

Abstract
FOXO transcription factors are considered bona fide tumor suppressors; however, recent studies showed FOXOs are also required for tumor survival. Here, we identify FOXOs as transcriptional activators of IDH1. FOXOs promote IDH1 expression and thereby maintain the cytosolic levels of α-ketoglutarate and NADPH. In cancer cells carrying mutant IDH1, FOXOs likewise stimulate mutant IDH1 expression and maintain the levels of the oncometabolite 2-hydroxyglutarate, which stimulates cancer cell proliferation and inhibits TET enzymes and histone demethylases. Combined, our data provide a new paradigm for the paradoxical role of FOXOs in both tumor suppression and promotion.
Keywords: FOXO, IDH1, oncometabolite, PI3K
Introduction
The forkhead box(O) (FOXO) family of transcription factors includes four members, FOXO1, FOXO3a, FOXO4, and FOXO6; of these, FOXO6 is expressed predominantly in the brain, whereas the other three members are more ubiquitously expressed. FOXOs integrate various environmental signals into specific transcriptional programs related to cell proliferation, metabolism, and reactive oxygen species (ROS) detoxification 1. The activity of FOXOs is negatively regulated by growth factors through the PI3K–PKB (AKT) pathway 2, 3, whereas certain environmental stresses, such as ROS and hypoxia, positively regulate FOXO activity by promoting their nuclear retention 4, 5. In most types of human cancer, constitutive activation of PI3K signaling is observed and this is believed to result in inactivation of FOXOs in cancer. FOXOs are considered bona fide tumor suppressors, as conditional deletion of FOXO homologues in mice (FoxO1−/−; FoxO3−/−; FoxO4−/− mice) results in enhanced tumorigenesis 6. Importantly, FOXOs are increasingly recognized as critical homeostatic regulators, operating in both untransformed and cancer cells 1. This has led to the hypothesis that FOXOs function as tumor suppressors in the context of normal cells but within a tumor can contribute to cancer cell survival 7.
Reprogramming of cellular bioenergetics is an emerging hallmark of cancer 8. Cancer cells reprogram their metabolism to sustain high growth and proliferation rates 9, and the recent identification of cancer-associated mutations in key metabolic enzymes, including the isocitrate dehydrogenases (IDH) 1 and 2 10, 11, reinforced the notion that metabolic changes in cancer do not merely reflect a condition “sine qua non”, but that defined metabolic changes can also drive tumorigenesis 12. There are three IDH isoforms, namely IDH1 that resides in the cytoplasm and peroxisomes and the two mitochondrial isoforms IDH2 and IDH3. IDH1 and IDH2 convert isocitrate, generated in the TCA cycle, to alpha-ketoglutarate (α-KG) with concomitant generation of NADPH.
Mutations in IDH1 and IDH2 are found in over 70% of lower-grade gliomas, as well as in glioblastomas and acute myeloid leukemias (AML) 10, 11, 13. Mutant IDH1 and IDH2 proteins have altered enzymatic activity and further convert α-KG to D-2-hydroxyglutarate (2-HG), a putative oncometabolite, as it was found to be sufficient in driving tumorigenesis under certain conditions 14, 15. The mechanism by which 2-HG drives tumorigenesis is still not fully understood; however, three potential mechanisms have been proposed, namely interference with normal mitochondrial function, dysregulation of cellular redox, and, the most prominent one, competitive inhibition of dioxygenase enzymes (for a recent review 16). In human, over 80 different dioxygenases have been identified. Ten-eleven translocation enzymes (TET1, TET2, and TET3) and the JmjC-domain histone demethylases are involved in DNA and histone demethylation, respectively. Importantly, inhibition of TET enzymes by 2-HG results in epigenetic alterations similar to those commonly found in cancer 17, 18.
Previous gene expression analyses to identify FOXO-regulated genes were performed either with constitutively active FOXO mutants or in the setting of a stable full FOXO knockout. However, constitutive FOXO overexpression or prolonged depletion may result in the activation of compensatory mechanisms and thus may introduce a bias in the analysis. Here, we describe gene expression analysis after a transient FOXO depletion with shRNA. We identify several metabolic genes to be regulated by FOXO-dependent transcription, among them IDH1. We show that FOXO transcription factors directly regulate the transcription of IDH1 and thereby regulate the levels of cytosolic α-KG and NADPH. Consequently, in cells carrying mutant IDH1, FOXOs regulate the transcription of the mutant IDH1 and thereby maintain the level of the oncometabolite 2-HG. In agreement with the proposed role of 2-HG in cell growth 19, we find the loss of FOXO expression in these cells results in changes in DNA and histone demethylation and affects their growth. In this setting, FOXOs are required for the regulation of oncogenic IDH1 and therefore can be considered to act oncogenic, in contrast to their widely considered role as tumor suppressors.
Results
FOXOs regulate IDH1 levels
To identify genes regulated by the endogenous FOXOs, we performed a global gene expression analysis in HeLa cells stably expressing a doxycycline-inducible shRNA targeting FOXO1 and FOXO3 and further transfected with siRNA targeting FOXO4. We used HeLa cells to circumvent the contribution of p53-dependent transcription in our analysis, considering that FOXOs and p53 are regulated by similar upstream pathways and share a number of transcriptional targets 1. Comparative analysis of control and FOXO knockdown cells identified multiple genes involved in metabolic processes as FOXO-regulated targets (Fig1A, Supplementary Fig S1A and Supplementary Table S1), and we focused on IDH1 for further analysis. To corroborate our finding that endogenous FOXOs regulate the transcription of IDH1, and to examine whether this regulation is specific to cancer cells, or also occurs in untransformed cells, we compared untransformed retina epithelium cells (RPE cells) with HeLa cells both expressing doxycycline-inducible shRNA targeting FOXO1 and FOXO3 (RPEshFOXO and HeLashFOXO cells, respectively). FOXO depletion (> 90%) resulted in a > 60% reduction in IDH1 mRNA transcripts in both cell lines (Fig1B and Supplementary Fig S1A). IDH1 protein levels were also significantly reduced in both FOXO-depleted cell lines (Fig1C and Supplementary Fig S1A). Thus, regulation of IDH1 by FOXOs is not restricted to cancer cells.
Figure 1.
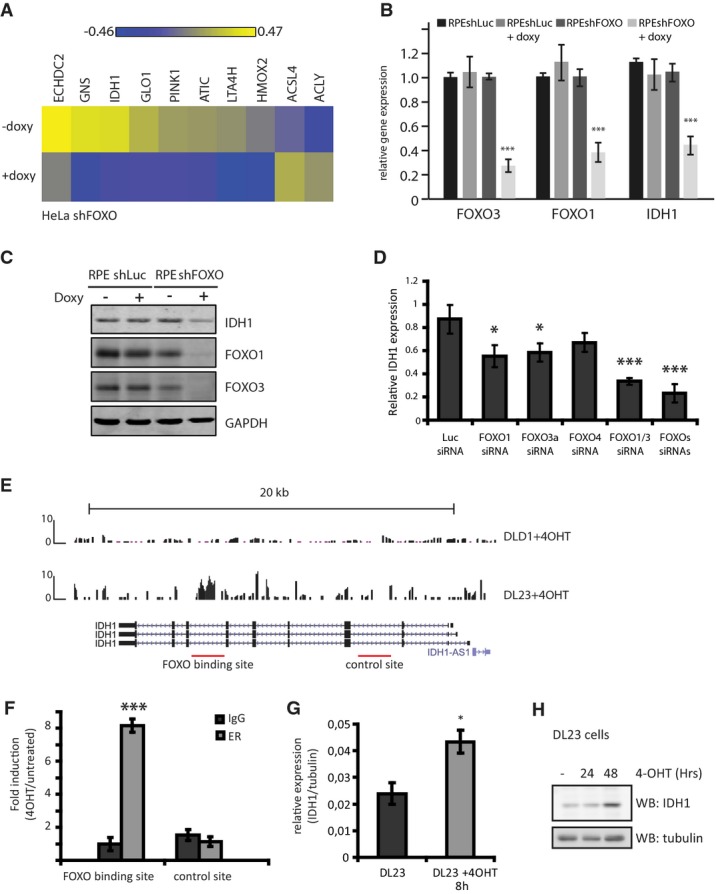
- Heat map of selected metabolic genes showing differential expression in FOXO-depleted HeLashFOXO cells. HeLashFOXO cells were grown in the presence or absence of doxycycline for 72 h before RNA was collected and used for gene expression analysis. Results are represented as LIMMAS, comparing each sample against a common reference.
- Relative FOXO3, FOXO1, and IDH1 mRNA levels in RPEshLuc and RPEshFOXO depleted or not of FOXOs. RPEshFOXO cells were cultured in the presence or absence of doxycycline for 72 h before sample collection (n = 3).
- IDH1, FOXO1, and FOXO3 protein levels in RPEshLuc and RPEshFOXO depleted or not of FOXOs. Cells were grown in the presence or absence of doxycycline for 72 h before sample collection. Results are representative of at least three independent experiments.
- Relative mRNA levels of IDH1 in RPE cells transfected with the indicated siRNAs. RNA samples were collected 72 h post-siRNA transfection. IDH1 expression was normalized by tubulin expression (n = 3).
- ChIP-sequencing analysis identified a FOXO3 binding site in the IDH1 genomic region in DL23 cells.
- ChIP–qPCR in DL23 cells 4 h after FOXO3 activation. The IDH1 (+) and the IDH1 (−) introns correspond to the FOXO binding site and the negative site in IDH1 (n = 3).
- Relative IDH1 mRNA levels in DL23 cells. DL23 cells were induced with 4-OHT for 4 h before sample collection (n = 3).
- IDH1 protein levels in DL23 cells grown in the presence of 4-OHT for different time points before sample collection. Result is representative of three independent experiments.
To identify whether IDH1 is a common target gene for all FOXOs or a specific trait of one isoform, we analyzed the contribution of each individual FOXO member on IDH1 transcription. After siRNA-mediated silencing of each FOXO member separately or combined in RPE cells, we found that knockdown of either FOXO1 or FOXO3a significantly reduced IDH1 expression (Fig1D). There was also a small contribution of FOXO4 to IDH1 expression, albeit that this appeared weaker than the FOXO1 and/or FOXO3 contribution (Fig1D). Taken together, these results show that FOXOs act redundantly to regulate IDH1 expression.
To assess whether endogenous FOXO activity is able to regulate IDH1, we used NIH3T3 cells overexpressing the insulin receptor (A14 cells 20). In these cells, FOXOs are inhibited by PI3K/PKB(AKT) signaling after insulin addition. Following treatment of A14 cells with insulin for 4 h, a significant decrease in IDH1 levels was observed, indicating that not only shRNA-mediated FOXO knockdown but also inhibition of endogenous FOXO activity through canonical PI3K/PKB(AKT) signaling regulates IDH1 expression (Supplementary Fig S1B). Evaluation of the other IDH isoforms (IDH2, IDH3A, IDH3B, and IDH3G) revealed that their levels remained unchanged or even increased following prolonged FOXO knockdown (Supplementary Fig S1C), suggesting that compensatory mechanisms are activated in response to a decrease in IDH1 expression.
Previously, we have used DL23 cells (DLD1 colon carcinoma cells expressing a 4-hydroxytamoxifen (4-OHT)-inducible FOXO3(A3)ER construct) to investigate genome occupancy of FOXO3a(A3) by chromatin immunoprecipitation (ChIP)-sequencing analysis 21. Data analysis identified a FOXO3 binding site in an IDH1 intron, following FOXO3(A3) induction with 4-OHT, which appeared specific as it was not identified in DLD1 control cells after 4-OHT treatment (Fig1E). Therefore, we performed a FOXO3 ChIP in DL23 cells after FOXO3(A3) activation. Subsequent analysis of IDH1 genomic regions verified the FOXO3 occupancy selectively on this region (Fig1F). FOXO3 binding appears to be linked to IDH1 transactivation, as FOXO3(A3) activation in DL23 cells significantly increased the IDH1 mRNA (Fig1G) and protein (Fig1H) levels. Taken together, our data indicate that FOXOs regulate IDH1 transcription directly in untransformed and cancer cell lines.
Reduced levels of α-KG and NADPH in FOXO-depleted cells
IDH1 functions in the cytoplasm to convert isocitrate to α-KG. To determine whether the reduced level of IDH1 expression in FOXO-depleted cells affects this conversion, we measured α-KG levels in RPEshFOXO cells after FOXO knockdown. Levels of α-KG were reduced by 50% after FOXO depletion (Fig2A). This reduction in α-KG levels was similar to that following siRNA-mediated IDH1 depletion (Supplementary Fig S2A). Importantly, α-KG levels remained unaffected following FOXO knockdown when cells ectopically expressed IDH1 (Fig2A) indicating that the reduced α-KG level is the consequence of IDH1 expression regulation by FOXOs.
Figure 2.

- α-KG levels in RPEshLuc, RPEshFOXO, and RPEshFOXO-IDH1 cells depleted or not of FOXOs (+doxy). Cells were cultured in the presence or absence of doxycycline for 96 h before sample collection and analysis by LC-MS (n = 5).
- Metabolite levels in RPEshLuc, RPEshFOXO, and RPEshFOXO-IDH1 depleted or not of FOXOs. Cells were cultured in the presence or absence of doxycycline for 96 h before sample collection and analysis by LC-MS. Metabolite levels were normalized by the non-treated condition. Data were obtained from four biological replicates. Schematic representation of the effect of FOXOs depletion on metabolites. Metabolites in blue and green correspond to no-change and decreased under FOXO depletion, respectively. Metabolites in black were not measured.
- NADPH/NADP+ ratio in RPEshLuc, RPEshFOXO, and RPEshFOXO-IDH1 cells depleted or not of FOXOs. To deplete FOXOs, cells were cultured in the presence or absence of doxycycline for 48 h and, thereafter, the medium was changed to medium with no glucose, supplemented with pyruvate and glutamine for another 48 h before sample collection (n = 3).
- Relative mRNA expression levels of various NADPH-generating enzymes (G6PD, ME1, ME2, IDH2) in RPEshLuc and RPEshFOXO cells depleted or not of FOXOs. Cells were cultured in the presence or absence of doxycycline for 72 h before sample collection (n = 3).
To analyze whether FOXO depletion affects α-KG specifically, we also analyzed the levels of other metabolites that impinge on α-KG metabolism (Fig2B). We observed isocitrate/citrate and glutamine to be reduced following FOXO knockdown, whereas other TCA metabolites (succinate, fumarate, malate) as well as glutamate remained unchanged. α-KG levels can also be maintained through glutaminolysis. However, the glutamate level does not change. Additionally, the α-KG level is recovered by IDH1 add-back and combined this indicates that the decrease in α-KG is due to FOXO regulation of IDH1. Previously, we have shown that FOXOs regulate glutamine synthase and this likely underlies the reduction in glutamine after FOXO knockdown 22. Besides α-KG and glutamine, also isocitrate/citrate levels are reduced. Reductive carboxylation involves these metabolites, and concomitant regulation of these metabolites by FOXO may indicate a role for FOXO under conditions, for example hypoxia, in which reductive carboxylation occurs.
Conversion of isocitrate to α-KG catalyzed by IDH1 also results in the generation of NADPH, and in addition to the pentose phosphate pathway (PPP), this provides a major source of NADPH for cells 23, 24. Thus, we analyzed the ratio of NADPH over NADP+ in cells depleted of FOXOs. To circumvent the contribution of the PPP to NADPH generation in our analysis, we grew cells in medium without glucose but supplemented with glutamine and pyruvate to maintain mitochondrial metabolism. We found that under these conditions, the NADPH/NADP+ ratio was significantly reduced in the FOXO-depleted cells (Fig2C), similar to cells transfected with siRNA targeting IDH1 (Supplementary Fig S2B). Importantly, this reduction in NADPH/NADP+ ratio was due to FOXO-mediated IDH1 regulation, as IDH1 add-back prevented NADPH/NADP+ reduction following FOXO knockdown (Fig2C). Furthermore and in agreement with the additional role of the PPP in NADPH generation, we observed a reduction in NADPH, albeit to a lesser extent when cells were grown in medium containing also glucose rather than only pyruvate-/glutamine-supplemented medium (Supplementary Fig S2C). Expression of other enzymes that generate NADPH remained either unchanged (G6PD) or even increased following FOXO knockdown (ME1, ME2, IDH2), suggesting compensatory mechanisms (Fig2D).
Cytoplasmic NADPH is utilized to reduce GSSG (glutathione disulfide, oxidized) to GSH (glutathione, reduced), which is subsequently used for ROS detoxification 25. FOXOs are well known for their contribution in reducing intracellular ROS levels 6, 26; therefore, we assessed the ratio of GSH/GSSG in cells depleted of FOXOs and grown under conditions that should inhibit any PPP contribution. We found a small but significant reduction in the levels of reduced glutathione in the FOXO-depleted cells (Supplementary Fig S2D).
FOXOs regulate the expression of IDH1 R132C and the levels of 2-HG in HT1080 cells
Correlative studies in gliomas revealed a negative correlation between mutations that support FOXO inactivation (PTEN mutation, EGFR amplification) and IDH1 mutation 13. We therefore investigated whether FOXOs also regulate the expression of mutant IDH1. To this end, we used HT1080 cells, a fibrosarcoma-derived cell line that endogenously carries the IDH1 R132C mutation. In these cells, we introduced the doxycycline-inducible shRNA targeting FOXOs and established HT1080shFOXO polyclonal cell lines. Endogenous FOXO is not excluded from the nucleus in these cells, is still controlled by PI3K signaling (Supplementary Fig S3A), and is transcriptionally active as we observed that upon FOXO depletion, IDH1 transcription is significantly reduced (Fig3A). This reduction is also reflected in the IDH1 protein levels (Fig3B). We further evaluated the levels of 2-HG, which were reduced by approximately 70% in the FOXO-depleted HT1080 cells (Fig3C). Again, 2-HG reduction by FOXO depletion was similar to that observed by siRNA-mediated IDH1 knockdown (Supplementary Fig S3B).
Figure 3.
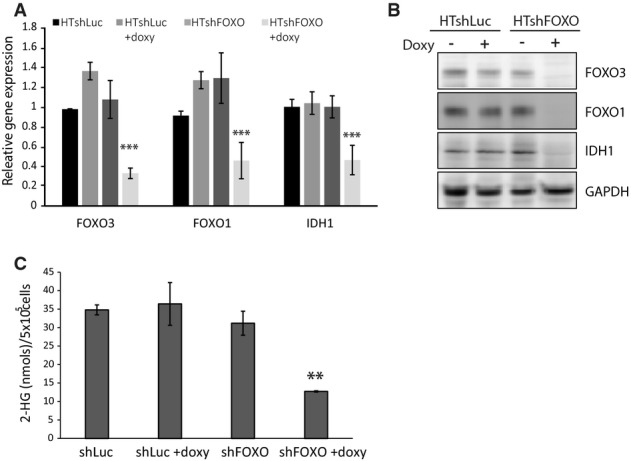
- Relative FOXO3, FOXO1, and IDH1 mRNA levels in HT1080shLuc and HT1080shFOXO. Cells were cultured in the presence or absence of doxycycline for 72 h before sample collection.
- FOXO3, FOXO1, and IDH1 protein levels in HT1080shLuc and HT1080shFOXO. Cells were grown in the presence or absence of doxycycline for 72 h before sample collection.
- 2-HG levels in HT1080shLuc and HT1080shFOXO cells depleted or not of FOXOs. HT1080shFOXO cells were cultured in the presence or absence of doxycycline for 72 h before sample collection and analysis by LC-MS. Data are from 4 biological replicates (n = 4).
FOXOs are required for the oncogenic potential of HT1080 cells
A potential mechanism by which mutant IDH1 mediates its oncogenic effect is by inhibition of α-KG-dependent dioxygenases, through competitive binding of 2-HG to their active center. These enzymes include histone demethylases and TET 5-methylcytosine (5 mc) hydroxylases [converting 5 mc to 5-hydroxymethylcytosine (5 hmc)] 16. We first assessed the effects of FOXOs depletion on the levels of histone H3 lysine 4 trimethylation (H3K4me3) and H3K9me3, which are histone marks reported to be regulated by the enzymatic activity of mutant IDH1 27. FOXO depletion, similar to silencing of IDH1, resulted in increased demethylation of both H3K4me3 and H3K9me3 (Fig4A). We next assessed the effects of FOXO depletion on the DNA methylation status of HT1080shFOXO cells. To this end, we isolated genomic DNA following FOXO or IDH1 depletion and assessed the relative levels of 5 mc and 5 hmc. FOXO depletion in HT1080 cells appears to restore the function of TET proteins, as it is accompanied by a concomitant decrease in 5 mc and increase in 5 hmc levels (Fig4B). Because in cancer cells TET enzyme activity is usually low, we ectopically expressed TET1 in HT1080 cells. Again, a reduction in FOXO levels is accompanied by an increase in 5 hmc. Importantly, exogenous treatment of HT1080 with 500 μM cell-permeable 2-HG ((R)-TFMB-2HG 15) rendered TET enzyme activity insensitive to FOXO depletion, suggesting that indeed FOXOs regulate TET activity through the regulation of 2-HG (Supplementary Fig S4A). Collectively, these data suggest a model in which FOXO-dependent transcription of IDH1 R132C is required for maintaining a high level of 2-HG and therefore contributes to 2-HG-dependent tumor-promoting epigenetic changes.
Figure 4.

- Analysis of histone methylation levels in HT1080shFOXO cells grown in the presence or absence of doxycycline for 72 h before sample collection. A representative Western blot from three independent experiments is shown. IDH1 silencing by siRNA serves as a positive control. Numbers indicate average decrease compared to control. Standard deviation for H3K4me3 and H3K9me3 was 0.07 and 0.09, respectively.
- Analysis of DNA methylation and hydroxymethylation levels in HT1080shFOXO cells grown in the presence or absence of doxycycline for 72 h before sample collection. A representative dot-blot from three independent experiments is shown. IDH1 silencing by siRNA serves as a positive control.
- Colony formation of HT1080shFOXO cells in soft agar. A total of 20,000 cells were plated in a 0.35% top layer, on top of a 0.5% bottom layer. Doxycycline was added to the upper layer when appropriate. Colonies were allowed to grow for 21 days.
- Growth curves of HT1080shFOXO transfected with siSCR or siIDH1 and treated or not with doxycycline. A total of 20,000 cells were plated and allowed to grow for several days; cells were collected and counted every 24 h for 6 days.
- Growth curves of HT1080shLuc and HT1080shFOXO cells grown in the presence or absence of doxycycline. A total of 20,000 cells were plated and allowed to grow for several days; cells were collected and counted every 24 h for 6 days.
- Determination of apoptosis in RPEshLuc, RPEshFOXO, RPEshFOXO siIDH1. RPEshLuc, RPEshFOXO, RPEshFOXO transfected with siIDH1, and RPEshFOXO cultured in the presence of doxycycline during 5 days. Cells were harvested and incubated with fluorescent annexin antibody. Positive cells were counted by FACS analysis. Where indicated, the cells were grown in the presence of 500 μM of 2-hydroxyglutarate (2-HG).
Considering that mutant IDH1 drives tumor progression, we assessed the contribution of FOXOs in this process. Initially, we observed a strong decrease in the colony-forming ability of HT1080 cells depleted of FOXOs (Fig4C), similar to what was reported for IDH1 depletion 19. To study the underlying mechanism and the contribution of IDH1, we next measured cell growth over time. FOXO, as well as IDH1 depletion in HT1080 cells, resulted in a reduction in cell number over time (Fig4D), albeit that IDH1 depletion reduced to a lesser extent compared to FOXO knockdown. In agreement with this result, the reduction in cell number by FOXO knockdown is in part rescued by exogenous treatment of cells with 500 μM (R)-TFMB-2HG (Fig4E). Cell number is determined by the combined rate of proliferation and apoptosis, and FOXOs are known to affect both processes. We therefore determined whether the reduction in cell number was due to the induction of apoptosis. Loss of FOXO in HT1080 cells induced apoptosis as shown by annexin V staining (Fig4F) and the appearance of cleaved PARP and cleaved caspase 3 (Supplementary Fig S4B). Importantly, IDH1 depletion in HT1080 did not affect apoptosis (Fig4F). FOXOs can regulate proliferation at multiple levels and by multiple mechanisms, and taken together, these data suggest that IDH1 regulation by FOXO in HT1080 cells in part contributes to the regulation of proliferation by FOXOs rather than apoptosis. This is in agreement with a recent report showing that inhibiting mutant IDH1 enzymatic activity or reducing the levels of mutant IDH1 delays growth 19. Collectively, these results suggest that cells carrying mutant IDH1 require FOXO to sustain IDH1 levels and this thereby contributes in part to their proliferative and oncogenic potential.
Discussion
Here, we provide evidence that FOXOs directly regulate the expression of IDH1 and thereby mediate metabolic functions of cancer and untransformed cells. FOXOs bind to an IDH1 intron sequence and induce transactivation of the gene. FOXO gene regulation by binding intron sequences has been shown before 28. By regulating the levels of IDH1, we find FOXOs to mediate different responses in normal and cancer cells expressing mutant IDH1 (schematically depicted in Fig5). In untransformed cells, FOXOs mediate the maintenance of cellular α-KG levels, thereby contributing to the function of α-KG-dependent dioxygenases in cellular differentiation and tumor suppression 17, 27, 29. Moreover, regulation of IDH1 by FOXOs contributes to the cytoplasmic levels of NADPH and thereby of GSH, providing yet another layer of protection against ROS and genomic instability. In cancer cells carrying IDH1 mutation, FOXOs are required for the maintenance of mutant IDH1 levels to sustain the high level of the oncometabolite 2-HG. In contrast to normal cells, FOXOs now inhibit the α-KG-dependent dioxygenases, such as TET enzymes and histone demethylases, resulting in de-differentiation and tumor progression.
Figure 5.
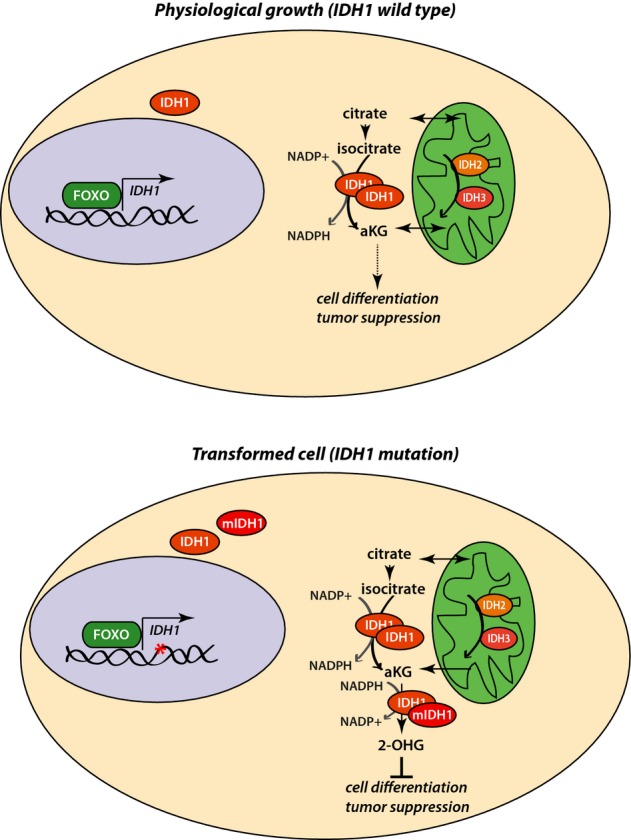
FOXOs mediate tumor-suppressive/tumor-promoting functions by regulating IDH1 transcription
Model depicting the functions mediated by FOXOs in different genomic and physiological settings, by the regulation of IDH1 transcription (for more information, refer to text).
Our data support a dual role of FOXOs in mediating both tumor-suppressive and tumor-promoting functions, by the regulation of IDH1 (wild-type and mutant) transcription. This appears at odds with the established role of FOXOs as bona fide tumor suppressors 6. However, several lines of evidence implicate a role for FOXOs in cancer progression due to their role in maintaining cellular homeostasis both in normal and in cancer cells. For example, FOXOs are involved in ROS detoxification by regulating the transcription of several ROS scavengers, including MnSOD and Sestrin 3 26, 30; cancer cells that inherently have high levels of intracellular ROS and an ensuing increased instability are known to benefit from antioxidants 31. FOXOs are also involved in the survival and maintenance of adult stem cells 30, 32, 33, 34, and recently, FOXO3 was shown to be required for the maintenance of leukemia initiating cells in chronic myeloid leukemia 35. In acute myeloid leukemia (AML), inhibition of FOXO results in myeloid differentiation and subsequent AML cell death 7. AML also shows a high prevalence for IDH1/IDH2 mutations, and interestingly, the role of 2-HG in the maintenance of glioma has been attributed to its role in blocking differentiation 19. Thus, literature suggests both FOXO and mutant IDH1/IDH2 contribute to maintaining a de-differentiated state in cancer. In HT1080 cancer cells, we observe that FOXO inhibition results in tumor cell loss and this involves at least in part inhibition of IDH1 expression and reduced production of the “onco-metabolite” 2-HG (Fig3C). All taken together suggest that FOXOs through regulation of mutant IDH1 may maintain a “de-differentiated” state in HT1080 cells. It will be therefore of interest to study in various tumor settings whether FOXOs contribute to the de-differentiated state and that, therefore, loss of FOXO in tumor cells results in the induction of differentiation and apoptosis and to what extent this involves IDH1 regulation.
The metabolic rewiring of cancer cells results in the acquisition of unique characteristics compared to untransformed cells and thereby, in principal, allows the selective targeting of transformed cells. Better understanding of cancer metabolism will thus provide new opportunities for the treatment of certain cancers 36, 37. In particular, in the case of mutant IDH1, specific small-molecule inhibitors have been developed that efficiently target the mutant enzymatic activity 19, 38 and hold promise for the treatment of lower-grade gliomas and AML. Identifying upstream components of IDH1 regulation could potentially be equally beneficial, especially in cases that resistance mechanisms develop. In this study, we identified FOXOs as regulators of IDH1 levels and thereby as important cellular components for the metabolic rewiring of cancer cells. FOXO function is in turn linked to their subcellular localization and efficient targeting of the molecular pathways that are responsible for FOXO nuclear retention in transformed cells could potentially prove beneficial for the treatment of cancer types related to IDH1.
Materials and Methods
Cell culture and transfections
The human retina epithelial cells RPE were maintained in DMEM/Ham F12 (Gibco) supplemented with 10% FBS (Gibco), glutamine (Lonza), and penicillin–streptomycin. DL23 cells were cultured in RPMI (Lonza) supplemented with 10% FBS and penicillin–streptomycin. HeLa, A14, and HT1080 cells were maintained in DMEM (Lonza) supplemented with 10% FBS, 2 mM glutamine (Lonza), and penicillin–streptomycin. DL23 and A14 cells were previously described 20, 28.
To establish the HeLashFOXO, RPEshFOXO, and the HT1080shFOXO cells, HeLa, RPE, and HT1080 cells, respectively, were virally transduced with pH1tet-flex/FH1t(FOXO1/3)UTG and the positive clones were selected with FACS. RPEshLuc and HT1080shLuc were generated by viral transfection with pH1tet-flex/FH1t(Luc)UTG as previously described by Paddison et al 39. RPEshFOXO-IDH1 add-back cell line was generated by viral transfection of RPEshFOXO with a plasmid containing the coding sequence of IDH1 tagged with V5 epitope and containing puromycin resistance. Polyclonal cell lines were derivatized after puromycin selection.
IDH1 and SCR smart pools were purchased from Dharmacon. siRNA transfections were performed with HiPerFect (Qiagen), and plasmid transfections were performed with FuGENE HD (Promega), according to the manufacturer's instructions. Cell-permeable 2-hydroxyglutarate ((R)-TFMB-2HG) was purchased from Looper Laboratories (U.S.) and was used in a final concentration of 500 μM.
RNA extraction and quantitative PCR (Q-PCR)
RNA was extracted with the RNeasy kit (Qiagen), with on-column DNase treatment (Qiagen), according to the manufacturer's instructions. RNA was reverse-transcribed with oligodT primers and the iScript cDNA synthesis kit (Biorad). Quantitative PCR was performed with FastStart SYBR Green Master mix (Roche). The primers used are listed in the Supplementary Information.
Protein extraction and Western blot
Total proteins were collected by direct lysis in Laemmli sample buffer. Proteins were run in SDS–PAGE and transferred to Polyscreen PVDF transfer membranes (PerkinElmer) or to Protran Nitrocellulose Membranes (GE Healthcare Life Sciences). Antibodies used were the following: anti-IDH1 (Cell Signaling), anti-FOXO1 and anti-FOXO3 (Santa Cruz), anti-H3K4me3 (Abcam), anti-H3K9me3 (Abcam), anti-H3 (Abcam), and tubulin (Calbiochem).
α-KG, 2-HG, and other metabolites measurement
For the determination of intracellular metabolites, including α-KG and 2-HG, cells that were cultured in DMEM either in the presence or absence of doxycycline for 96 h were plated in 6-well plates (400,000 cells per well) and grown for another 16 h. Thereafter, cells were washed with 10% NaCl, collected in ice-cold methanol and H2O and, after addition of chloroform, were allowed to extract at 4°C for 20 min. Subsequently, the organic and aqueous phases were separated by centrifugation and the aqueous phase was collected and evaporated to dryness. The residue was dissolved in 50 μl 0.1% formic acid of which 10 μl was injected for analysis. Analysis was performed employing a Waters Acquity UPLC system (Etten-Leur, The Netherlands), operated at a flow rate 300 μl/min, and an Acquity HSS T3 column (2.1 × 100 mm, 1.8 μm). The mobile phase consisted of 1% acetonitrile in water also containing 0.1% formic acid. The column outlet was coupled to a Waters Xevo triple quadrupole mass spectrometer equipped with an electrospray ion source operated in the negative mode.
NADPH/NADP+ and GSH/GSSG measurements
For the determination of NADPH/NADP+ and GSH/GSSG ratio, RPEshFOXO cells were grown in DMEM containing or not doxycycline for 48 h. Subsequently, the growth medium was switched to DMEM minus glucose (Gibco) supplemented with 10 mM pyruvate and 2 mM glutamine (Lonza) and cells were grown for another 48 h. NADP+ and NADPH were measured with the NADP/NADPH Quantitation Colorimetric kit (BioVision), according to the manufacturer's instructions. GSH and GSSG were measured with the Glutathione (total) detection kit (Enzo).
Growth rates and soft agar assays
To determine the differences in growth rates in the HT1080shFOXO cells after FOXO depletion, 20,000 cells per well were plated in 6-well plates and, subsequently, doxycycline was added in half of the wells. After 48 h and every 24 h thereafter, cells were collected and counted using the Countess Automated Cell Counter (Life Technologies). For the soft agar assay, 20,000 HT1080shFOXO cells were plated in a 0.35% top layer (DMEM, 10% FBS), on top of a 0.5% bottom layer (DMEM, 10% FBS). Where appropriate, the upper layer was supplemented with doxycycline. The cells were fed every 4 days with DMEM 10% FBS, and doxycycline was refreshed. Cells were allowed to grow for 3 weeks, and images were acquired in a Zeiss Axioskop 40 microscope.
Isolation of genomic DNA and 5-methylcytosine blot
HT1080shFOXO cells were grown in DMEM containing or not doxycycline for 96 h. Genomic DNA was isolated with the DNA mini kit (Qiagen), according to the manufacturer's instructions. Subsequently, 100, 50, 25, 10, and 5 ng of genomic DNA were spotted on Hybond N membrane (GE Healthcare) and then blotted with anti-methylcytosine antibody (Millipore) or anti-hydroxylmethylcytosine antibody (Active motif).
Acknowledgments
The authors wish to thank Dr. Arjan Brenkman and all other members of the Burgering laboratory for discussion. We thank Dr Ryan Looper (University of Utah, USA) for providing cell-permeable 2-HG. This work was financially supported by the Center of Biomedical Genetics (CBG), Cancer Genomics Center (CGC.nl), and the Dutch Cancer Foundation (KWF).
Author contributions
PC and MR-C designed and performed experiments and co-wrote the manuscript, JG and NMVD measured and analyzed metabolites, MvT and MH performed experiments, MGK performed the microarray analysis, FH analyzed microarray results, FH, NMVD, and RL provided important reagents and discussed results, and BMTB designed experiments and co-wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Information
Review Process File
References
- Eijkelenboom A, Burgering BMT. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- Kops GJPL, Ruiter NDD, De Vries-Smits AMM, Powell DR, Bos JL, Burgering BMT. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Bakker WJ, Harris IS, Mak TW. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol Cell. 2007;28:941–953. doi: 10.1016/j.molcel.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Essers MAG, Weijzen S, de Vries-Smits AMM, Saarloos I, de Ruiter ND, Bos JL, Burgering BM. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J-H, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, Ferraro F, Mercier F, Singh H, Brumme KM, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146:697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Ward Patrick S, Thompson Craig B. Metabolic reprogramming: a cancer hallmark even warburg Did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones SN, Zhang X, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene. 2010;29:6409–6417. doi: 10.1038/onc.2010.444. [DOI] [PubMed] [Google Scholar]
- Bhagwat N, Levine RL. Metabolic syndromes and malignant transformation: where the twain shall meet. Sci Transl Med. 2010;2:54ps50. doi: 10.1126/scitranslmed.3001669. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakheja D, Medeiros LJ, Bevan S, Chen W. The emerging role of d-2-hydroxyglutarate as an oncometabolite in hematolymphoid and central nervous system neoplasms. Front Oncol. 2013;3:169. doi: 10.3389/fonc.2013.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman J-A, Kaelin WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgering BM, Medema RH, Maassen JA, van de Wetering ML, van der Eb AJ, McCormick F, Bos JL. Insulin stimulation of gene expression mediated by p21ras activation. EMBO J. 1991;10:1103–1109. doi: 10.1002/j.1460-2075.1991.tb08050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, van Triest MH, van Boxtel R, Schulze A, de Laat W, Cuppen E, et al. Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol Syst Biol. 2013;9:638. doi: 10.1038/msb.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol. 2012;14:829–837. doi: 10.1038/ncb2536. [DOI] [PubMed] [Google Scholar]
- Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Shin JY, Kim M, Hann S-K, Oh SH. Expression of cytosolic NADP+-dependent isocitrate dehydrogenase in melanocytes and its role as an antioxidant. J Dermatol Sci. 2012;65:118–125. doi: 10.1016/j.jdermsci.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- Kops GJPL, Dansen TB, Polderman PE, Saarloos I, Wirtz KWA, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJPL, Medema RH, Glassford J, Essers MAG, Dijkers PF, Coffer PJ, Lam EW, Burgering BM. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol. 2002;22:2025–2036. doi: 10.1128/MCB.22.7.2025-2036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo H, Lyssiotis CA, Zhang Y, Ying H, Asara JM, Cantley LC, Paik JH. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J. 2013;32:2589–2602. doi: 10.1038/emboj.2013.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm AM, Khalturin K, Anton-Erxleben F, Hemmrich G, Klostermeier UC, Lopez-Quintero JA, Oberg HH, Puchert M, Rosenstiel P, Wittlieb J, et al. FoxO is a critical regulator of stem cell maintenance in immortal Hydra. Proc Natl Acad Sci USA. 2012;109:19697–19702. doi: 10.1073/pnas.1209714109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yalcin S, Lee DF, Yeh TY, Lee SM, Su J, Mungamuri SK, Rimmelé P, Kennedy M, Sellers R, et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat Cell Biol. 2011;13:1092–1099. doi: 10.1038/ncb2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, Hirao A, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676–680. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Information
Review Process File