contributed reagents; TRK conceived and implemented the technology for developing L. acidophilus mutant strains; MM conceived the project, supervised the immunologic and bacteriologic aspects of the work, designed experiments, and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Review Process File
References
- Ahern PP, Faith JJ, Gordon JI. Mining the human gut microbiota for effector strains that shape the immune system. Immunity. 2014;40:815–823. doi: 10.1016/j.immuni.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- Cheng SX, Lightfoot YL, Yang T, Zadeh M, Tang L, Sahay B, Wang GP, Owen JL, Mohamadzadeh M. Epithelial CaSR deficiency alters intestinal integrity and promotes proinflammatory immune responses. FEBS Lett. 2014;588:4158–4166. doi: 10.1016/j.febslet.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, Maloy KJ. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med. 2012;209:1595–1609. doi: 10.1084/jem.20111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers S. DC-SIGN and mannosylated surface structures of Mycobacterium tuberculosis: a deceptive liaison. Eur J Cell Biol. 2010;89:95–101. doi: 10.1016/j.ejcb.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Engering A, Geijtenbeek TB, van Kooyk Y. Immune escape through C-type lectins on dendritic cells. Trends Immunol. 2002;23:480–485. doi: 10.1016/s1471-4906(02)02296-2. [DOI] [PubMed] [Google Scholar]
- Eriksson M, Johannssen T, von Smolinski D, Gruber AD, Seeberger PH, Lepenies B. The C-type lectin receptor SIGNR3 binds to fungi present in commensal microbiota and influences immune regulation in experimental colitis. Front Immunol. 2013;4:196. doi: 10.3389/fimmu.2013.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13:3–10. doi: 10.1016/j.autrev.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Goh YJ, Azcárate-Peril MA, O'Flaherty S, Durmaz E, Valence F, Jardin J, Lortal S, Klaenhammer TR. Development and application of a upp-based counterselective gene replacement system for the study of the S-layer protein SlpX of Lactobacillus acidophilus NCFM. Appl Environ Microbiol. 2009;75:3093–3105. doi: 10.1128/AEM.02502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hold GL, Smith M, Grange C, Watt ER, El-Omar EM, Mukhopadhya I. Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years? World J Gastroenterol. 2014;20:1192–1210. doi: 10.3748/wjg.v20.i5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology. 2011;140:957–965. doi: 10.1053/j.gastro.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C, Kostic AD, Xavier RJ. Inflammatory bowel disease as a model for translating the microbiome. Immunity. 2014;40:843–854. doi: 10.1016/j.immuni.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M, Rotter JI, Wang HL, McGovern DP, Brown GD, Underhill DM. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Honda K. Intestinal commensal microbes as immune modulators. Cell Host Microbe. 2012;12:496–508. doi: 10.1016/j.chom.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B, Selle K, O'Flaherty S, Goh YJ, Klaenhammer T. Identification of extracellular surface-layer associated proteins in Lactobacillus acidophilus NCFM. Microbiology. 2013;159:2269–2282. doi: 10.1099/mic.0.070755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper JB, Nataro JP, Mobley HLT. Pathogenic Escherichia coli. Nat Rev Micro. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- Khazaie K, Zadeh M, Khan MW, Bere P, Gounari F, Dennis K, Blatner NR, Owen JL, Klaenhammer TR, Mohamadzadeh M. Abating colon cancer polyposis by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc Natl Acad Sci USA. 2012;109:10462–10467. doi: 10.1073/pnas.1207230109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinov SR, Smidt H, de Vos WM, Bruijns SC, Singh SK, Valence F, Molle D, Lortal S, Altermann E, Klaenhammer TR, van Kooyk Y. S layer protein A of Lactobacillus acidophilus NCFM regulates immature dendritic cell and T cell functions. Proc Natl Acad Sci USA. 2008;105:19474–19479. doi: 10.1073/pnas.0810305105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kooyk Y, Geijtenbeek TB. DC-SIGN: escape mechanism for pathogens. Nat Rev Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Lebeis SL, Powell KR, Merlin D, Sherman MA, Kalman D. Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun. 2009;77:604–614. doi: 10.1128/IAI.00907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre L, Lugo-Villarino G, Meunier E, Valentin A, Olagnier D, Authier H, Duval C, Dardenne C, Bernad J, Lemesre JL, Auwerx J, Neyrolles O, Pipy B, Coste A. The C-type lectin receptors dectin-1, MR, and SIGNR3 contribute both positively and negatively to the macrophage response to Leishmania infantum. Immunity. 2013;38:1038–1049. doi: 10.1016/j.immuni.2013.04.010. [DOI] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Lightfoot YL, Mohamadzadeh M. Tailoring gut immune responses with lipoteichoic acid-deficient Lactobacillus acidophilus. Front Immunol. 2013;4:25. doi: 10.3389/fimmu.2013.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfoot YL, Yang T, Sahay B, Zadeh M, Cheng SX, Wang GP, Owen JL, Mohamadzadeh M. Colonic immune suppression, barrier dysfunction, and dysbiosis by gastrointestinal bacillus anthracis infection. PLoS ONE. 2014;9:e100532. doi: 10.1371/journal.pone.0100532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luperchio SA, Newman JV, Dangler CA, Schrenzel MD, Brenner DJ, Steigerwalt AG, Schauer DB. Citrobacter rodentium, the causative agent of transmissible murine colonic hyperplasia, exhibits clonality: synonymy of Citrobacter rodentium and mouse-pathogenic Escherichia coli. J Clin Microbiol. 2000;38:4343–4350. doi: 10.1128/jcm.38.12.4343-4350.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of enterobacteriaceae. Cell Host Microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nat Immunol. 2011;12:5–9. doi: 10.1038/ni0111-5. [DOI] [PubMed] [Google Scholar]
- McDermott AJ, Huffnagle GB. The microbiome and regulation of mucosal immunity. Immunology. 2014;142:24–31. doi: 10.1111/imm.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamadzadeh M, Pfeiler EA, Brown JB, Zadeh M, Gramarossa M, Managlia E, Bere P, Sarraj B, Khan MW, Pakanati KC, Ansari MJ, O'Flaherty S, Barrett T, Klaenhammer TR. Regulation of induced colonic inflammation by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc Natl Acad Sci USA. 2011;108(Suppl. 1):4623–4630. doi: 10.1073/pnas.1005066107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. 2005;7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- Napolitano LM, Koruda MJ, Meyer AA, Baker CC. The impact of femur fracture with associated soft tissue injury on immune function and intestinal permeability. Shock. 1996;5:202–207. doi: 10.1097/00024382-199603000-00006. [DOI] [PubMed] [Google Scholar]
- Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- Osorio F, Reis e Sousa C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity. 2011;34:651–664. doi: 10.1016/j.immuni.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Powlesland AS, Ward EM, Sadhu SK, Guo Y, Taylor ME, Drickamer K. Widely divergent biochemical properties of the complete set of mouse DC-SIGN-related proteins. J Biol Chem. 2006;281:20440–20449. doi: 10.1074/jbc.M601925200. [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho D, Reis e Sousa C. Sensing of cell death by myeloid C-type lectin receptors. Curr Opin Immunol. 2013;25:46–52. doi: 10.1016/j.coi.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders SP, Barlow JL, Walsh CM, Bellsoi A, Smith P, McKenzie AN, Fallon PG. C-type lectin SIGN-R1 has a role in experimental colitis and responsiveness to lipopolysaccharide. J Immunol. 2010;184:2627–2637. doi: 10.4049/jimmunol.0901970. [DOI] [PubMed] [Google Scholar]
- Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A, Gaggar A, Shastry S, Rowe SM, Shim YM, Hussell T, Blalock JE. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science. 2010;330:90–94. doi: 10.1126/science.1190594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Seksik P, Rigottier-Gois L, Lay C, Lepage P, Podglajen I, Marteau P, Doré J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:106–111. doi: 10.1097/01.MIB.0000200323.38139.c6. [DOI] [PubMed] [Google Scholar]
- Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, Dougan G, von Mering C, Hardt WD. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, Barratt MJ, VanArendonk LG, Zhang Q, Province MA, Petri WA, Ahmed T, Gordon JI. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature. 2014;509:417–421. doi: 10.1038/nature13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanne A, Ma B, Boudou F, Tailleux L, Botella H, Badell E, Levillain F, Taylor ME, Drickamer K, Nigou J, Dobos KM, Puzo G, Vestweber D, Wild MK, Marcinko M, Sobieszczuk P, Stewart L, Lebus D, Gicquel B, Neyrolles O. A murine DC-SIGN homologue contributes to early host defense against Mycobacterium tuberculosis. J Exp Med. 2009;206:2205–2220. doi: 10.1084/jem.20090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin DM, Vary JC, Jr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, Hagge DA, Khadge S, King MC, Hawn TR, Moens CB, Ramakrishnan L. The lta4 h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140:717–730. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature. 2014;510:152–156. doi: 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Review Process File