

Abstract
Major efforts are invested to characterize the factors controlling the proliferation of neural stem cells. During mammalian corticogenesis, our group has identified a small pool of genes that are transiently downregulated in the switch of neural stem cells to neurogenic division and reinduced in newborn neurons. Among these switch genes, we found Tox, a transcription factor with hitherto uncharacterized roles in the nervous system. Here, we investigated the role of Tox in corticogenesis by characterizing its expression at the tissue, cellular and temporal level. We found that Tox is regulated by calcineurin/Nfat signalling. Moreover, we combined DNA adenine methyltransferase identification (DamID) with deep sequencing to characterize the chromatin binding properties of Tox including its motif and downstream transcriptional targets including Sox2, Tbr2, Prox1 and other key factors. Finally, we manipulated Tox in the developing brain and validated its multiple roles in promoting neural stem cell proliferation and neurite outgrowth of newborn neurons. Our data provide a valuable resource to study the role of Tox in other tissues and highlight a novel key player in brain development.
Keywords: brain development, DamID sequencing, HMG-box transcription factors, neural stem cells, Tox
See also: M Karow & B Berninger (April 2015)
Introduction
During embryonic development of the mammalian brain, neural stem and progenitor cells progressively switch from proliferative divisions that expand the stem cell pool to differentiative divisions that generate neurons and glia (Kriegstein & Alvarez-Buylla, 2009; Taverna et al, 2014). Understanding the molecular mechanisms underlying this switch is a major goal in developmental and stem cells biology both for basic research and for potential applications in regenerative therapy (Goldman, 2005; Lindvall & Kokaia, 2006). Over the years, several transcription factors and signalling molecules have been characterized that are instrumental in the regulation of corticogenesis (Guillemot, 2007; Pinto & Gotz, 2007; Paridaen & Huttner, 2014). Yet, many other factors remain elusive or await further characterization.
Next-generation sequencing has recently provided the field with a powerful tool to identify novel factors involved in corticogenesis (Han et al, 2009; Ayoub et al, 2011; Fietz et al, 2012; Yao et al, 2012), but major limitations remain in the identification and sorting of individual cell types for comparative analyses. Specifically, in the rodent brain the two germinal layers of the ventricular and sub-ventricular zone (VZ and SVZ, respectively) consist of a varying proportion of intermingled progenitors undergoing proliferative or differentiative divisions (Kriegstein & Alvarez-Buylla, 2009; Taverna et al, 2014). The vast majority of proliferative progenitors (PP) are confined to the VZ where they divide at its apical boundary and are for this reason also referred to as apical progenitors. In contrast, differentiating progenitors (DP) migrate to the basal boundary of the VZ to form the SVZ and are for this reason also referred to as basal progenitors (Kriegstein & Alvarez-Buylla, 2009; Taverna et al, 2014). While apical and basal progenitors represent, overall, the vast majority of PP and DP, respectively, during development an increasing proportion of apical progenitors switch their fate to become DP and generate basal progenitors or neurons (Haubensak et al, 2004; Miyata et al, 2004; Noctor et al, 2004). Conversely, a small proportion of basal progenitors can become PP to expand their pool within the SVZ (Attardo et al, 2008; Ochiai et al, 2009). Finally, newborn neurons generated in the VZ or SVZ by apical or basal DP, respectively, migrate through the intermediate zone (IZ) to form the cortical plate (CP) (Kriegstein & Alvarez-Buylla, 2009; Taverna et al, 2014). Hence, difficulties in the isolation of intermingled PP and DP, and identification of either progenitor type from neurons, limited the use of next-generation sequencing to comparisons of developmental stages and portions of tissues or species rather than individual cell types (Han et al, 2009; Ayoub et al, 2011; Fietz et al, 2012; Yao et al, 2012).
Our group has recently overcome this limitation by generating a dual-reporter, Btg2RFP/Tubb3GFP mouse line in which PP (RFP−/GFP−), DP (RFP+/GFP−) and neurons (GFP+, irrespective from RFP) were identified by the combinatorial expression of two fluorescent reporters (Aprea et al, 2013). Transcriptome sequencing of the three cell types led to the identification of ∽200 transcripts being upregulated solely in DP and downregulated in both PP and neurons (on-switch genes). Conversely, a similarly abundant pool of genes was downregulated in DP and upregulated in both PP and neurons (off-switch genes). Supporting the notion that switch genes characterize the signature of neurogenic commitment, it was found that essentially all neurogenic markers were on-switches (Aprea et al, 2013). Moreover, confirming that a remarkably high proportion of switch genes are functionally relevant in corticogenesis, manipulation of novel or uncharacterized on/off-switch transcripts in vivo led to the identification of several genes with hitherto unknown functions in brain development (Aprea et al, 2013, 2015; B. Artegiani, J. Aprea, S. Bragado Alonso, F. Calegari, unpublished data).
Surprisingly, and in stark contrast to on-switch genes, our group has found that only a remarkably small number of off-switches have ever been studied in the context of cortical development (Aprea et al, 2013). This bias and under-representation of off-switch genes with functional roles in brain development was puzzling since it can be inferred that both on- and off-switches should be equally important. Hence, to challenge our assumption, we next sought to characterize novel off-switch genes with unknown roles in neural stem cell commitment.
Among these, we found Tox (thymocyte selection-associated HMG-box) that is downregulated 2.7-fold in the switch from PP to DP and upregulated 3.0-fold in newborn neurons relative to DP (Aprea et al, 2013). Tox is a highly conserved transcription factor belonging to the family of HMG-box proteins and being expressed in the thymus, liver and brain (Wilkinson et al, 2002; Aliahmad et al, 2012). Tox downstream targets and its binding motif are currently unknown but, like Sox proteins, its single HMG-box domain is expected to confer sequence specificity in DNA binding (Stros et al, 2007). Yet, analysis of Tox amminoacidic sequence has led to the opposite conclusion suggesting that its binding is structure, rather than sequence, specific (O'Flaherty & Kaye, 2003).
Until now, Tox was only studied in the context of the immune system where it is required for lymphoid tissue organogenesis and regulation of T-cell and natural killer cell development during hematopoiesis (Wilkinson et al, 2002; Aliahmad et al, 2004, 2010; Yun et al, 2011). Expression pattern and functional relevance of this transcription factor in any other tissue, including the nervous system, are completely uncharacterized. For these reasons, we decided to characterize: (i) the upstream molecular mechanisms controlling Tox expression during development; (ii) its downstream targets; and (iii) functional role during the fate switch of PP to DP and maturation of postmitotic neurons during mammalian cortical development.
Results
Tox expression during corticogenesis recapitulates the gradients of neurogenesis
First, we analysed Tox expression by immunohistochemistry at the onset, mid and end of cortical neurogenesis as well as in the adult brain. At embryonic day (E) 9.5, Tox displayed a rostral-high/caudal-low and lateral-high/medial-low gradient of expression (Fig1A, top-left) that closely resembled the known gradients of differentiation at the onset of cortical neurogenesis (Caviness et al, 2009). Accordingly, the proportion of Tox+ neuroepithelial cells varied from nearly 100% to essentially 0%. Moreover, the very few Tubb3+ neurons that could be found at this stage were Tox− (Supplementary Fig S1A), suggesting that newborn neurons do not express Tox. Similar results were obtained 3 days later at E12.5, again showing a rostral-high/caudal-low and lateral-high/medial-low gradient of expression within the neuroepithelium with, additionally, Tox+ neurons particularly at the level of the prospective olfactory bulb and mid/hindbrain regions (Fig1A, top-right; and data not shown).
Figure 1.

- A-C Fluorescence pictures of WT (A and B) or Btg2RFP (C) mouse brains at E9.5, E12.5, E15.5 or E18.5 (as indicated) after immunohistochemistry for Tox (A-C; red), Tubb3 (B; green), Sox2 (C; white) or RFP (C; magenta) and DAPI counterstaining (A; blue).
- D Quantification of Sox2+ or Btg2RFP+ cells within the Tox+ population (left; n = 3; error bars = SD) and immunoreactivity levels (au = arbitrary units) of each of the two makers relative to Tox represented as a linear correlation (middle; n = 79) or box plot (right; n ≥ 24), respectively. ***P < 0.001. Scale bars, 50 μm (A) or 40 μm (B).
At E15.5, the rostro-caudal gradient of Tox expression essentially disappeared with virtually all cells in the VZ being Tox+ (Fig1A, bottom-left). In contrast, a lateral-medial gradient was still detectable (Fig1A, bottom-right) that was reminiscent of the gradients of expression reported for the neurogenic markers Tbr2 and Ngn2 (Englund et al, 2005; Britz et al, 2006). Although generally abundant in progenitors of the VZ, Tox was undetectable in basal progenitors of the SVZ and was reinduced in most neurons of all areas analysed (Fig1A, bottom and Supplementary Fig S1B). With regard to the latter cell pool, we observed that immature, migrating neurons in the IZ displayed a much lower, if any, expression of Tox as compared to more differentiated neurons in the CP that were essentially all Tox+ (Fig1B). This observation is consistent with analyses at E9.5 and E12.5 showing that Tox expression is restricted within the subset of more mature neurons. Overall, Tox immunoreactivity recapitulated its mRNA levels previously assessed by transcriptome analysis of PP, DP and neurons (Aprea et al, 2013) although the overlap with the neurogenic gradients and difference among IZ and CP neurons could not be detected by pooling together cortical areas and neuron subtypes.
Co-labelling with cellular markers identifying the different classes of neural progenitors was performed to characterize the identity of Tox+ cells at mid-corticogenesis at E15.5. In the VZ, essentially all (95.8 ± 2.6%) Tox+ cells were positive for the proliferative marker Sox2 (Fig1C and D, left). Conversely, many Sox2+ cells did not express Tox due to its gradients of expression already mentioned (Supplementary Fig S1B). Interestingly, quantification of Sox2 immunoreactivity within Tox+ nuclei of individual cells showed a clear positive correlation with the levels of Tox (Fig1D, middle). This held true in different regions of the VZ despite the fact that each region varied with regard to the proportion of Tox+ cells (Supplementary Fig S1B and data not shown). To identify VZ progenitors committed to differentiative divisions, we next used the Btg2RFP reporter mouse that revealed not only that in the VZ only a small proportion of Tox+ cells were Btg2+ (14.5 ± 2.4%) but also that in Btg2+ cells the level of Tox was significantly lower than in Btg2− cells (Fig1D). In contrast, the second germinal layer, the SVZ, was virtually deficient of Tox+ cells in all regions analysed (Fig1C), thus indicating a remarkably tight control in Tox protein levels in different progenitor types.
We next analysed Tox expression at the end of the neurogenic interval at E18.5. Tox was maintained in the germinal layers, and at this stage, the lateral-medial gradient spread much further than at E15.5 and was almost undetectable with essentially all cells in the VZ of the lateral cortex being Tox+ (Fig1A, bottom-right). The only germinal zone in which Tox+ cells were undetectable was the prospective hippocampus that is known to have a prolonged neurogenic interval extended to postnatal life. Again, no significant expression of Tox was observed within the residual SVZ being detectable at this stage (data not shown). With regard to neurons, Tox was enriched in deeper layer neurons of the CP with its expression broadly overlapping with the deep layer marker Ctip2 and detected also in a proportion of double-positive Ctip2+/Satb2+ neurons (Fig1A, bottom-right, Supplementary Fig S1C and further discussed below).
Finally, we found that Tox in the adult brain was maintained in the sub-ventricular zone but, surprisingly, not in the second adult neurogenic niche of the hippocampus that was never found to express this transcription factor throughout development (Supplementary Fig S1D and D′, and data not shown). In addition, Tox in the adult cortex was still expressed in the majority (71.7 ± 10.2%) of Ctip2+ neurons (Supplementary Fig S1D and D′′), suggesting that Tox in the neuronal compartment identifies a specific sub-class of cortical neurons during both embryonic and postnatal life.
All together, these data show that Tox expression in progenitors cells of the developing VZ strongly correlates with the temporal and spatial gradients of neurogenesis. Perhaps counterintuitively, Tox expression within neurogenic areas was found to be higher in cells undergoing proliferative, rather than neurogenic, division such as Sox2+ and/or Btg2− cells and apical, rather than basal, progenitors. The substantial decrease in Tox expression in DP of the VZ and SVZ and reinduction in CP neurons further indicated that this transcription factor is tightly regulated at the transcriptional and, possibly, post-translational level. In fact, analysis of Tox protein sequence identified four sites of ubiquitination suggesting that this transcription factor is actively degraded in the switch from PP to DP (data not shown). Its reinduction in a specific sub-class of neurons, primarily Ctip2+, may in turn suggest that Tox plays different roles in progenitors and neurons and, possibly, in different neurogenic niches of the adult brain.
Tox is regulated by the calcineurin/Nfat signalling pathway
To gain insight into the molecular mechanisms regulating Tox expression during corticogenesis, we took advantage of previous evidence linking calcineurin activity to Tox levels in thymocytes (Aliahmad et al, 2004) as well as our own analysis by NetPhos predicting several phosphorylation sites in the Tox sequence (data not shown). Calcineurin is a Ca2+-dependent phosphatase involved in several biological functions including stem cell commitment and brain function (Crabtree & Olson, 2002; Horsley et al, 2008; Mukherjee & Soto, 2011; Kujawski et al, 2014). Calcineurin acts by controlling nuclear trafficking of target proteins including several transcription factors, among which the Nfat family members are some of the most characterized (Crabtree & Olson, 2002; Horsley et al, 2008). Hence, we first sought to investigate whether Tox was a direct target of calcineurin.
To this aim, we administered the highly specific, placenta-permeable calcineurin inhibitor cyclosporin A (CsA) (Liu & Janeway, 1991; Graef et al, 2003) by repetitive injections in pregnant females at E13.5 and collected embryos 48 h later (Fig2A). Western blot analyses of nuclear versus cytosolic extracts derived from either vehicle or CsA-treated mice did not show either a change in the cellular localization of Tox nor a shift in its apparent molecular weight resulting from hyper-phosphorylation (Fig2B). In contrast, we observed a 30% decrease in Tox expression levels upon CsA treatment as compared to controls (Fig2B and B′), suggesting that Tox is indirectly regulated by calcineurin rather than being a direct target of dephosphorylation and nuclear transport.
Figure 2.
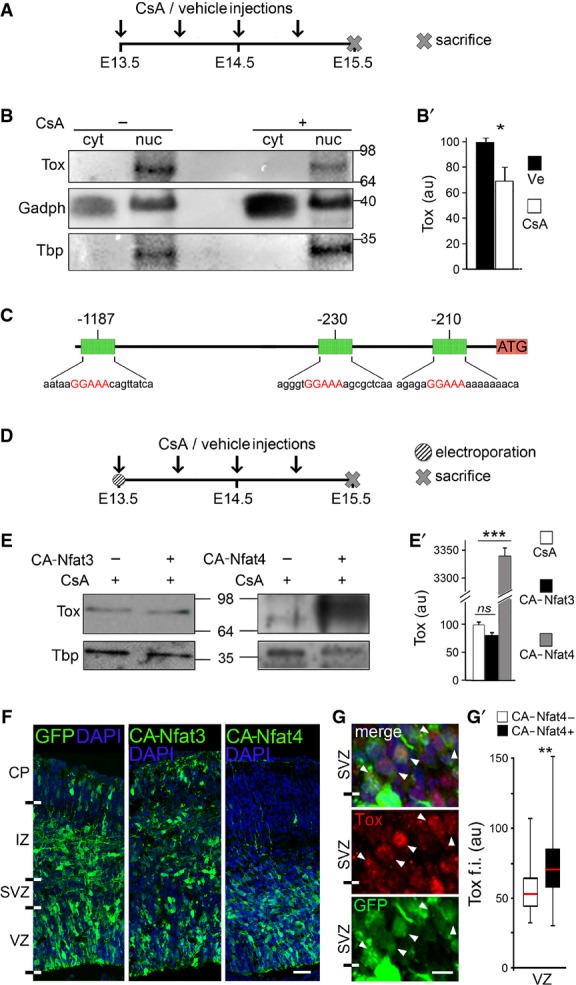
- A-B′ Layout of CsA, or vehicle as control, administration during embryonic development (A) and Western blot analysis (B) of Tox levels in cytoplasmic (cyt) or nuclear (nuc) fractions of E15.5 brains. Quantifications (B′) were performed upon internal normalization for Gadph or Tbp and referred to treatment with vehicle. n = 3; error bars = SD; *P < 0.05.
- C Map of Tox locus and Nfat-binding recognition sites (red) identified by MatInspector.
- D-E′ Layout of CsA or vehicle administration and Western blot analysis as in (A–B′) upon in utero electroporation with CA-Nfat3 or CA-Nfat4 vectors at E13.5. Tox in extracts of E15.5 brains was quantified normalized to Tbp and referred to CsA treatment. n = 3; error bars = SD; ***P < 0.001.
- F-G′ Fluorescent pictures of the mouse cortex upon electroporation with GFP, CA-Nfat3 or CA-Nfat4 (as indicated) followed by GFP (green) or Tox (G; red) immunolabelling and DAPI counterstaining (blue). Tox upregulation after CA-Nfat4 overexpression was assessed within nuclei of GFP+ cells (n = 30) in the VZ and represented as a box plot relative to GFP− cells (n = 30) used as internal control (G′). Tox ectopic expression within the SVZ is indicated (G; arrowheads). Scale bar, 50 μm (F) and 5 μm (G); **P < 0.01.
To identify calcineurin-dependent transcription factors responsible for the regulation of Tox expression by calcineurin, we then analysed its 5′-region by MatInspector and found with high-confidence three binding sites for Nfat (Fig2C). Furthermore, transcriptome analysis of PP, DP and neurons (Aprea et al, 2013) indicated that only two of the four calcineurin-dependent Nfat members are expressed at any significant level during corticogenesis allowing us to focus our attention on Nfat3(c4) and Nfat4(c3) as two candidate regulators of Tox expression through calcineurin. To address each, we next used in utero electroporation and overexpressed calcineurin-independent, constitutively active versions of either of the two Nfat members and investigated whether this manipulation rescued the downregulation of Tox induced by CsA.
Electroporation was performed at E13.5 with plasmids encoding either a constitutively active (CA) Nfat3 or Nfat4 and using a co-injected GFP vector as a reporter of targeted cells. Subsequently, pregnant mice were administered CsA for 48 h (Fig2D), embryos collected at E15.5 and the GFP+ portion of the cortex microdissected together with its contralateral side serving as an internal negative control. Western blot analyses of nuclear extracts showed unchanged levels, hence no rescue, of Tox upon overexpression of CA-Nfat3 while, in contrast, overexpression of CA-Nfat4 triggered a massive upregulation of Tox sufficient not only to rescue the effects of CsA but also to raise its levels by 30-fold (Fig2E and E′).
Consistent with the lack of effect of CA-Nfat3 on Tox, and as previously reported (Kurabayashi & Sanada, 2013), we found that the distribution of GFP+ cells across the cortical layers of the E15.5 brain upon CA-Nfat3 electroporation was undistinguishable from that of brains electroporated with control plasmids (Fig2F), indicating that overexpression of CA-Nfat3 had no obvious consequence on brain development. In contrast, interestingly, electroporation of CA-Nfat4 showed an almost complete lack of GFP+ cells in the IZ/CP (Fig2F), showing that this Nfat family member plays crucial roles in neurogenic commitment, neuronal maturation, migration and/or survival, some of which may, at least in part, depend on its effect on Tox.
Finally, we sought to corroborate our biochemical assessment of Tox upregulation induced by CA-Nfat4 (Fig2E and E′) at the cellular level and quantified Tox immunoreactivity in nuclei of randomly chosen GFP+ cells after electroporation while using GFP− cells as an internal negative control. This showed that CA-Nfat4 induced an increase by ∽35% in Tox immunoreactivity within the VZ (Fig2G and G′) and that within the SVZ several GFP+ cells started to be characterized by levels of Tox immunoreactivity comparable to VZ cells (Fig2G). The latter increase of GFP+ relative to GFP− cells in the SVZ, however, could not be assessed mathematically given the fact that the latter do not express any detectable level of Tox (Fig1C). Notwithstanding the many differences in specificity, sensitivity and saturation of signal inherent in the two methods, we conclude that the increase in Tox levels by Nfat4 as assessed by Western blot (Fig2E′) results from the combined effects observed by immunohistochemistry in the VZ and SVZ (Fig2G). Hence, these data suggest that Tox expression during corticogenesis is controlled by the calcineurin/Nfat signalling pathway and that among Nfat family members Nfat4 is the main upstream regulator of Tox.
DamID-Seq identifies the features of Tox binding to chromatin and its downstream targets
We next sought to identify the downstream transcriptional targets of Tox. However, commercially available Tox antibodies were not validated for ChIP and, in our conditions, we did not achieve immunoprecipitation with any of the antibodies tested (data not shown). Hence, as an alternative method, we decided to use DNA adenine methyltransferase identification (DamID) to characterize DNA binding of proteins by their fusion to the prokaryotic Dam-methylase resulting in adenine methylation of GATC sequences (van Steensel & Henikoff, 2000; Vogel et al, 2006, 2007; Southall & Brand, 2007) (Supplementary Fig S2A). In this system, biases in the identification of targets are minimized by the fact that Dam methylation can occur at a considerable distance from the transcription factor binding site, up to about ±2 kb, while GATC motifs have a median occurrence every 460 nucleotides in the human genome (own bioinformatic analysis), that is about eight motifs within methylation range. Furthermore, differences in chromatin accessibility, untargeted binding and methylation are controlled for by the use of unfused Dam. Of note, even recently DamID is typically combined with microarray analysis (van Bemmel et al, 2013; Southall et al, 2014), which is known to provide a lower spatial resolution than ChIP-Seq in defining binding domains (Shimbo et al, 2013). Therefore, inspired by reports on nuclear envelop proteins (Wu & Yao, 2013), we explored the use of high-throughput sequencing combined with DamID (DamID-Seq) to increase both the spatial resolution and quantitative read-out of transcription factor binding to chromatin regions.
DamID was performed in HEK-293T and Neuro-2a cells in parallel since both were expected to provide specific advantages. In particular, we considered HEK-293T as a system that should in principle allow the identification of Tox targets independently of any bias towards a specific lineage. Conversely, Neuro-2a may have provided an in vitro model for the identification of neural-specific Tox targets. As a recognized key factor for the successful use of DamID (Southall & Brand, 2007; Vogel et al, 2007), we first confirmed that fusion to Dam did not perturb the nuclear localization of Tox as assessed upon its overexpression by transfection with a constitutive Tox-Dam expression vector in vitro (Supplementary Fig S2B). Moreover, DamID relies on the minimal expression of the transgenes, which is necessary for a highly specific methylation pattern without saturation. Therefore, we used highly diluted lentiviruses expressing Dam or Tox-Dam under two inducible promoters neither of which was ever induced and resulting in undetectable levels of Tox-Dam expression derived from double-leakiness. Genomic DNA was collected 2 days later, methylated-GATC fragments isolated, and high-throughput DNA sequencing performed (Supplementary Fig S2A).
Pearson's correlation analysis revealed the high reproducibility of the two biological replicates and the diversity between samples obtained after infection of HEK-293T cells with Dam or Tox-Dam viral suspensions (Fig3A). In contrast, surprisingly, Neuro-2a cells did not show any significant difference between conditions, neither using the same viral titre applied to HEK-293T cells nor a 10-fold diluted suspension (data not shown). We do not know the reason for this discrepancy among cell lines, but it is reasonable to assume that the expression of endogenous Tox will compete with Tox-Dam for DNA binding. This is unlikely to occur in HEK-293T cells since Tox is not expressed at any detectable level in this cell line (Supplementary Fig S2B). Hence, we continued our study using data obtained from HEK-293T cells.
Figure 3.

- A Pearson's correlation analysis between Tox-Dam versus Dam reads of HEK-293T cells sampling the genome in windows of 10 kb.
- B Meta-analysis of normalized Tox-Dam at the transcription start site (TSS) of Ensembl-annotated genes.
- C Profile of normalized Tox-Dam reads on cell-specific enhancers (colours; categories ordered according to enrichment). Background (grey) was assessed based on the Tox signal at random genomic locations.
- D Relationship between the fold-change of Tox peaks and their distance to the TSS. Note the enrichment in peaks at the boundary of ±15 kb from TSS and the correlation between their fold-change and proximity to the TSS.
- D′ Distribution of Tox peaks within regions of loci (as indicated). The relative length of each genomic region was chosen to represent its average length across the human genome and circles distributed to represent the proportion of Tox peaks mapping on that given region (1 circle = 1%).
- E PhastCons scores of 100 vertebrate genomes averaged for the Tox peak locations (blue) and for a matched genomic background (grey) showing higher conservation within Tox peaks (start-end).
- F Putative 10-mer binding motif of Tox identified with gimmeMotifs.
We next pooled the HEK-293T replicates together and mapped reads in each condition to assess their local enrichment. Selection of Tox-Dam peaks (P < 0.01) that were 2.5-fold enriched relative to Dam resulted in the identification of ∽13,000 chromatin regions (Supplementary File S1). Among these, as expected for a transcription factor, we found that Tox-Dam peaks were characterized by a substantial increase in the coverage of regions in proximity of transcriptional start sites (TSS) relative to Dam controls (Fig3B).
Interestingly, enrichment in Tox-Dam peaks mapping on active enhancers of different tissues (Andersson et al, 2014) was revealed to be even more prominent in enhancers of the telencephalon, neural stem cells and neurons than those of the immune system, such as macrophages and T cells (Fig3C) in which Tox function was first characterized. Furthermore, we found a strong, positive correlation between the fold-change of Tox-Dam peaks and their vicinity to a TSS (Fig3D). This feature, together with a drastic increase in the number of Tox-Dam peaks within a boundary of ±15 kb from a TSS (Fig3D), suggested that Tox has a higher affinity and/or specificity of binding within this region. Hence, we identified annotated genes within ±15 kb from a Tox-Dam peak resulting in 9,368 loci encoding potential Tox targets (Supplementary File S1). Both the number of loci and the distribution of peaks within intronic, exonic and untranslated regions of annotated genes (Fig3D′) compared well with previous characterizations of transcription factor binding by either ChIP or DamID followed by either microarray or sequencing analysis (Chen et al, 2008; Southall & Brand, 2009; Shimbo et al, 2013).
Consistent with the high evolutionary conservation of Tox family members (O'Flaherty & Kaye, 2003; and our own analysis, data not shown), we found that Tox-Dam peaks were characterized by a substantial increase in non-exonic nucleotide conservation across 100 vertebrates species (Fig3E). Finally, discovery of over-represented sequences within the 1,000 Tox peaks with the highest fold-change and up to 1 kb in length allowed us to identify a 10-mer binding motif of Tox that is rich in GC sequences (Fig3F).
Altogether, the use of DamID-Seq gave us a pipeline for studying the features of chromatin binding of this transcription factor together with a list of its putative targets, both of which were essential for further dissecting its biological function.
Tox can act as a repressor or activator of genes critical for corticogenesis
We next investigated whether the 9,368 putative targets of Tox were proportionally over-represented in functional, gene ontology (GO) terms for: (i) cellular compartment, (ii) molecular function and (iii) biological process. Using DAVID and clustering genes with redundant terms together, we found a substantial enrichment in (i) nucleoplasm and cellular protrusions (Fig4A, left), (ii) DNA binding, transcription factors, kinases and cytoskeletal proteins (Fig4A, middle) and (iii) axonogenesis, CNS development, neurogenesis and regulation of transcription (Fig4A, right). This enrichment in biological processes closely related to brain development and neuronal protrusions is particularly striking if one considers that the use of HEK-293T cells, contrary to Neuro-2a, did not imply a priori any bias towards the expression of neuronal genes.
Figure 4.
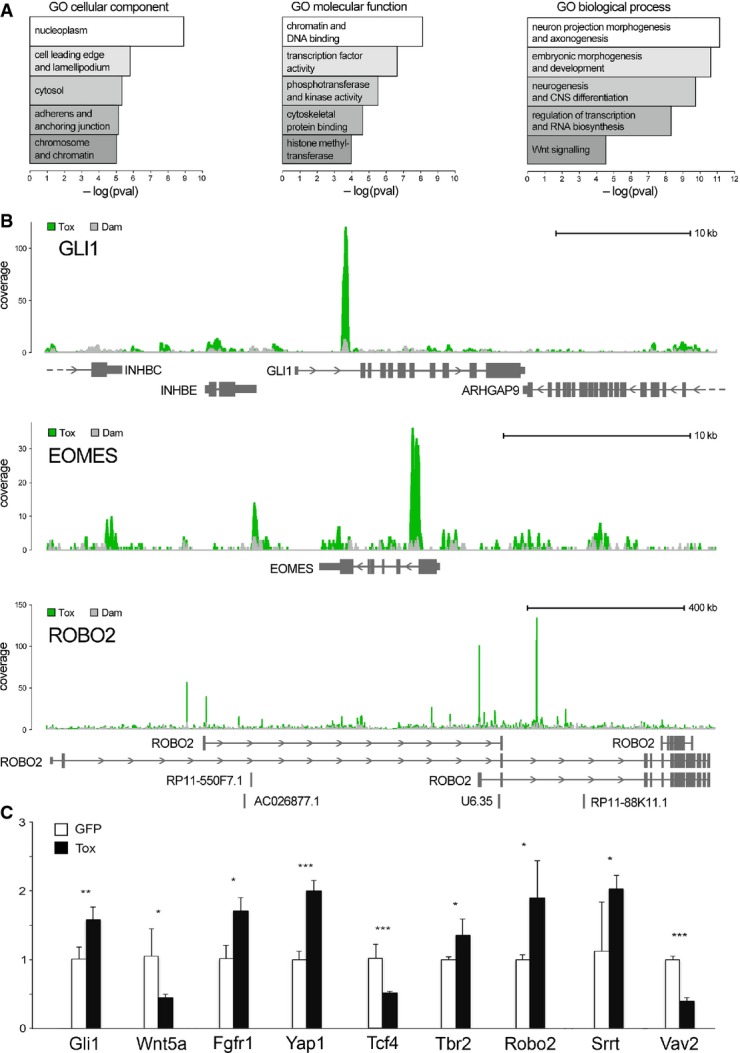
- Functional enrichment scores for cellular compartment, molecular function and biological process (left to right).
- Examples of Tox-Dam (green) or Dam (grey) peaks (superimposed in each panel) for Gli1, Eomes/Tbr2 and Robo2 (top to bottom) to represent factors involved in signalling, neurogenic commitment and neurite outgrowth, respectively.
- qRT–PCRs of GFP+ FAC-sorted cells from the E15.5 mouse brain upon electroporation at E13.5 with control (white) or Tox (black) constructs. Primers (Supplementary Table S1) were selected to amplify multi-exon transcripts (indicated) to represent the classes of genes that would explain functional annotations in (A). n ≥ 3; error bars = SD; *P < 0.05; **P < 0.01; ***P < 0.001.
Beyond genomewide analyses and clustering by GO terms, we next sought to manually inspect individual Tox peaks for at least some of the genes that are known to play key roles in CNS development including proliferation versus differentiation of neural progenitors and maturation and specification of postmitotic neurons. These included: (i) key members of the Shh, Wnt, Notch and Fgf signalling pathways (Gli1 is shown as an example in Fig4B; top); (ii) well-characterized regulators of neural stem cell fate such as Tbr2/Eomes, Sox2, Prox1, Foxp1/2 and Yap1 (e.g. Tbr2/Eomes: Fig4B; middle); and (iii) genes involved in neuronal maturation and specification such as Robo2, Tbr1, Satb2 and Erbb4 (e.g. Robo2: Fig4B; bottom). In almost all cases analysed, we found that Tox-Dam peaks were characterized by very sharp and defined increases in coverage relative to Dam controls and that the vast majority of these peaks well exceeded the 2.5-fold threshold of confidence originally considered to identify the 9,368 putative target of Tox (Supplementary File S1).
These data revealed the high resolution and confidence achieved by DamID-Seq for transcription factor binding to chromatin regions. Yet, this did not provide us with any information about the effect of this binding on the expression of those targets in tissue. In order to achieve this, and validate our approach in vivo, we performed expression profiling of the putative Tox targets mentioned above by qRT–PCR upon Tox overexpression in developing mouse embryos. For this purpose, only multi-exon targets were chosen to discriminate transcripts from genomic DNA contamination after PCR amplification.
In utero electroporation was performed at E13.5 with control or Tox-expressing vectors, each encoding GFP to identify targeted cells. Embryos were collected 48 h later and the targeted area dissected to isolate GFP+ cells by FAC sorting. RNA extraction was followed by qRT–PCR using primers (Supplementary Table S1) spanning across at least two exons of putative Tox targets whose up/downregulation could potentially explain the high enrichment in GO terms related to neurogenesis, axonogenesis, CNS developmental and regulation of transcription (Fig4A).
Validating DamID-Seq in vivo, we found that the abundance of all transcripts being investigated was significantly up- or downregulated upon Tox overexpression compared to GFP controls (Fig4C). This further indicated that Tox can act both as a transcriptional activator or repressor of its targets.
Tox inhibits the differentiation of cortical progenitors
To investigate the function of Tox during cortical development, we next decided to manipulate its expression by in utero electroporation. Vectors encoding Tox together with GFP, or GFP alone as control, were delivered at E13.5 and embryos collected 48 h later. Distribution of GFP+ electroporated cells and their progeny revealed that Tox overexpression increased the proportion of cells in the SVZ while reducing those in the CP relative to control (12.6 ± 1.6% versus 19.6 ± 3.9% in SVZ and 12.9 ± 1.9% versus 3.8 ± 2.8% in CP). This occurred without any substantial change in the proportion of cells in the VZ and IZ (Fig5A). Reinforcing the link between Tox and calcineurin/Nfat signalling, distribution of GFP+ cells upon Tox overexpression closely resembled that observed upon overexpression of CA-Nfat4 (Figs2F and 5A). Moreover, as an additional control of our DamID-Seq analysis, electroporation with Tox or Tox-Dam vectors induced essentially identical phenotypes (Fig5A and Supplementary Fig S2C) making it unlikely that chromatin binding was perturbed by the fusion of Tox to Dam.
Figure 5.
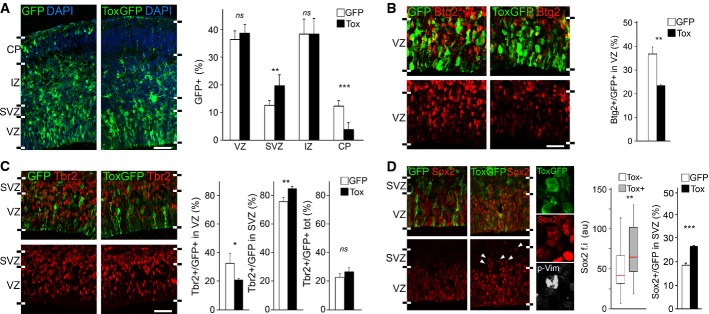
- A-D Fluorescence pictures (left) and quantifications (right) of GFP+ (green) cells of the E15.5 lateral cortex positive for Btg2RFP (B), Tbr2 (C), Sox2 (D) (red) or p-Vim (D; white) upon electroporation with control or Tox (white or black bars, respectively) plasmids at E13.5. n ≥ 3; error bars = SD (except box plot in D, middle; n ≥ 50); ns = not significant; *P < 0.05; **P < 0.01; ***P < 0.001. Scale bars, 50 μm.
Being Tox an off-switch gene downregulated in DP, we first investigated whether its overexpression altered the proportion of DP. To this aim, we performed electroporation in Btg2RFP reporter mice and found a reduction of Btg2RFP+ cells in the VZ by 30% in Tox-overexpressing brains relative to control (from 37.7 ± 3.1% to 26.4 ± 4.9%, respectively) (Fig5B). Moreover, and consistent with a decrease in DP, we observed that Tox overexpression reduced the proportion of Tbr2+ newborn basal progenitors in the VZ by 40% while, conversely, increasing it in the SVZ by 10% (32.6 ± 7.1% versus 20.8 ± 0.7% in VZ and 75.9 ± 2.9% versus 85.2 ± 1.4% in SVZ), such that the two effects ultimately compensated each other (Fig5C). Finally, we found that Tox-overexpressing cells in the VZ expressed significantly higher levels of the proliferative marker of apical progenitors Sox2 (Fig5D, left). This increase in Sox2 immunoreactivity is intriguing since (i) Tox and Sox2 expression positively correlated in physiological conditions (Fig1C and C′) and (ii) DamID-Seq identified Sox2 as a direct target of Tox, although this single-exon transcript could not be assessed by qRT–PCR.
These data show that Tox overexpression inhibits the differentiation of apical progenitors in the VZ. Yet, its overexpression increased primarily progenitors in the SVZ, and not in the VZ (Fig5A). To address this counterintuitive finding, we inspected the SVZ to assess whether overexpression of Tox promoted the mislocalization of PP from the VZ into the SVZ. As a marker of apical progenitors, we again used Sox2 and found an increase by almost 50% (18.2 ± 1.1% versus 26.4 ± 0.4%) in Sox2+ progenitors in the SVZ (Fig5D, right). Moreover, considering that cortical progenitors include not only apical and basal progenitors but also a small fraction of basal radial glia (Shitamukai et al, 2011; Wang et al, 2011), we next investigated whether Tox overexpression altered the proportion of one specific progenitor type as identified by their bi-polar, a-polar and uni-polar morphology in mitosis, respectively. To address this, we acquired high-resolution, Z-stack images of p-Vim+ mitotic cells in the SVZ/IZ upon electroporation with control or Tox-expressing vectors. This showed that Tox-overexpressing cells in these layers were neither apical progenitors with extended interkinetic nuclear migration nor newly generated basal radial glia because the almost totality (34/36; i.e. 94%) maintained an a-polar morphology as it was the case in both GFP− unmanipulated cells or GFP+ electroporated with control plasmids (together 132/138; i.e. 96%) (Fig5D and data not shown). This was further confirmed by the analysis of interphase, GFP+ cells in the SVZ, with the vast majority displaying a-polar morphology, thus, showing that the increase in the proportion of Sox2+ cells in the SVZ is primarily due to its ectopic expression in basal progenitors (Fig5D and data not shown).
Altogether, these data show that Tox controls neural stem cell fate by inhibiting their switch from PP to DP and decreasing neurogenesis.
Tox affects neurite outgrowth and neural specification
Given the enrichment in functional terms related to axonogenesis and neurite outgrowth resulting from DamID-Seq data (Fig4A; right), we next investigated whether Tox overexpression had any effect on neuronal migration or neurite growth. To address the former, we performed electroporation at E13.5 followed 24 h later by administration of a single dose of BrdU to label progenitors in S phase (Fig6A). Embryos were collected at E15.5 and proportion and distribution of GFP+/BrdU+ cells assessed. First, we found that the distribution of GFP+/BrdU+ cells in five equidistant bins within the IZ, hence, the pattern of migration of Tox-overexpressing neurons generated 24 h prior to dissection, was essentially identical after GFP or Tox overexpression (Fig6B and B′, left). Moreover, this experiment allowed us to assess the proportion of neurons generated from a pool of progenitors labelled with BrdU 24 h earlier. Reinforcing our observations on cell fate change (Fig5), we found a significant decrease in the proportion of GFP+/BrdU+ neurons in Tox-overexpressing brains relative to control by 30% (Fig5B′, right) (from 50.9 ± 2.1% to 36.6 ± 0,7%).
Figure 6.
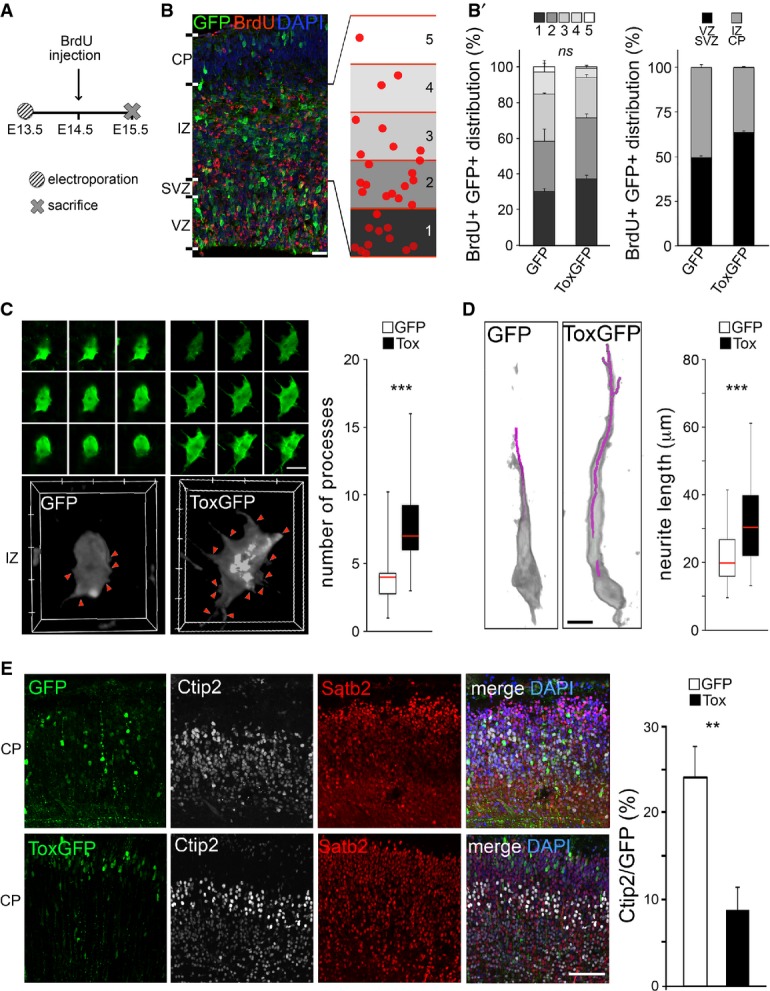
- A-B′ Experimental layout (A), fluorescence pictures (B) and quantifications (B′) used to assess neuronal migration and output upon electroporation with GFP or Tox (B′; as indicated) plasmids at E13.5 followed by BrdU exposure and sacrifice as shown in (A). Neuronal migration was assessed by calculating the distribution of GFP+/BrdU+ cells within five equidistant bins of the IZ (B and B′; left), while output was quantified as the proportion of GFP+/BrdU+ in the IZ and CP relative to all GFP+ cells (B'; right). n = 2; error bars = SEM. Scale bar, 50 μm.
- C, D 3D reconstruction (left) and quantification (right) of newborn (C) or polarized (D) neurons in the IZ or CP, respectively. Individual Z-stacks (0.1 μm optical thickness; green) and computer reconstructions (grey) were used to count cell protrusions (C; arrowheads) or neurite length (D; purple line) that were statistically evaluated (box plots; right, n ≥ 49; ***P < 0.001). Scale bars, 10 μm.
- E Fluorescence pictures (left) of the E18.5 mouse cortex upon electroporation of GFP or Tox plasmids at E13.5 followed by immunohistochemistry for GFP (green), Ctip2 (white), Satb2 (red) and DAPI counterstaining (blue). Quantification of Ctip2+/GFP+ cells is shown in either condition (right). n = 3; **P < 0.01; error bars = SD. Scale bar, 50 μm.
Having excluded an effect on neuronal migration by Tox, we next investigated its possible effects on process outgrowth. To this aim, we acquired high-resolution Z-stacks images of randomly chosen GFP+ cells within the IZ or CP of GFP or Tox electroporated brains and 3D-reconstructed individual cells to assess their protrusions (Fig6C and D). Newly born neurons in the IZ are primarily characterized by relatively short processes being dynamically extended and retracted (Kriegstein & Noctor, 2004; Barnes & Polleux, 2009). In contrast, more mature and polarized neurons are characterized by a trailing and a leading process. After reaching the CP, these neurons grow an unipolar dendrite directed towards the marginal zone and begin axonogenesis (Kriegstein & Noctor, 2004; Barnes & Polleux, 2009). Looking for these two features of newborn and polarized neurons, we found that Tox overexpression induced a doubling (from 3.6 ± 1.6 to 8.0 ± 2.8) in the number of processes of non-polarized neurons in the IZ (Fig6C). Moreover, independently from the fact that remarkably few neurons could be found in the CP upon Tox overexpression (Fig5A), the few that were found displayed a substantially longer dendrite (from 21.2 ± 7.2 to 32.10 ± 11.6 μm) (Fig6D).
Finally, we investigated whether Tox-overexpressing neurons were able to undergo full maturation and specification irrespective of their change in numbers and altered formation of processes. To this aim, we electroporated E13.5 embryos with control or Tox-expressing vectors as previously described but collecting brains at a longer survival time at E18.5. This showed that the majority of Tox electroporated neurons reached the CP (Fig6E), supporting our conclusion about a lack of phenotype on migration. However, the proportion of GFP+ cells in the CP that expressed Ctip2 decreased after Tox overexpression by ∽60% compared to control (from 24.1 ± 3.6% to 8.7 ± 2.6%). In contrast, the overwhelming majority of GFP+ cells in upper and deeper layers remained Satb2+ in either condition. Importantly, this reduction in the proportion of Ctip2+ neurons induced by Tox can be explained either as a consequence of a delayed differentiation of PP or as a cell-intrinsic effect on neuronal specification.
Hence, we propose that Tox is a multifunctional factor involved in corticogenesis by promoting not only proliferative divisions of neural progenitors but also neurite outgrowth and fate of newborn neurons. These functions were predicted by DamID-Seq and likely due to the regulation of different transcriptional targets of Tox in different cell types.
Discussion
Here, we found that the expression of Tox at the tissue level recapitulates the temporal and spatial gradients of cortical neurogenesis. At the cellular level, Tox positively correlated with markers of proliferation while, conversely, markers of differentiation were less abundant in Tox+ cells. Possibly underlying these highly specific patterns of expression in tissues and individual cells, we found that Tox is regulated by calcineurin/Nfat4 signalling. Interestingly, both calcineurin and Nfat4 are known as key players implicated in stem cell commitment and regeneration (Crabtree & Olson, 2002; Horsley et al, 2008; Mukherjee & Soto, 2011; Kujawski et al, 2014). Moreover, and possibly underlying the coordination of calcineurin/Nfat signalling, VZ progenitors were described to undergo spontaneous Ca2+ oscillations during development (Owens & Kriegstein, 1998). In this frame, our study provides one additional link between Ca2+ and neurogenesis (Aprea & Calegari, 2012) in which at least some of these effects may act through Nfat4-dependent upregulation of Tox.
Until now, Tox has been studied solely in the immune system and during hematopoiesis (Wilkinson et al, 2002; Aliahmad et al, 2004, 2010; Yun et al, 2011), but even in this context, its downstream targets were unknown and it was unclear whether this transcription factor binds DNA in a sequence- or structure-dependent manner (O'Flaherty & Kaye, 2003; Stros et al, 2007). To address this, we combined powerful experimental and bioinformatical tools developed by other groups (van Steensel & Henikoff, 2000; Vogel et al, 2006, 2007; Southall & Brand, 2007; Wu & Yao, 2013) and performed the first DamID-Seq profiling of a transcription factor. This revealed a level of resolution comparable to ChIP-Seq with the great advantage of avoiding the use of antibodies and giving us not only important information about the properties of DNA binding of Tox, including its motif, but also a comprehensive list of its downstream targets that should be devoid of biases towards any specific lineage. Here we extended the use of DamID as pioneered by other groups (van Steensel & Henikoff, 2000; Orian et al, 2003; Bianchi-Frias et al, 2004; Southall et al, 2013, 2014) and developed DamID-Seq for a transcription factor providing a powerful tool to identify the features of, in principle, any DNA-binding protein. In addition, our list of Tox targets represents an important resource for the study of this transcription factor in, virtually, any tissue.
Since Tox is an off-switch gene, we expected that its targets would also be either off- or on-switches. Yet, we found among Tox targets a mixture of on- and off-switches as well as genes up- or downregulated in PP, DP and/or neurons. This in turn suggests that different genes are differentially regulated by Tox in different cell types, which is supported by the fact that Tox overexpression in the developing cortex could either inhibit or induce its different targets. Corroborating this, DamID-Seq analyses revealed that in addition to the generic terms “transcription factors” and “CNS development” the primary and most significant functional terms associated with Tox targets were “neurogenesis” and “dendrite outgrowth”, which are related to stem cell fate and neuronal maturation, respectively. From this, we propose that Tox is involved in the regulation of two classes of downstream targets in the two cell types, which might depend on the expression of different cofactors in each population (Fig7).
Figure 7.
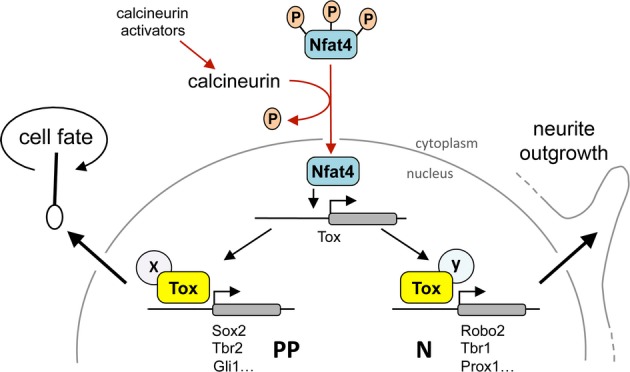
Model of Tox activity
Model summarizing the regulation and induced molecular effects of Tox as deduced from the literature (red arrows) and our own experiments (black arrows). We propose that activation of calcineurin signalling promotes Tox expression through nuclear transport of Nfat4, which results in Tox-induced transcriptional regulation of different sets of genes (some indicated) in PP or neurons (N) to ultimately control stem cell fate (left) or neurite outgrowth (right) through yet to be characterized cell-specific co-factors (X and Y).
Finally, we overexpressed Tox by in utero electroporation and investigated its effects during corticogenesis. We found that Tox inhibited the differentiation of neural progenitors resulting in a decrease in neurogenesis. Concerning this, DamID-Seq and qRT–PCR revealed that in certain cases Tox overexpression had a seemingly counterintuitive effect on the expression of specific transcription factors, specifically Tbr2; in that Tox was found to upregulate Tbr2 despite the fact that this neurogenic factor is expected to be downregulated in a context of decreased neurogenesis. This, however, is easily explained by the fact that the effect of Tox overexpression is the result of the concerted action of all of its targets simultaneously. Hence, upregulation of potent proliferative factors such as Gli1, Yap1 and Sox2 may substantially overcome the concomitant effect on neurogenic factors such as Tbr2. Since Tox expression recapitulates the gradients of neurogenesis in physiological conditions, we propose that Tox upregulation in neurogenic cortical areas is important to maintain a pool of proliferating progenitors among these areas. In contrast, cortical areas in which the neurogenic output is physiologically low, such as caudal/medial cortical regions in early embryos, may not require Tox upregulation, at least until the gradients of differentiation have reached those areas.
Beyond progenitor cells, we observed that newborn neurons migrating through the IZ to reach the CP were characterized by an increase in the number and length of protrusion(s), respectively. Tox was additionally involved in neuronal fate with a decrease in the proportion of deeper layer neurons. This effect can be due to a delay in neurogenesis resulting from the increased expansion of progenitors and/or by a cell-intrinsic affect in neuronal maturation, which is reminiscent of other transcription factors, such as Brn2 and Cux2 expressed, similar to Tox, in progenitors and subsequently a subset of neurons (Franco et al, 2012; Dominguez et al, 2013).
All together, here we identified Tox as a multifunctional, off-switch transcription factor controlling brain development, neural stem cell differentiation and dendritogenesis. Interestingly, Tox was found among the group of transcription factors regulated by active enhancers of the developing forebrain (Visel et al, 2013; Pattabiraman et al, 2014) and another Tox family member, Tox3, was found to be differentially regulated in neural cell types of the adult sub-ventricular zone (Beckervordersandforth et al, 2010). Hence, these data point to the identification of the Tox family as novel regulator of neural stem cells commitment.
Material and Methods
Constructs
Tox cDNA was generated by RT–PCR of E13.5 cortices (Supplementary Table S1) and cloned into pLgwV5EcoDam (Vogel et al, 2006) or pBI-CMV1 (Clontech) vectors, the latter also used to express GFP from its bidirectional promoter, and used for DamID or in utero electroporation, respectively. The vectors pcDNA3.T7-Δ-Nfat3 (CA-Nfat3) (Addgene plasmid 28224) (Fougere et al, 2010) and pEGFP-RHR4 (CA-Nfat4) (kind gift of Dr. S. Matsuda) (Matsuda et al, 2000) were co-electroporated together with pBI-CMV1-GFP.
Manipulation in mice
In utero electroporation was performed as previously described (Artegiani et al, 2012) by injecting ∽1–5 μg of DNA into C57BL/6J or Btg2RFP mice followed by the delivery of 6, 50 ms long, electric pulses of 30 V with intervals of 1 s. Embryos were electroporated at E13.5 and sacrificed 48 h later eventually upon intraperitoneal administration of 1 mg BrdU (1 pulse at E14.5) or 0.5 mg of CsA (two injections per day) (dissolved in 100 μl PBS or 400 μl of NaCl 0.9%, respectively).
Biochemistry
Electroporated areas of the brain were dissociated (MACS dissociation kit; Miltenyi), cells washed with PBS and incubated 15 min at 4°C in hypotonic solution (Hepes 10 mM, MgCl2 1.5 mM, KCl 10 mM, DTT 0.5 mM and Complete Protease Inhibitors Cocktail (CPIC; Roche)), and extracts centrifuged at 16,000 g for 15 min at 4°C to separate cytoplasmic and nuclear fractions. The latter was further resuspended in nuclear extraction buffer (Hepes 20 mM, MgCl2 1.5 mM, NaCl 420 mM, EDTA 0.2 mM, DTT 0.5 mM, glycerol 25% and CPIC) for 30 min at 4°C and centrifuged as above. Extracts were stored at −80°C and 20 μg analysed after boiling in LDS buffer (Invitrogen) using NuPage 4–12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose. Membranes were blocked 1 h at room temperature in 3% BSA in TBS 0.3% Tween and incubated overnight with 1:1,000 Tox (Abcam), Gadph (NovusBiological) or Tbp (Abcam) antibodies followed by washing, incubation with HRP-conjugated secondary antibody for 2 h and chemiluminescence reaction (ECL Dura or Pico, Pierce). Non-saturated radiograms were analysed using “Gels” on Fiji 9 software.
Immunohistochemistry
Immunohistochemistry was performed as described (Lange et al, 2009; Artegiani et al, 2011). Briefly, after fixation (4% PFA), cryoprotection (30% sucrose) and cryosectioning, slices (10 μm thick) were permeabilized and blocked (0.3% Triton X-100 and donkey serum 10% in PBS) (IHC buffer) for 2 h at room temperature. All primary antibodies are described in Nonaka-Kinoshita et al (2013) except for anti-Tox (Atlas Antibodies 1:500) and incubated overnight at 4°C in IHC followed by 2-h washing in PBS and 3-h incubation with DyLight-secondary antibodies (1:1,000; Jackson Laboratory) at room temperature. DAPI was used to counterstain nuclei. ApoTome (Zeiss) composite images were acquired and processed by AxioVision or Zen (Zeiss). Assessment of immunoreactivity within individual cells was performed by outlining randomly chosen, DAPI-labelled nuclei and quantifying pixels' density in the relevant channel. 3D reconstruction of randomly selected neurons was performed using 40 or 63× objectives and 0.1 μm Z-stacks. Analyses were performed on digital images using Photoshop (Adobe) and/or Fiji 9 (http://fiji.sc/Fiji).
DamID-Seq
DamID was performed according to Vogel et al (2007) using lentiviruses obtained as previously described (Artegiani et al, 2012, 2013) but without concentration. HEK-293T or Neuro-2a cells were infected with viral supernatants diluted 1:2 (or even 1:10 in the case of Neuro-2a), and 48 h later, their genomic DNA was extracted, digested with DpnI, ligated to adaptors, further digested with DpnII and PCR-amplified. Sequencing libraries were prepared as described (Aprea et al, 2013) and subjected to 75-bp single-read sequencing on a HiSeq 2000 platform (Illumina), resulting in ∽20 million reads per sample. Sequencing raw data were deposited in GEO.
Bioinformatics
Reads alignment to the human genome (Ensembl67) was performed with bowtie (v0.12.7) using “–best” and “-m 1” to report uniquely mapped reads. Replicate reproducibility was tested by bamCorrelate from deepTools (Ramirez et al, 2014) and alignments of replicates pooled before peak calling. Genomic regions of Tox-Dam enrichment were identified by SICER (Zang et al, 2009; Wu & Yao, 2013) considering genes within 15 kb upstream of the TSS, or inside the transcript body. Peak annotation with genomic features was done by ChIPseeker (Bioconductor). For Tox read profile around the TSS, aligned reads were normalized to Dam using bamCompare (Ramirez et al, 2014) setting the parameters: “–ratio ratio; –normalizeTo1x 2451960000; and –scaleFactorsMethod readCount”. Cell-specific enhancers (Andersson et al, 2014) were converted from hg18 to hg19 using liftOver (UCSC) and Tox read density calculated by computeMatrix scale regions (GitHub), scaling to the average size of enhancers (3 kb). Conservation scores for placental species (phastCons46way) were downloaded from the UCSC genome browser and exonic regions masked. Matched random genomic locations to the Tox peaks were created with shuffleBed (bedTools) using the parameters: “-seed 927442958; -chrom; -noOverlapping”. Tox putative binding motif was predicted by gimmeMotifs clustering results from several other softwares (van Heeringen & Veenstra, 2011) and using 1,000 peaks with ≥ 2.5-fold enrichment and ≤ 1 Kb.
qRT–PCR
Electroporated cells were dissociated as described above and GFP+ cells sorted using a FACSAria III (Becton Dickinson). RNA was extracted using the RNeasy micro kit (QIAGEN) and cDNA generated by Superscript kit (Invitrogen) starting with 100 ng of RNA and using 1 μl of cDNA diluted 1:10 for qPCRs. Primers (Supplementary Table S1) were designed to span across exons to distinguish cDNA from genomic DNA.
Statistical analysis
All the analyses presented in this work were performed with at least three biological replicates (n ≥ 3) except for DamID-Seq (n = 2). Values obtained were used to calculate mean, standard deviation and significance by two-tailed Student's t-test, assuming 0.05 as a threshold for significance. Quantifications of individual cells were performed using a minimum of 24–80 cells as indicated in each figure panel.
Acknowledgments
We thank the Biomedical Services of the MPI-CBG, the FACS facility of the CRTD and the histology facility of the BIOTEC for technical support. We are very grateful to all people providing us with reagents, advise and sharing protocols, particularly Drs. Bas van Steensel, Pompeo Macioce, Joerg Mansfeld, Gabor Bakos, Satoshi Matsuda, Shigeo Koyasu, Sebastian Jaulian and Tony Southall. This work was supported by the CRTD, the TU-Dresden the DFG (CA 893/9-1) and Collaborative Research Center SFB655 (subproject A20).
Author contributions
BA and FC planned the experiments and wrote the manuscript; BA performed all experiments supported by SBA, EB and SM; and AMJD performed all bioinformatic analyses supported by AD. All authors approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Legends
Supplementary File S1
Review Process File
References
- Aliahmad P, O'Flaherty E, Han P, Goularte OD, Wilkinson B, Satake M, Molkentin JD, Kaye J. TOX provides a link between calcineurin activation and CD8 lineage commitment. J Exp Med. 2004;199:1089–1099. doi: 10.1084/jem.20040051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat Immunol. 2010;11:945–952. doi: 10.1038/ni.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliahmad P, Seksenyan A, Kaye J. The many roles of TOX in the immune system. Curr Opin Immunol. 2012;24:173–177. doi: 10.1016/j.coi.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, Ntini E, Arner E, Valen E, Li K, Schwarzfischer L, Glatz D, Raithel J, Lilje B, Rapin N, Bagger FO, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprea J, Calegari F. Bioelectric state and cell cycle control of Mammalian neural stem cells. Stem Cells Int. 2012;2012:816049. doi: 10.1155/2012/816049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprea J, Prenninger S, Dori M, Ghosh T, Monasor LS, Wessendorf E, Zocher S, Massalini S, Alexopoulou D, Lesche M, Dahl A, Groszer M, Hiller M, Calegari F. Transcriptome sequencing during mouse brain development identifies long non-coding RNAs functionally involved in neurogenic commitment. EMBO J. 2013;32:3145–3160. doi: 10.1038/emboj.2013.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprea J, Lesche M, Massalini S, Prenninger S, Alexopoulou D, Dahl A, Hiller M, Calegari F. Identification and expression patterns of novel long non-coding RNAs in neural progenitors of the developing mammalian cortex. Neurogenesis. 2015 doi: 10.1080/23262133.2014.995524. doi: 10.1080/23262133.2014.995524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artegiani B, Lindemann D, Calegari F. Overexpression of cdk4 and cyclinD1 triggers greater expansion of neural stem cells in the adult mouse brain. J Exp Med. 2011;208:937–948. doi: 10.1084/jem.20102167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artegiani B, Lange C, Calegari F. Expansion of embryonic and adult neural stem cells by in utero electroporation or viral stereotaxic injection. J Vis Exp. 2012:e4093. doi: 10.3791/4093. . doi: 10.3791/4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artegiani B, Calegari F. Lentiviruses allow widespread and conditional manipulation of gene expression in the developing mouse brain. Development. 2013;140:2818–2822. doi: 10.1242/dev.093823. [DOI] [PubMed] [Google Scholar]
- Attardo A, Calegari F, Haubensak W, Wilsch-Brauninger M, Huttner WB. Live imaging at the onset of cortical neurogenesis reveals differential appearance of the neuronal phenotype in apical versus basal progenitor progeny. PLoS ONE. 2008;3:e2388. doi: 10.1371/journal.pone.0002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub AE, Oh S, Xie Y, Leng J, Cotney J, Dominguez MH, Noonan JP, Rakic P. Transcriptional programs in transient embryonic zones of the cerebral cortex defined by high-resolution mRNA sequencing. Proc Natl Acad Sci USA. 2011;108:14950–14955. doi: 10.1073/pnas.1112213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AP, Polleux F. Establishment of axon-dendrite polarity in developing neurons. Annu Rev Neurosci. 2009;32:347–381. doi: 10.1146/annurev.neuro.31.060407.125536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckervordersandforth R, Tripathi P, Ninkovic J, Bayam E, Lepier A, Stempfhuber B, Kirchhoff F, Hirrlinger J, Haslinger A, Lie DC, Beckers J, Yoder B, Irmler M, Gotz M. In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell. 2010;7:744–758. doi: 10.1016/j.stem.2010.11.017. [DOI] [PubMed] [Google Scholar]
- van Bemmel JG, Filion GJ, Rosado A, Talhout W, de Haas M, van Welsem T, van Leeuwen F, van Steensel B. A network model of the molecular organization of chromatin in Drosophila. Mol Cell. 2013;49:759–771. doi: 10.1016/j.molcel.2013.01.040. [DOI] [PubMed] [Google Scholar]
- Bianchi-Frias D, Orian A, Delrow JJ, Vazquez J, Rosales-Nieves AE, Parkhurst SM. Hairy transcriptional repression targets and cofactor recruitment in Drosophila. PLoS Biol. 2004;2:E178. doi: 10.1371/journal.pbio.0020178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britz O, Mattar P, Nguyen L, Langevin LM, Zimmer C, Alam S, Guillemot F, Schuurmans C. A role for proneural genes in the maturation of cortical progenitor cells. Cereb Cortex. 2006;16(Suppl 1):i138–i151. doi: 10.1093/cercor/bhj168. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Nowakowski RS, Bhide PG. Neocortical neurogenesis: morphogenetic gradients and beyond. Trends Neurosci. 2009;32:443–450. doi: 10.1016/j.tins.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Dominguez MH, Ayoub AE, Rakic P. POU-III transcription factors (Brn1, Brn2, and Oct6) influence neurogenesis, molecular identity, and migratory destination of upper-layer cells of the cerebral cortex. Cereb Cortex. 2013;23:2632–2643. doi: 10.1093/cercor/bhs252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fietz SA, Lachmann R, Brandl H, Kircher M, Samusik N, Schroder R, Lakshmanaperumal N, Henry I, Vogt J, Riehn A, Distler W, Nitsch R, Enard W, Paabo S, Huttner WB. Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self-renewal. Proc Natl Acad Sci USA. 2012;109:11836–11841. doi: 10.1073/pnas.1209647109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fougere M, Gaudineau B, Barbier J, Guaddachi F, Feugeas JP, Auboeuf D, Jauliac S. NFAT3 transcription factor inhibits breast cancer cell motility by targeting the Lipocalin 2 gene. Oncogene. 2010;29:2292–2301. doi: 10.1038/onc.2009.499. [DOI] [PubMed] [Google Scholar]
- Franco SJ, Gil-Sanz C, Martinez-Garay I, Espinosa A, Harkins-Perry SR, Ramos C, Müller U. Fate-restricted neural progenitors in the mammalian cerebral cortex. Science. 2012;337:746–749. doi: 10.1126/science.1223616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S. Stem and progenitor cell-based therapy of the human central nervous system. Nat Biotechnol. 2005;23:862–871. doi: 10.1038/nbt1119. [DOI] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–670. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Guillemot F. Cell fate specification in the mammalian telencephalon. Prog Neurobiol. 2007;83:37–52. doi: 10.1016/j.pneurobio.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Han X, Wu X, Chung WY, Li T, Nekrutenko A, Altman NS, Chen G, Ma H. Transcriptome of embryonic and neonatal mouse cortex by high-throughput RNA sequencing. Proc Natl Acad Sci USA. 2009;106:12741–12746. doi: 10.1073/pnas.0902417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci USA. 2004;101:3196–3201. doi: 10.1073/pnas.0308600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heeringen SJ, Veenstra GJ. GimmeMotifs: a de novo motif prediction pipeline for ChIP-sequencing experiments. Bioinformatics. 2011;27:270–271. doi: 10.1093/bioinformatics/btq636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley V, Aliprantis AO, Polak L, Glimcher LH, Fuchs E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell. 2008;132:299–310. doi: 10.1016/j.cell.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawski S, Lin W, Kitte F, Bormel M, Fuchs S, Arulmozhivarman G, Vogt S, Theil D, Zhang Y, Antos CL. Calcineurin regulates coordinated outgrowth of zebrafish regenerating fins. Dev Cell. 2014;28:573–587. doi: 10.1016/j.devcel.2014.01.019. [DOI] [PubMed] [Google Scholar]
- Kurabayashi N, Sanada K. Increased dosage of DYRK1A and DSCR1 delays neuronal differentiation in neocortical progenitor cells. Genes Dev. 2013;27:2708–2721. doi: 10.1101/gad.226381.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell. 2009;5:320–331. doi: 10.1016/j.stem.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Kokaia Z. Stem cells for the treatment of neurological disorders. Nature. 2006;441:1094–1096. doi: 10.1038/nature04960. [DOI] [PubMed] [Google Scholar]
- Liu Y, Janeway CA., Jr Monoclonal antibodies against T cell receptor/CD3 complex induce cell death of Th1 clones in the absence of accessory cells. Adv Exp Med Biol. 1991;292:105–113. doi: 10.1007/978-1-4684-5943-2_12. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Shibasaki F, Takehana K, Mori H, Nishida E, Koyasu S. Two distinct action mechanisms of immunophilin-ligand complexes for the blockade of T-cell activation. EMBO Rep. 2000;1:428–434. doi: 10.1093/embo-reports/kvd090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M. Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development. 2004;131:3133–3145. doi: 10.1242/dev.01173. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Soto C. Role of calcineurin in neurodegeneration produced by misfolded proteins and endoplasmic reticulum stress. Curr Opin Cell Biol. 2011;23:223–230. doi: 10.1016/j.ceb.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Nonaka-Kinoshita M, Reillo I, Artegiani B, Martinez-Martinez MA, Nelson M, Borrell V, Calegari F. Regulation of cerebral cortex size and folding by expansion of basal progenitors. EMBO J. 2013;32:1817–1828. doi: 10.1038/emboj.2013.96. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai W, Nakatani S, Takahara T, Kainuma M, Masaoka M, Minobe S, Namihira M, Nakashima K, Sakakibara A, Ogawa M, Miyata T. Periventricular notch activation and asymmetric Ngn2 and Tbr2 expression in pair-generated neocortical daughter cells. Mol Cell Neurosci. 2009;40:225–233. doi: 10.1016/j.mcn.2008.10.007. [DOI] [PubMed] [Google Scholar]
- O'Flaherty E, Kaye J. TOX defines a conserved subfamily of HMG-box proteins. BMC Genom. 2003;4:13. doi: 10.1186/1471-2164-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orian A, van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, Williams E, Loo LW, Cowley SM, Yost C, Pierce S, Edgar BA, Parkhurst SM, Eisenman RN. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17:1101–1114. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Patterns of intracellular calcium fluctuation in precursor cells of the neocortical ventricular zone. J Neurosci. 1998;18:5374–5388. doi: 10.1523/JNEUROSCI.18-14-05374.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paridaen JT, Huttner WB. Neurogenesis during development of the vertebrate central nervous system. EMBO Rep. 2014;15:351–364. doi: 10.1002/embr.201438447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman K, Golonzhka O, Lindtner S, Nord AS, Taher L, Hoch R, Silberberg SN, Zhang D, Chen B, Zeng H, Pennacchio LA, Puelles L, Visel A. Rubenstein JL. Transcriptional regulation of enhancers active in protodomains of the developing cerebral cortex. Neuron. 2014;82:989–1003. doi: 10.1016/j.neuron.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L, Gotz M. Radial glial cell heterogeneity–the source of diverse progeny in the CNS. Prog Neurobiol. 2007;83:2–23. doi: 10.1016/j.pneurobio.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimbo T, Du Y, Grimm SA, Dhasarathy A, Mav D, Shah RR, Shi H, Wade PA. MBD3 localizes at promoters, gene bodies and enhancers of active genes. PLoS Genet. 2013;9:e1004028. doi: 10.1371/journal.pgen.1004028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitamukai A, Konno D, Matsuzaki F. Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J Neurosci. 2011;31:3683–3695. doi: 10.1523/JNEUROSCI.4773-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southall TD, Brand AH. Chromatin profiling in model organisms. Brief Funct Genomic Proteomic. 2007;6:133–140. doi: 10.1093/bfgp/elm013. [DOI] [PubMed] [Google Scholar]
- Southall TD, Brand AH. Neural stem cell transcriptional networks highlight genes essential for nervous system development. EMBO J. 2009;28:3799–3807. doi: 10.1038/emboj.2009.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southall TD, Gold KS, Egger B, Davidson CM, Caygill EE, Marshall OJ, Brand AH. Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev Cell. 2013;26:101–112. doi: 10.1016/j.devcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southall TD, Davidson CM, Miller C, Carr A, Brand AH. Dedifferentiation of neurons precedes tumor formation in Lola mutants. Dev Cell. 2014;28:685–696. doi: 10.1016/j.devcel.2014.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol. 2000;18:424–428. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- Stros M, Launholt D, Grasser KD. The HMG-box: a versatile protein domain occurring in a wide variety of DNA-binding proteins. Cell Mol Life Sci. 2007;64:2590–2606. doi: 10.1007/s00018-007-7162-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna E, Gotz M, Huttner WB. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol. 2014;30:465–502. doi: 10.1146/annurev-cellbio-101011-155801. [DOI] [PubMed] [Google Scholar]
- Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, McKinsey GL, Pattabiraman K, Silberberg SN, Blow MJ, Hansen DV, Nord AS, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer-Frick I, Shoukry M, Afzal V, Kaplan T, et al. A high-resolution enhancer atlas of the developing telencephalon. Cell. 2013;152:895–908. doi: 10.1016/j.cell.2012.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel MJ, Guelen L, de Wit E, Peric-Hupkes D, Loden M, Talhout W, Feenstra M, Abbas B, Classen AK, van Steensel B. Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res. 2006;16:1493–1504. doi: 10.1101/gr.5391806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel MJ, Peric-Hupkes D, van Steensel B. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat Protoc. 2007;2:1467–1478. doi: 10.1038/nprot.2007.148. [DOI] [PubMed] [Google Scholar]
- Wang X, Tsai JW, LaMonica B, Kriegstein AR. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci. 2011;14:555–561. doi: 10.1038/nn.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson B, Chen JY, Han P, Rufner KM, Goularte OD, Kaye J. TOX: an HMG box protein implicated in the regulation of thymocyte selection. Nat Immunol. 2002;3:272–280. doi: 10.1038/ni767. [DOI] [PubMed] [Google Scholar]
- Wu F, Yao J. Spatial compartmentalization at the nuclear periphery characterized by genome-wide mapping. BMC Genom. 2013;14:591. doi: 10.1186/1471-2164-14-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao MJ, Chen G, Zhao PP, Lu MH, Jian J, Liu MF, Yuan XB. Transcriptome analysis of microRNAs in developing cerebral cortex of rat. BMC Genom. 2012;13:232. doi: 10.1186/1471-2164-13-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S, Lee SH, Yoon SR, Kim MS, Piao ZH, Myung PK, Kim TD, Jung H, Choi I. TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol Lett. 2011;136:29–36. doi: 10.1016/j.imlet.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. doi: 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Legends
Supplementary File S1
Review Process File