

Abstract
Despite the success of genome-wide association studies (GWAS) in detecting a large number of loci for complex phenotypes such as rheumatoid arthritis (RA) susceptibility, the lack of information on the causal genes leaves important challenges to interpret GWAS results in the context of the disease biology. Here, we genetically fine-map the RA risk locus at 19p13 to define causal variants, and explore the pleiotropic effects of these same variants in other complex traits. First, we combined Immunochip dense genotyping (n = 23,092 case/control samples), Exomechip genotyping (n = 18,409 case/control samples) and targeted exon-sequencing (n = 2,236 case/controls samples) to demonstrate that three protein-coding variants in TYK2 (tyrosine kinase 2) independently protect against RA: P1104A (rs34536443, OR = 0.66, P = 2.3x10-21), A928V (rs35018800, OR = 0.53, P = 1.2x10-9), and I684S (rs12720356, OR = 0.86, P = 4.6x10-7). Second, we show that the same three TYK2 variants protect against systemic lupus erythematosus (SLE, Pomnibus = 6x10-18), and provide suggestive evidence that two of the TYK2 variants (P1104A and A928V) may also protect against inflammatory bowel disease (IBD; Pomnibus = 0.005). Finally, in a phenome-wide association study (PheWAS) assessing >500 phenotypes using electronic medical records (EMR) in >29,000 subjects, we found no convincing evidence for association of P1104A and A928V with complex phenotypes other than autoimmune diseases such as RA, SLE and IBD. Together, our results demonstrate the role of TYK2 in the pathogenesis of RA, SLE and IBD, and provide supporting evidence for TYK2 as a promising drug target for the treatment of autoimmune diseases.
Introduction
Human genetics has the potential to identify biological pathways that lead to complex diseases such as rheumatoid arthritis (RA). Meta-analyses of multi-ethnic genome-wide association studies (GWAS) in RA have now identified more than 100 loci associated to risk of disease [1]. Despite the success of GWAS, the associated loci usually include several genes in the region of linkage disequilibrium (LD), thus providing limited information to incriminate the causal genes. Cis-eQTL effects of SNPs that are in LD with the index SNPs have been reported in immune cell types, often describing association with variation of expression of several genes in the locus. Additionally, only a few RA loci harbour missense variants [1,2]. Even then, however, it is not clear if these SNPs are responsible for the signal of association, thus illustrating the challenges of interpreting GWAS findings to provide insights into the disease biology [3,4].
Several studies have described genes in GWAS loci that harbour multiple independent functional mutations associated with a disease, providing genetic evidence for causality and, by extension, insight into disease pathogenesis [5–18]. This highlights the critical need for detailed analyses combining dense genotyping and sequencing to pinpoint causal genes with an allelic series of associated protein-coding functional variants. In addition, this approach has the potential to clarify disease mechanisms and to identify novel therapeutic targets to guide drug discovery [19,20].
Allelic pleiotropy, where one genetic variant influences several distinct phenotypes, is increasingly recognized as a common phenomenon from GWAS findings [21], especially in the field of immune-mediated diseases [22]. Investigation of pleiotropic effects can inform disease biology and predict potential adverse events of targets derived from human genetics [20–22]. One approach to comprehensively investigate pleiotropy is through genotype data linked to clinical data derived from electronic medical records (EMR) [23]. This unbiased approach, called phenome-wide association study (PheWAS), allows for genotypes of interest to be tested for association to hundreds of clinically-relevant phenotypes [24]. We and others have demonstrated the value of this approach in successfully replicating results from GWAS and assessing pleiotropic effects [25–29].
One locus that has emerged from GWAS in RA is on chromosome 19p13 [2]. This locus spans 286 kb (LD region r2>0.5) and contains 11 genes, including TYK2 (tyrosine kinase 2), a member of the Janus kinase (JAK) family of proteins that mediates signalling downstream of several cytokine receptors [30,31], and intercellular adhesion molecule (ICAM)–coding genes (ICAM1, ICAM3, ICAM4, ICAM5), which are part of the immunoglobulin superfamily. Based on the biology of RA alone [32], TYK2 and ICAM genes are equally likely candidate genes responsible for the signal of association. In support of TYK2, the signal of association is driven by a low-frequency missense variant in TYK2, with a reported odds ratio (OR) of 0.62, protective for RA [2]. The variant, rs34536443 (p.P1104A), is predicted to be damaging using function prediction tools, and has been reported to be loss-of-function (LOF), affecting TYK2 kinase activity in primary T cells, fibroblast cell lines and B cell lines [33,34].
Genetic variation in the 19p32/TYK2-ICAM locus has also been associated with several other autoimmune diseases, including psoriasis, multiple sclerosis (MS), Type 1 diabetes (T1D), Crohn’s disease (CD), ulcerative colitis, and systemic lupus erythematosus (SLE) [35–42]. However, the leading signals of association differ in terms of implicated SNPs, effect sizes and directions of effect (protection vs risk). Whether these differences in association signals in autoimmune diseases refer to distinct causal variants and/or causal genes, or pleiotropic effects of the same variants remains unclear.
In the current study, we performed a detailed analysis of the 19p32/TYK2-ICAM locus to comprehensively investigate the contribution of common and rare protein-coding variants to RA susceptibility using 1) dense genotyping of the locus with the Immunochip and Exomechip genotyping platforms and 2) exon sequencing of all 11 genes within this locus. We then used Exomechip data to investigate the association of TYK2 missense variants with two additional autoimmune diseases, SLE and IBD, both of which have been reported previously to harbour associations to genetic variants in this locus [35–38,40]. Finally, we linked our findings with electronic medical records (EMR) to comprehensively assess pleiotropic effects of the RA-associated TYK2 missense variants.
Results
Three independent TYK2 protein-coding variants protect against RA
To fine-map the 19p32/TYK2-ICAM locus, we first performed a stepwise conditional analysis using Immunochip genotype data available for 7,222 ACPA+ RA cases and 15,870 controls of European ancestry (S1 Table) [2]. In this analysis, we applied no minor allele frequency (MAF) cut-off, in order to investigate all variants at the locus. The strongest signal was at the previously reported TYK2 missense variant rs34536443 (P1104A, minor allele frequency [MAF] = 3.4%, OR = 0.62, P = 2.2x10-14) (Fig 1A and S2 Table) [2]. After conditioning on the P1104A variant, we observed a significant association at P = 4.0x10-9 with an OR = 0.42, driven by the rare TYK2 missense variant A928V (rs35018800, MAF = 0.8%) (Fig 1B and S2 Table). Conditional on both TYK2 P1104A and A928V variants, we observed a third signal of association at P = 5.4x10-4 (OR = 0.87), driven by the TYK2 missense variant I684S (rs12720356; MAF = 8%) (Fig 1C and S2 Table) [2]. After conditioning on TYK2 P1104A, A928V and I684S variants, we observed no association at P<0.01 (Fig 1D). We used the genotypes from the three TYK2 variants P1104A, A928V and I684S to build haplotypes. The haplotype model confirmed independence of the variants, with the minor alleles of the three variants lying on different haplotypes (Fig 2A). All three missense variants were predicted to be damaging using Polyphen-2 and SIFT [43,44].
Fig 1. Results from stepwise conditional analysis of the TYK2 locus.

We fine-mapped the TYK2 locus using Immunochip data available for 7,222 ACPA+ RA cases and 15,870 controls (MAF>0). (A) In the meta-analysis, the best signal of association was at the TYK2 missense variant P1104A (rs34536443).(B) Conditional on P1104A, the best signal of association was at the TYK2 missense variant A928V (rs35018800). (C) Conditional on P1104A and A928V variants, the best signal of association is at the TYK2 missense variant I684S (rs12720356). (D) Conditional on the 3 RA-protective variants in TYK2, we observed no additional signal of association at the locus (best signal is rs3176768, P = 0.01). P-values from meta-analyses of logistic regressions results from 6 Immunochip collections are shown. The three TYK2 missense variants predicted to be damaging and independently associated with RA risk are highlighted in green.
Fig 2. Contribution of 3 independent TYK2 protein-coding variants to protection from RA.
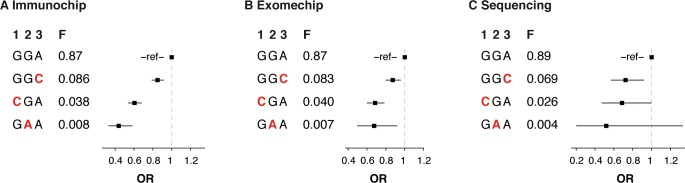
(A) Three variants with MAF>0.5% predicted to be damaging and protecting from RA (P1104A, A928V and I684S) were identified using Immunochip data for 7,222 ACPA+ RA cases and 15,870 controls of European ancestry. (B) The three variants were genotyped in an independent dataset on the Exomechip (4,726 RA cases, 13,683 controls). (C) The three variants genotypes were also available from exon sequencing of TYK2 in 1,118 RA cases, 1,118 matched controls of European ancestry. Frequencies of the independent haplotypes and odds ratios (OR) relative to the most frequent haplotype are shown. Minor alleles of the variants are highlighted in red. H, haplotypes; F, haplotype frequency; 1, P1104A; 2, A928V; 3, I684S.
To replicate the association signals at the three TYK2 missense variants, we used Exomechip genotype data in an independent set of 4,726 RA cases and 13,683 controls of European ancestry. We also directly sequenced the coding exons of TYK2 in an independent set of 1,118 RA cases and 1,118 matched controls of European ancestry (S1 Table). In both the Exomechip and the sequencing data, the effect sizes of the protective haplotypes built using the P1104A, A928V and I684S variants were highly similar to the effect sizes observed in the Immunochip data, and were significantly associated with protection from RA (Pomnibus = 4.6x10-8 in Exomechip; Pomnibus = 0.0058 in sequencing) (Fig 2B and 2C). Meta-analysis of the Immunochip, Exomechip and sequencing datasets confirmed replication of the three independent association signals in TYK2 (S2 Table). While both the P1104A and A928V variants reached genome-wide significance (PMETA = 2.3x10-21 and PMETA = 1.2x10-9, respectively), we estimated that a sample size of >20,000 RA cases would be required to observe an association at the I684S variant at genome-wide significance (P<5x10-8), based on the frequency and estimated effect size of the variant.
Together, these data implicate TYK2 rather than one of the ICAM (or other) genes in the region of LD as the most likely causal gene responsible for the signal of association.
Contribution of rare TYK2 protein-coding variants to RA
To comprehensively investigate the contribution of rare protein-coding variants, we analysed exon-sequencing data available for the 11 genes in the 19p32/TYK2-ICAM locus in 1,118 RA cases and 1,118 matched controls of European ancestry (S1 and S3 Tables). We restricted the analysis to protein-coding variants (nonsense or missense) with MAF<0.5%, thus excluding the TYK2 P1104A, A928Vand I684S variants.
We first performed gene-based association tests for each of the 11 genes (assessing significance using 10,000 permutations of case-control status), using 4 different methods: the burden test (BURDEN, one-sided test), the frequency weighted test (WT, one-sided test), the variable threshold test (VT, one-sided test), and SKAT-O (two-sided test) [45–47]. Using the one-sided methods, we performed two tests, to assess the accumulation of rare variants in cases and controls, respectively. We found no gene harbouring rare variants significantly associated with RA (at P<0.004, 0.05/11 genes), in either gene-based tests including all protein-coding variants or restricted to variants predicted to be damaging (P>0.01; Fig 3 and S4 Table).
Fig 3. TYK2 protein-coding variants identified by exon-sequencing of RA cases and controls.
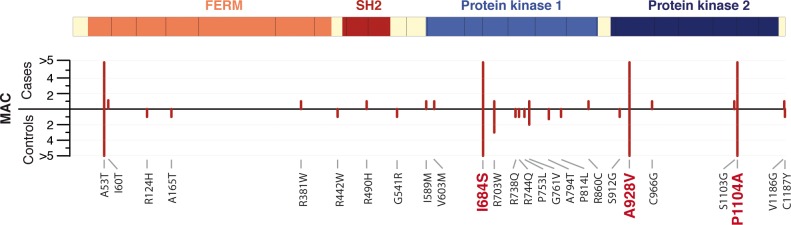
Using dense genotyping, we demonstrate that three TYK2 protein-coding variants predicted to be damaging, P1104A, A928V, and I684S, protect against RA (highlighted in red). By exon-sequencing in 1,118 RA cases and 1,118 controls, we identified 23 additional missense variants predicted to be damaging (PolyPhen-2 and SIFT), with no strong evidence of association to RA in gene-based association tests. The TYK2 coding exons, the protein domains, and the minor allele count (MAC) of the rare variants (MAC<5) in cases and controls are shown.
We then performed a sliding-window test. For each position in the protein-coding sequence of the 11 genes, we extracted all rare missense and nonsense variants in a 500 bp window centered on the position, and performed a window-based association test using SKAT-O (assessing significance using 1,000 permutations of case-control status; S1 Fig). We only observed a suggestive association at TYK2 resulting from an accumulation of rare missense variants predicted to be damaging (7 variants, including 5 singletons) in controls in the protein kinase 1 domain–coding region (P = 0.016) (Fig 3, S1 Fig, S4 and S5 Tables).
We also investigated the contribution of all TYK2 protein-coding variants genotyped in the Exomechip in our collection of 4,726 RA cases and 13,683 controls. We observed no additional single variant associated to RA beyond P1104A, A928Vand I684S (P>0.05; S2 Fig), and no aggregate signal of association driven by TYK2 variants with MAF<0.5% using SKAT-O (P = 0.80).
Together, these results support the finding that the TYK2 protein-coding variants P1104A, A928V and I684S variants are responsible for the signal of association, and that protein-coding variants in other genes in the TYK2 locus do not contribute to RA susceptibility.
Pleiotropic effects of RA-associated TYK2 variants in other autoimmune diseases
Loci implicated in risk of RA are also associated with risk of other autoimmune diseases [1,22]. TYK2 protein-coding variants have been associated with several autoimmune diseases, including systemic lupus erythematosus (SLE) and inflammatory bowel disease (IBD) [35–38,40]. In SLE, the reported association is with the common TYK2 missense variant V362F predicted to be benign (rs2304256, MAF = 23%, OR = 0.70) [35]; in IBD, the reported association is with the TYK2 variant I684S (OR = 1.12) [36], which we demonstrate as protecting against RA. Accordingly, we explored whether the three RA-associated TYK2 variants contributed to risk of SLE and IBD or whether the published variants provide a better genetic explanation for the signal of association.
Using Exomechip genotype data available for 3,053 SLE cases and 13,687 controls of European ancestry, we observed that the three RA-protecting TYK2 variants P1104A, A928V, and I684S protected against SLE (Pomnibus = 6x10-18), with effect sizes similar to the effect sizes in RA (Fig 4A). In this dataset, the TYK2 missense variant V362F previously reported to be associated with SLE [35,37,40] showed a protective effect (OR = 0.85 [0.79–0.92]) at P = 1.8x10-5. Importantly, the haplotype analysis highlighted that the V362F association was driven by imperfect LD to the three RA-associated missense variants P1104A, A928V, and I684S (Pomnibus, 3df = 6x10-18). Indeed, we found no effect of the haplotype carrying only the minor allele of the V362F variant (Fig 4A).
Fig 4. Association of the three RA-associated TYK2 missense variants with SLE and IBD.
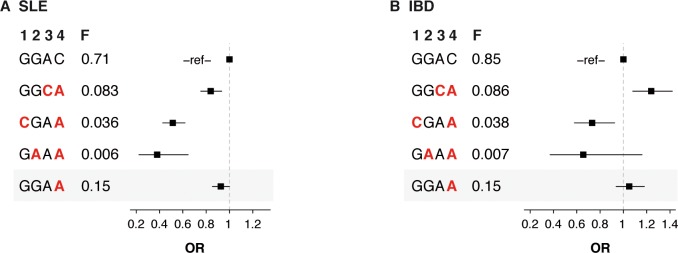
We used Exomechip data from 3,053 SLE cases and 13,687 controls (A) and 1,346 IBD cases and 13,687 controls (B) to built haplotypes using the RA-associated TYK2 variants P1104A, A928V and I684S. In the haplotype model, we also included the TYK2 SNP V362F, which has previously been reported to be associated with SLE (highlighted in gray). Frequencies of the independent haplotypes and odds ratios (OR) relative to the most frequent haplotype are shown. Minor alleles of the variants are highlighted in red. H, haplotypes; F, haplotype frequency; 1, P1104A; 2, A928V; 3, I684S; 4, V362F.
We also analysed Exomechip genotype data available for 1,346 IBD cases and 13,687 controls of European ancestry (Fig 4B). Consistent with previous reports in Crohn’s disease, the TYK2 I684S variant was associated with increased risk of IBD in our dataset (OR = 1.26 [1.10–1.43], P = 8x10-4) [36]. Interestingly, however, we observed a protective effect of the TYK2 P1104A variant (OR = 0.75 [0.60–0.93], P = 0.008). The point estimate of effect size at the TYK2 A928V variant was also consistent with protection against IBD (0.64 [0.37–1.1], P = 0.11), although our analysis was underpowered to detect an association at P< 0.05 at this SNP. While additional studies are required to definitively fine-map the TYK2 locus in IBD (as we have done in RA), our data suggest that TYK2 protein-coding variants contribute to IBD susceptibility.
Using the Exomechip data available for RA, SLE and IBD, we found no additional TYK2 variants associated at P<0.05, in either the disease-specific analyses or a diseases-combined analysis (based on the hypothesis that independent genetic variants contribute to susceptibility in all three autoimmune diseases combined) (S2 Fig). Finally, in gene-based association test (SKAT-0), we observed no aggregate signal of association driven by rare TYK2 protein-coding variants predicted to be damaging, in either SLE or IBD (P>0.05).
Comprehensive investigation of pleiotropic effects of RA-associated TYK2 variants using electronic medical records
We next sought to investigate whether the TYK2 P1104A, A928V and I684S variants protecting against RA and reported or predicted to be LOF were associated with other clinical diagnoses. To that end, we used two independent EMR clinical datasets linked to genotype data: 1) 3,005 individuals of European ancestry from the Informatics for Integrating Biology and the Bedside (i2b2) center [48], and 2) 26,372 individuals of European ancestry from Vanderbilt University Medical Center’s BioVU EMR-linked DNA biobank [49]. We performed an unbiased PheWAS for all common EMR-linked binary traits (N = 502 phenotypes), followed by an analysis focused on clinical traits related to TYK2 biology (N = 30 phenotypes).
In the PheWAS testing the association between the TYK2 variants and the 502 common binary phenotypes (phenotype frequency>1%), we only observed a significant association (P<1x10-4) between the P1104A variant and RA (OR = 0.65, PMETA = 2.3x10-5), with an effect size consistent to the effect size observed in the Immunochip RA case-control cohort (Fig 5A) [2]. We observed no PheWAS phenotype associated with the I684S or A928V variants at PMETA<10–4 (Fig 5B and S3 Fig). (The 502 PheWAS phenotypes tested did not include SLE and IBD, which both had a frequency <1% in the i2b2 collection.) The list of PheWAS phenotypes associated at PMETA<0.05 with consistent effect sizes in both EMR datasets (with PMETA<P BioVU and PMETA<Pi2b2) is shown in S7 Table. Together, these results provided no evidence of association with strong increased risk (OR≥1.3, based on our power calculations; S4–S6 Figs) between the RA-protecting TYK2 variants and any of the PheWAS phenotypes tested.
Fig 5. Investigation of pleiotropic effects of RA-protecting TYK2 variants using electronic medical records.

We first tested association of the P1104A (A) and I684S (B) variants to 502 PheWAS phenotypes with frequency>1% in two independent EMR collections including 3,005 and 26,372 individuals of European ancestry, respectively. Pvalues of each PheWAS phenotype in meta-analysis of the two EMR collections are shown. We also tested association of the TYK2 P1104A and I684S variants with low-density lipoproteins (LDL) levels (C), and white blood cell counts (WBC) (D). Effect sizes and confidence intervals in each EMR collection are shown. Pvalues from meta-analysis of the two EMR collections are indicated. Association results from SNPs previously reported to be associated with each quantitative trait (indicated by their respective rsIDs) are also shown.
Complete LOF of TYK2 leads to human primary immunodeficiency, which is caused by rare autosomal recessive null mutations in TYK2 and results in increased risk of severe infections (bacterial, viral and fungal) [50,51]. To investigate the hypothesis that individuals carrying the P1104A, A928V or I684S variants might be at increased risk of serious infection due to partial inhibition of TYK2, we used a comprehensive set of infection-related ICD9 codes developed and validated elsewhere [52,53]. We found no group of infection significantly associated with either of the P1104A, A928V or I684S variants (P<1x10-4) (S8 Table). We observed suggestive evidence of association between the A928V variant and increased risk of pneumonia (OR = 1.48, P = 0.011 in the BioVU dataset; OR = 2.59, P = 0.079 in the i2b2 dataset; ORMETA = 1.54, PMETA = 0.004 in meta-analysis), but the signal did not surpass significance thresholds after multiple hypotheses testing correction
Finally, we tested the association of the P1104A, A928V and I684S variants with two quantitative traits available in the EMR: white blood cell counts (WBC) and low-density lipoprotein (LDL) levels (Fig 5C and 5D, S3 Fig, and S9 Table). We selected these two phenotypes as a drug, tofacitinib, targets a pathway related to TYK2—the JAK signalling pathway—and patients treated with JAK-inhibitors have lower levels of WBCs/neutrophils and elevated levels of LDL cholesterol [54,55]. We observed only suggestive association of the TYK2 P1104A variant with increased LDL levels (BETAMETA = +3.4 mg/dL, PMETA = 0.005 in the meta-analysis) (Fig 5C and S9 Table) and no association of the TYK2 variants with WBC. As a positive control, we showed significant association to WBC and LDL levels of known associated SNPs from previous GWAS investigating these two traits [56–60], demonstrating that our analysis using EMR had the power to detect associations to these traits (Fig 5C and 5D, and S10 Table).
Discussion
Previous studies had identified one TYK2 missense variant, P1104A (rs34536443), as associated with RA susceptibility [2]. Here, through dense genotyping, haplotype analyses and deep sequencing, we demonstrate that three independent TYK2 missense variants (P1104A, A928V [rs35018800] and I684S [rs12720356]) unequivocally protect against RA (Figs 1, 2, and 3). In aggregate, the 3 TYK2 variants account for 0.25% of the phenotypic variance of RA. Together with the lack of convincing association to protein-coding variants in ICAM genes and other genes from the 19p32.3 locus, our results provide multiple lines of evidence implicating TYK2, rather than another nearby gene, as a causal gene involved in RA disease susceptibility.
TYK2 is a member of the JAK family. The four JAK proteins (JAK1, JAK2, JAK3 and TYK2) selectively associate with various cytokine receptors [30,31]. While JAK1 and JAK2 have broad functions, JAK3 and TYK2 are primarily important for immune responses. TYK2 associates with receptor chains utilized by a large number of cytokines, including IL6-R, which is the target of tocilizumab, an anti-IL6R monoclonal antibody used in the treatment of RA [32,61]. Two of the variants associated with RA in our study, P1104A and I684S, have recently been shown to affect TYK2 function in primary T cells, fibroblasts and B cell lines and impair pro-inflammatory cytokines signalling, providing evidence that both variants are LOF mutations and that LOF mutations in TYK2 alter immune-mediated pathways [33,34].
We have previously proposed three features of human genetics that can be applied to drug discovery [20]: (1) identification of targets that, when perturbed in a manner that mimics trait-associated alleles, demonstrate efficacy in treating complex human diseases—as illustrated by recent studies highlighting the increased success rate of targets supported by human genetics [19]; (2) identification of alternative clinical indications through genetic associations of related diseases for drug repurposing [62]; and (3) prediction of potential on-target adverse drug events via pleiotropic associations [7,63]. In the present study, we explored all three features as it pertains to the TYK2 locus.
First, the observation of multiple independent RA-protecting variants (Figs 1, 2, and 3) provides an accumulation of evidence that a drug that mimics the effect of TYK2 alleles may be effective at treating RA (proxy for drug efficacy). This concept is consistent with the overlap between human genetics and drug discovery in other diseases [62], as exemplified by Mendelian randomization studies on variants in PCSK9 and IL6R [7,20,63]. In addition, the recent development of drugs inhibiting the TYK2-related proteins JAK1, JAK2 and JAK3 for the treatment of RA (including the drug tofacitinib, recently approved by the food and drug administration [FDA]), further support TYK2 as an appealing candidate drug target [54,64].
Second, the protective effect of the three TYK2 variants in SLE observed in our study (Fig 4A) highlights that a drug that mimics the effect of the RA-protecting TYK2 alleles may also be effective at treating SLE, and potentially other autoimmune diseases such as IBD. We note that the relatively small sample size in IBD (n = 1,346 IBD cases) limits our ability to perform detailed fine-mapping of the TYK2 locus in IBD, and that additional studies are required.
As a third feature, we used electronic medical records to comprehensively investigate pleiotropy of RA-associated TYK2 variants that could predict potential adverse events, including risk of serious infections, decreased WBC or neutrophil counts, or increased LDL levels, which are major adverse drug events in RA drug development that have been observed in clinical trials of tofacitinib [54,55,64] (Fig 5 and S3 Fig). In this analysis, we observed no strong evidence of a phenotype at increased risk in carriers of the TYK2 RA protecting-variants P1104A, A928V or I684S. However, we did observe several phenotypes with suggestive evidence of association (PMETA<0.05) and consistent direction of effect in two independent EMR datasets (Fig 5, S7 and S8 Tables), including association of the P1104A variant with increased LDL levels (Beta = +3.4 mg/dL, PMETA = 0.005), and association of the rare A928V variant with risk of pneumonia (OR = 1.5, PMETA = 0.004). These observations will require further investigation in very large collections to predict whether serious infections and/or hypercholesterolemia might be a potential on-target adverse event of a drug mimicking the effect of these alleles.
There are limitations to our study. It is possible that RA-associated TYK2 variants have pleiotropic associations with other phenotypes (e.g., infection), but that our EMR-based approach was not able to detect these associations at the level of statistical significance in our study. For example, EMR diagnostic codes are comprehensive but imprecise, which limits accurate estimations of effect size of associations from EMR data alone. Further, our EMR analysis had limited power to identify individual diagnoses associated with TYK2 LOF, considering the sample size and prevalence of the individual diagnoses. However, the use of EMR has been shown to successfully detect associations with a large spectrum of phenotypes [25]. In the present study, we were able to replicate previously published associations (Fig 5 and S10 Table). Of note, GWAS have not yet proven to be successful at identifying loci contributing to risk of infection with reproducible results, which limits our ability to include infection-related variants as a positive control in our PheWAS.
In conclusion, although previous studies have nominated TYK2 as a potential therapeutic target [41,65], our study provides compelling human genetic data demonstrating that TYK2 alleles with partial loss-of-function (1) protect against RA, SLE and potentially other autoimmune diseases such as IBD; and (2) are tolerated in the general population, as there are no obvious detrimental associations in our PheWAS. Our results also highlight the potential of investigating in details the different biological effects of the three TYK2 variants to inform drug efficacy, toxicity and repurposing at early stages of drug development. In theory, the same approach linking human genetics with “real life data” like EMR should be applicable to other complex diseases, thereby providing an estimate of drug efficacy and toxicity at the time of target validation.
Methods
Samples and ethics statement
A detailed description of the samples included in this study is provided in S1 and S2 Tables, and in the related Methods sections. Our study was approved by the Institutional Review Board of Brigham & Women's Hospital. All the enrolled subjects provided written informed consent for the participation of the study. Blood samples were collected according to protocols approved by local institutional review boards.
RA case-control Immunochip dataset
To fine-map the TYK2 locus and investigate independent signals of association to RA, we used 7,222 ACPA+ RA cases and 15,870 controls genotyped on the Immunochip platform as part of the Rheumatoid Arthritis Consortium International (S1 Table) [2]. Quality control and initial data filtering were performed as described previously [2]. Briefly, genotype calling was performed on all samples as a single project using the GenomeStudio Data Analysis software. SNPs with low cluster separation, call rate <0.99, or departure from Hardy-Weinberg equilibrium (P HWE < 5.7x10−7) were excluded from each of the collections. Samples with a call rate <0.99 were excluded. Principal component analysis (PCA) was performed using EIGENSOFT v4.2 [66] with HapMap phase 2 samples as reference populations, and non-Caucasians samples were excluded. A second PCA was performed to exclude outliers and calculate the principal components (PCs) to include as covariates in the logistic regressions. We build haplotypes using BEAGLE, and tested for association of the genotypes and haplotypes with risk of RA, using PLINK (including 10 PCs as covariates) [67,68].
Autoimmune diseases—case control Exomechip datasets
To replicate association signals to RA, we used an unpublished dataset of 4,726 RA cases and 13,683 controls genotyped on the Exomechip (S1 Table). This dataset included samples collected from the Dutch Rheumatoid Arthritis Monitoring (DREAM) registry [69], the Informatics for Integrating Biology and the Bedside (i2b2) cohort [48], the North American Rheumatoid Arthritis Consortium (NARAC) family cohort [70], the Study of New Onset Rheumatoid Arthritis (SONORA) cohort [71], the Veteran's Affairs Rheumatoid Arthritis registry (VARA) [72].
We also tested the association of TYK2 variants to SLE and IBD using Exomechip genotype data from 3,053 SLE cases and 1,346 IBD cases (S1 Table). The SLE dataset included samples from the Autoimmune Biomarkers Collaborative Network (ABCoN) [73], Genentech Clinical Trials, the Multiple Autoimmune Disease Genetics Consortium (MADGC) [74], the Oklahoma Medical Research Foundation (OMRF) [75], the University of California San Francisco (UCSF) [76], and the UK King’s College [77]. The IBD dataset included samples from the University of Dundee [78], the EMerging BiomARKers in Inflammatory Bowel Disease (EMBARK) study [79], and Genizon BioSciences, Inc [80].
Extensive quality control and data filtering were performed in each dataset. Samples were selected by excluding: 1) individuals with <95% complete Exomechip genotype data; 2) individuals with IBD [Pi-hat >0.125 using 10,000 higher frequency SNPs (MAF>0.05)], 3) non-European ancestry (>0.10, based on STRUCTURE (v2.3.3) [81] analyses using core sets of different continental groups including >100 subjects from each ancestry (European, East Asian, Amerindian, South Asian and West African) and genotypes from >2000 high frequency LD independent SNPs; and 4) PCA outliers (>5 SD for first 10 principal components). The PCA was performed in EIGENSOFT 4.2 [66] using 13,682 Exomechip SNPs that 1) were not in LD (r2<0.01); 2) >99% complete typing data and 3) were enriched for SNPs with minor allele frequencies >0.05 (>50% of SNPs). Independence of the samples from the Exomechip and Immunochip RA datasets was confirmed using overlapping SNPs.
We build haplotypes using BEAGLE, and tested for association of the genotypes and haplotypes with risk of RA, SLE and IBD, using PLINK (including 10 PCs as covariates) [67,68].
Exon sequencing
To investigate the contribution to RA of rare protein-coding variants at the TKY2 locus, we used exon-sequencing data available in 1,420 RA patients and 1,340 controls originating from Europe or the United States (S2 Table). A total of 10 collections from 5 countries were included: the Autoimmune Biomarkers Collaborative Network (ABCoN) [82], the Academic Medical Center (AMC) and VU University medical center (VUMC), the UK Biological in Rheumatoid arthritis Genetics and Genomics Study Syndicate (BRAGGSS) [83], the Consortium of Rheumatology Researchers of North America (CORRONA) [84], the Informatics for Integrating Biology and the Bedside (i2b2) center, the Leiden University Medical Center (LUMC) [85], the Dutch Rheumatoid Arthritis Monitoring registry (DREAM) and the Nijmegen Biomedical Study (NBS) [69], the French Research in Active Rheumatoid Arthritis (ReAct) [86], and the Rheumatic Diseases Portuguese Registry (Reuma.pt/ Biobanco-IMM) [87]. DNA libraries were prepared in sets of 96. The barcoded libraries from each set were then pooled together. Enrichment of the target genomic regions was performed using the NimbleGen Sequence Capture technology. After target capture, each pool was loaded on two lanes of the HiSeq sequencer. Reads were then aligned to the reference human genome (NCBI Build37/hg19) using BWA [88] and duplicate reads were excluded using Picard. In total, 95% of the samples reached an minimum average coverage of 20X in >70% of target regions, with 96% of the target regions in the samples passing this initial quality control (QC) covered at > = 20X coverage. Single nucleotide polymorphisms (SNPs) were called using Samtools v1.16 [89] and VarScan 2.2.9 [90] using stringent minimum coverage, mapping quality, and strandness filters. SNPs called from each sample using both calling algorithms were then merged and additional filters were applied (number and frequency of the reads supporting the variant, position in the reads). Finally, only variants passing filters in >50% of the samples were considered high-quality and included in the subsequent analysis. Sequencing, initial QC and SNP calling were performed at the Genome Institute. After applying stringent filters to remove individuals based on sequencing coverage and quality (N = 131 individuals excluded), and population stratification using case-control principal components (PC)-matching (N = 393 individuals excluded), a final set of 1,118 case-control matched pairs of European ancestry was included in the association tests (S1 Table). The transition:transversion ratio based on the variants passing QC was 2.5 (vs 2.6 for dbSNP SNPs in target space). MAF correlation of variants called in the sequenced controls and samples from the Exome Sequencing Project (ESP) was 98%. Concordance between sequencing genotype calls and Exomechip data available for 137 samples was calculated to further assess the quality of the sequencing data. Overall, we observed 99.7% concordance at 1,718 shared variants polymorphic in the 137 samples set. The variants were annotated using ANNOVAR [91]. We used PolyPhen-2 and SIFT to predict the function of the missense variants [43,44]. We then grouped the variants based on the prediction results from both software: 1) benign in both PolyPhen-2 and SIFT (that we considered as benign), 2) benign using one software and possibly/probably damaging using the other software (that we considered as benign), 3) possibly damaging in both PolyPhen-2 and SIFT or possibly damaging in one software and probably damaging using the second software (that we considered as “potentially damaging”), 4) probably damaging in both PolyPhen-2 and SIFT (that we considered as “potentially damaging”).
Gene-based association tests
We performed gene-based association tests to investigate the contribution of rare variants (MAF<0.5%) to protection from RA. For each of the 11 genes in the TYK2 locus (defined by the SNPs in linkage disequilibrium (LD, r2>0.5) to the strongest association to RA (driven by rs34536443, P1104A) [2], we investigated the overall contribution of 1) all rare (MAF<0.5%) missense variants, and 2) the rare nonsense variants and the missense variants predicted to be “potentially damaging” using Polyphen-2 and SIFT [43,44]. We used 3 published “one-sided” methods: (1) the classic burden test, (2) the frequency-weighted (FW) test and (3) the Variable-threshold (VT) test, all 3 tests implemented in PLINKSEQ [46,47]. We also used SKAT-O (“two-sided” method) [45]. We performed all 4 tests with 10,000 case-control permutations to assess empirical P-values.
In addition to the gene-based tests, we performed window-based tests to investigate the contribution of rare variants per 500 bp window of the genes coding sequence, using SKAT-O. and 10,000 case-control permutations to assess empirical P-values. Finally, we used the one-sided methods (BURDEN, WT, VT) to further assess the contribution of rare variants in the protein kinase 1 domain of TYK2 in domain-based tests.
Linking haplotypes with clinical diagnoses from Electronic Medical Records
To comprehensively investigate pleiotropic effects of TYK2 LOF variants, we used two independent electronic medical records (EMR) datasets linked to genotype data: 1) EMR data from 3,005 individuals of European ancestry from the Informatics for Integrating Biology and the Bedside (i2b2) center who received medical care within the Brigham and Women’s Hospital (BWH) and Massachusetts General Hospital (MGH) healthcare system linked to Immunochip genotype data [48,92], and 2) EMR data from 26,372 individuals of European ancestry from BioVU, the Vanderbilt University DNA biobank [49], linked to Exomechip genotype data (S1 Table). The i2b2 collection was initially optimized for RA genetic studies [48,92], resulting in a high frequency of patients with ICD9 code = 714 (Rheumatoid arthritis and other inflammatory polyarthropathies) and 714.0 (Rheumatoid arthritis) in this collection (S1 Table). The BioVU Exomechip cohort was primarily chosen based on longitudinal exposure in healthcare system, without a specific emphasis on RA or other autoimmune diseases. International Classification of Diseases 9th Revision (ICD9) codes were grouped into 1,570 clinically relevant phenotypes using the current version of the PheWAS codes, as described previously [25]. We restricted our analysis to PheWAS codes referring to ICD9 001–779 (ignoring signs and symptoms and injuries) and with a prevalence >1% in both EMR datasets, resulting in 502 PheWAS codes. For each PheWAS code, we considered individuals with at least two reported events as cases.
We first conducted a phenome-wide association study (PheWAS). In each EMR dataset, we tested for association of TYK2 variants with each PheWAS code in the additive model using logistic regressions adjusted for age, gender and PCs to correct for population stratification. In the analysis of the i2b2 EMR dataset, we further adjusted for RA status. We conducted an inverse-variance-weighted meta-analysis to combine the results from the two EMR datasets.
We then conducted an association study focused on ICD9 codes related to serious infections. We used two previously published sets of ICD9 codes for serious infections grouped by anatomical site and compiled based on expert consensus: 1) a ‘‘comprehensive” set that included a wide range of codes to maximize sensitivity; 2) a ‘‘restricted” set including more specific ICD9 codes [53].
Finally, we tested for association of the TYK2 variants with two quantitative traits available in the EMR: white blood cell counts (WBC), and low-density lipoprotein (LDL) levels. For WBC, we defined the primary outcome as the mean of all measurements available for each subject in the EMR. We tested the association between the TYK2 variants and the mean values, adjusted by age, gender, PCs and the number of measurements used to calculate the mean values. For LDL levels, the primary outcome was defined by each subject’s first LDL measurement in the EMR as described previously [93]. We excluded subjects with electronic prescription for an HMG-CoA reductase inhibitor (statin) prior to the LDL measurement to maximize the chance of selecting subjects prior to any lipid lowering intervention. We tested the association between the TYK2 variants and the first LDL measurement, adjusted by age at LDL measurement, gender and PCs. We also adjusted for year of LDL measurement, which has been shown to strongly contribute to the variability in LDL levels [93,94].
Estimation of the statistical power in the PheWAS
To estimate the power to detect a significant association (at P<1x10-4) in the PheWAS based on the sample size, the frequency of each code and the frequency of the SNPs tested, we used and R script adapted from the ldDesign R package to query the Genetic Power Calculator online tool [95]. We assessed power for a variant with MAF = 3.4% (corresponding to the P1104A variant MAF), MAF = 8.7% (corresponding to the I684S variant MAF) and MAF = 0.7% (corresponding to the A928V variant MAF). We assessed power for a set of code frequencies and ORs, in the models where 1) the RA-protecting variants increase risk (risk allele frequency [RAF] = 0.034, RAF = 0.087 and RAF = 0.007) or decrease risk (RAF = 0.966, RAF = 0.913 and RAF = 0.993).
Based on the case:control ratio for each phenotype, the PheWAS approach has significantly greater power to detect increased risk compared to protection (S4–S6 Figs). Estimations highlighted the statistical power to detect a significant association (P<1x10-4; P = 0.05/532 phenotypes tested) to the P1104A and I684S variants. For rs34536443, we estimated power to detect a significant association with: 1) OR≥2 at phenotype frequency = 1%, 2) OR≥1.4 at phenotype frequency = 10% in BioVu. For rs12720356, we estimated power to detect a significant association with: 1) OR≥1.6 at phenotype frequency = 1%, OR≥1.3 at phenotype frequency = 10% in BioVU (S4 and S5 Figs). However, we estimated limited power to detect a significant association to the rare variant A928V (MAF = 0.7%; S6 Fig).
Supporting Information
An accumulation of true rare missense variants (MAF<0.5%) predicted to be damaging was observed in the Protein kinase 1 domain of TYK2. Association results from 500 bp sliding window tests in SKAT-O restricted to nonsense variants (pink) and missense variants predicted to be damaging (red) are shown. Variants with MAF>1% (indicated by a star) were excluded in the test. In TYK2, we further excluded the A928V and A53T variants with 0.5%<MAF<1% (indicated by a star) that were independently investigated using Exomechip data. The light blue background highlights the coding sequence region with P<0.05.
(PDF)
Only the 3 variants with MAF>0.5% confirmed to be associated to RA in our study reached P<0.05, in either the disease-specific analyses or the diseases-combined analysis.
(PDF)
(A) We first tested association of the A928V variant to 502 PheWAS phenotypes with frequency>1% in two independent EMR collections. Pvalues of each PheWAS phenotype in meta-analysis of the two EMR collections are shown. We also tested association of the A928V variant with LDL levels (B), and white blood cell counts (WBC) (C). Effect sizes and confidence intervals in each EMR collection are shown.
(PDF)
We estimated the power to detect an association at P<1x10-4 for a variant with MAF = 3.4%, based on phenotype frequency in the EMR and estimated OR. (A) Power estimations for a sample size of 3,005 subjects. (B) Power estimations for a sample size of 26,372 subjects. The left panel shows results for the minor allele associated with increased risk. The right panel shows results for the minor allele with a protective effect. The dashed red line indicated a phenotype frequency of 1%. The barplots highlight the number of cases per phenotype in the EMR collections.
(PDF)
We estimated the power to detect an association at P<1x10-4 for a variant with MAF = 8.5%, based on phenotype frequency in the EMR and estimated OR. (A) Power estimations for a sample size of 3,005 subjects. (B) Power estimations for a sample size of 26,372 subjects. The left panel shows results for the minor allele associated with increased risk. The right panel shows results for the minor allele with a protective effect. The dashed red line indicated a phenotype frequency of 1%. The barplots highlight the number of cases per phenotype in the EMR collections.
(PDF)
We estimated the power to detect an association at P<1x10-4 for a variant with MAF = 0.7%, based on phenotype frequency in the EMR and estimated OR. (A) Power estimations for a sample size of 3,005 subjects. (B) Power estimations for a sample size of 26,372 subjects. The left panel shows results for the minor allele associated with increased risk. The right panel shows results for the minor allele with a protective effect. The dashed red line indicated a phenotype frequency of 1%. The barplots highlight the number of cases per phenotype in the EMR collections.
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(XLS)
(XLS)
(PDF)
Acknowledgments
We thank Lindsey Criswell (UCSF), Patrick Gaffney (OMRF), and Timothy Vyse (King’s College, UK) for contribution of SLE cases for exome array genotyping.
Data Availability
Detailed summary statistic levels data from our analyses are available in the manuscript and in supplementary data. De-identified genotype level data at the TYK2 locus cannot be made publicly available due to restrictions in the consent forms signed by participants. These data are available upon request. RA sequencing data should be requested to Dr Soumya Raychaudhuri (soumya@broadinstitute.org). RA Immunochip data should be requested to Dr Jane Worthington (Jane.Worthington@manchester.ac.uk), RA, SLE and IBD TYK2 Exomechip data should be requested to Dr Robert Graham (graham.robert@gene.com). i2b2 EMR-linked TYK2 genetic data should be requested to Dr Katherine Liao (kliao@partners.org). BioVU EMR-linked TYK2 genetic data should be requested to Dr Joshua Denny (josh.denny@Vanderbilt.Edu).
Funding Statement
DD was supported by grants from the National Institutes of Health (NIH) (U01-GM092691 and R01-AR063759). RMP was supported by grants from the NIH (R01-AR057108, R01-AR056768, U01-GM092691, R01-AR059648) and holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506: 376–381. 10.1038/nature12873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. 2012;44: 1336–1340. 10.1038/ng.2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Diogo D, Okada Y, Plenge RM. Genome-wide association studies to advance our understanding of critical cell types and pathways in rheumatoid arthritis: recent findings and challenges. Curr Opin Rheumatol. 2014;26: 85–92. 10.1097/BOR.0000000000000012 [DOI] [PubMed] [Google Scholar]
- 4. Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93: 779–797. 10.1016/j.ajhg.2013.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beaudoin M, Goyette P, Boucher G, Lo KS, Rivas MA, Stevens C, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013;9: e1003723 10.1371/journal.pgen.1003723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonnefond A, Clement N, Fawcett K, Yengo L, Vaillant E, Guillaume JL, et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. 2012;44: 297–301. 10.1038/ng.1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cohen JC, Boerwinkle E, Mosley TH Jr., Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354: 1264–1272. [DOI] [PubMed] [Google Scholar]
- 8. Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature. 2014;505: 550–554. 10.1038/nature12825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flannick J, Thorleifsson G, Beer NL, Jacobs SB, Grarup N, Burtt NP, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014. [DOI] [PMC free article] [PubMed]
- 10. Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40: 592–599. 10.1038/ng.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johansen CT, Wang J, McIntyre AD, Martins RA, Ban MR, Lanktree MB, et al. Excess of rare variants in non-genome-wide association study candidate genes in patients with hypertriglyceridemia. Circ Cardiovasc Genet. 2012;5: 66–72. 10.1161/CIRCGENETICS.111.960864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jordan CT, Cao L, Roberson ED, Duan S, Helms CA, Nair RP, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet. 2012;90: 796–808. 10.1016/j.ajhg.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8: e1002521 10.1371/journal.pgen.1002521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Momozawa Y, Mni M, Nakamura K, Coppieters W, Almer S, Amininejad L, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet. 2011;43: 43–47. 10.1038/ng.733 [DOI] [PubMed] [Google Scholar]
- 15. Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324: 387–389. 10.1126/science.1167728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pearce LR, Atanassova N, Banton MC, Bottomley B, van der Klaauw AA, Revelli JP, et al. KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell. 2013;155: 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43: 1066–1073. 10.1038/ng.952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. 2013;45: 1366–1370. 10.1038/ng.2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13: 419–431. 10.1038/nrd4309 [DOI] [PubMed] [Google Scholar]
- 20. Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12: 581–594. 10.1038/nrd4051 [DOI] [PubMed] [Google Scholar]
- 21. Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 2013;14: 483–495. 10.1038/nrg3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. 2013;14: 661–673. 10.1038/nrg3502 [DOI] [PubMed] [Google Scholar]
- 23. Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26: 1205–1210. 10.1093/bioinformatics/btq126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hebbring SJ. The challenges, advantages and future of phenome-wide association studies. Immunology. 2014;141: 157–165. 10.1111/imm.12195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31: 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hebbring SJ, Schrodi SJ, Ye Z, Zhou Z, Page D, Brilliant MH. A PheWAS approach in studying HLA-DRB1*1501. Genes Immun. 2013;14: 187–191. 10.1038/gene.2013.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kho AN, Hayes MG, Rasmussen-Torvik L, Pacheco JA, Thompson WK, Armstrong LL, et al. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome-wide association study. J Am Med Inform Assoc. 2012;19: 212–218. 10.1136/amiajnl-2011-000439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ritchie MD, Denny JC, Zuvich RL, Crawford DC, Schildcrout JS, Bastarache L, et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127: 1377–1385. 10.1161/CIRCULATIONAHA.112.000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shameer K, Denny JC, Ding K, Jouni H, Crosslin DR, de Andrade M, et al. A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum Genet. 2014;133: 95–109. 10.1007/s00439-013-1355-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36: 542–550. 10.1016/j.immuni.2012.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Strobl B, Stoiber D, Sexl V, Mueller M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front Biosci. 2011;16: 3214–3232. [DOI] [PubMed] [Google Scholar]
- 32. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365: 2205–2219. 10.1056/NEJMra1004965 [DOI] [PubMed] [Google Scholar]
- 33. Couturier N, Bucciarelli F, Nurtdinov RN, Debouverie M, Lebrun-Frenay C, Defer G, et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain. 2011;134: 693–703. 10.1093/brain/awr010 [DOI] [PubMed] [Google Scholar]
- 34. Li Z, Gakovic M, Ragimbeau J, Eloranta ML, Ronnblom L, Michel F, et al. Two rare disease-associated tyk2 variants are catalytically impaired but signaling competent. J Immunol. 2013;190: 2335–2344. 10.4049/jimmunol.1203118 [DOI] [PubMed] [Google Scholar]
- 35. Cunninghame Graham DS, Morris DL, Bhangale TR, Criswell LA, Syvanen AC, Ronnblom L, et al. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 2011;7: e1002341 10.1371/journal.pgen.1002341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42: 1118–1125. 10.1038/ng.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hellquist A, Jarvinen TM, Koskenmies S, Zucchelli M, Orsmark-Pietras C, Berglind L, et al. Evidence for genetic association and interaction between the TYK2 and IRF5 genes in systemic lupus erythematosus. J Rheumatol. 2009;36: 1631–1638. 10.3899/jrheum.081160 [DOI] [PubMed] [Google Scholar]
- 38. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491: 119–124. 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mero IL, Lorentzen AR, Ban M, Smestad C, Celius EG, Aarseth JH, et al. A rare variant of the TYK2 gene is confirmed to be associated with multiple sclerosis. Eur J Hum Genet. 2010;18: 502–504. 10.1038/ejhg.2009.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76: 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44: 1341–1348. 10.1038/ng.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet. 2010;42: 68–71. 10.1038/ng.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7: 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4: 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- 45. Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13: 762–775. 10.1093/biostatistics/kxs014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5: e1000384 10.1371/journal.pgen.1000384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Price AL, Kryukov GV, de Bakker PI, Purcell SM, Staples J, Wei LJ, et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010;86: 832–838. 10.1016/j.ajhg.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liao KP, Cai T, Gainer V, Goryachev S, Zeng-treitler Q, Raychaudhuri S, et al. Electronic medical records for discovery research in rheumatoid arthritis. Arthritis Care Res (Hoboken). 2010;62: 1120–1127. 10.1002/acr.20184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84: 362–369. 10.1038/clpt.2008.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kilic SS, Hacimustafaoglu M, Boisson-Dupuis S, Kreins AY, Grant AV, Abel L, et al. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr. 2012;160: 1055–1057. 10.1016/j.jpeds.2012.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25: 745–755. [DOI] [PubMed] [Google Scholar]
- 52. Curtis JR, Xie F, Chen L, Baddley JW, Beukelman T, Saag KG, et al. The comparative risk of serious infections among rheumatoid arthritis patients starting or switching biological agents. Ann Rheum Dis. 2011;70: 1401–1406. 10.1136/ard.2010.146365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Patkar NM, Curtis JR, Teng GG, Allison JJ, Saag M, Martin C, et al. Administrative codes combined with medical records based criteria accurately identified bacterial infections among rheumatoid arthritis patients. J Clin Epidemiol. 2009;62: 321–327, 327 e321–327. 10.1016/j.jclinepi.2008.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garber K. Pfizer's first-in-class JAK inhibitor pricey for rheumatoid arthritis market. Nat Biotechnol. 2013;31: 3–4. 10.1038/nbt0113-3 [DOI] [PubMed] [Google Scholar]
- 55. Kremer JM, Cohen S, Wilkinson BE, Connell CA, French JL, Gomez-Reino J, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64: 970–981. 10.1002/art.33419 [DOI] [PubMed] [Google Scholar]
- 56. Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41: 47–55. 10.1038/ng.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Crosslin DR, McDavid A, Weston N, Nelson SC, Zheng X, Hart E, et al. Genetic variants associated with the white blood cell count in 13,923 subjects in the eMERGE Network. Hum Genet. 2012;131: 639–652. 10.1007/s00439-011-1103-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41: 56–65. 10.1038/ng.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nalls MA, Couper DJ, Tanaka T, van Rooij FJ, Chen MH, Smith AV, et al. Multiple loci are associated with white blood cell phenotypes. PLoS Genet. 2011;7: e1002113 10.1371/journal.pgen.1002113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Waterworth DM, Ricketts SL, Song K, Chen L, Zhao JH, Ripatti S, et al. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2010;30: 2264–2276. 10.1161/ATVBAHA.109.201020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mima T, Nishimoto N. Clinical value of blocking IL-6 receptor. Curr Opin Rheumatol. 2009;21: 224–230. 10.1097/BOR.0b013e3283295fec [DOI] [PubMed] [Google Scholar]
- 62. Sanseau P, Agarwal P, Barnes MR, Pastinen T, Richards JB, Cardon LR, et al. Use of genome-wide association studies for drug repositioning. Nat Biotechnol. 2012;30: 317–320. 10.1038/nbt.2151 [DOI] [PubMed] [Google Scholar]
- 63. Hingorani AD, Casas JP. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379: 1214–1224. 10.1016/S0140-6736(12)60110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garber K. Pfizer's JAK inhibitor sails through phase 3 in rheumatoid arthritis. Nat Biotechnol. 2011;29: 467–468. 10.1038/nbt0611-467 [DOI] [PubMed] [Google Scholar]
- 65. Argiriadi MA, Goedken ER, Banach D, Borhani DW, Burchat A, Dixon RW, et al. Enabling structure-based drug design of Tyk2 through co-crystallization with a stabilizing aminoindazole inhibitor. BMC Struct Biol. 2012;12: 22 10.1186/1472-6807-12-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38: 904–909. [DOI] [PubMed] [Google Scholar]
- 67. Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81: 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kievit W, Fransen J, Oerlemans AJ, Kuper HH, van der Laar MA, de Rooij DJ, et al. The efficacy of anti-TNF in rheumatoid arthritis, a comparison between randomised controlled trials and clinical practice. Ann Rheum Dis. 2007;66: 1473–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gregersen PK. The North American Rheumatoid Arthritis Consortium—bringing genetic analysis to bear on disease susceptibility, severity, and outcome. Arthritis Care Res. 1998;11: 1–2. [DOI] [PubMed] [Google Scholar]
- 71. Sokka T, Willoughby J, Yazici Y, Pincus T. Databases of patients with early rheumatoid arthritis in the USA. Clin Exp Rheumatol. 2003;21: S146–153. [PubMed] [Google Scholar]
- 72. Mikuls TR, Fay BT, Michaud K, Sayles H, Thiele GM, Caplan L, et al. Associations of disease activity and treatments with mortality in men with rheumatoid arthritis: results from the VARA registry. Rheumatology (Oxford). 2011;50: 101–109. 10.1093/rheumatology/keq232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Petri M, Singh S, Tesfasyone H, Dedrick R, Fry K, Lal P, et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus. 2009;18: 980–989. 10.1177/0961203309105529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Criswell LA, Pfeiffer KA, Lum RF, Gonzales B, Novitzke J, Kern M, et al. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet. 2005;76: 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Guthridge JM, Lu R, Sun H, Sun C, Wiley GB, Dominguez N, et al. Two functional lupus-associated BLK promoter variants control cell-type- and developmental-stage-specific transcription. Am J Hum Genet. 2014;94: 586–598. 10.1016/j.ajhg.2014.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet. 2012;44: 1227–1230. 10.1038/ng.2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Morris DL, Fernando MM, Taylor KE, Chung SA, Nititham J, Alarcon-Riquelme ME, et al. MHC associations with clinical and autoantibody manifestations in European SLE. Genes Immun. 2014;15: 210–217. 10.1038/gene.2014.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Van Limbergen J, Russell RK, Nimmo ER, Kabakchiev B, Drummond HE, Satsangi J, et al. Haplotype-tagging analysis of common variants of the IL23R gene demonstrates gene-wide extent of association with IBD. Inflamm Bowel Dis. 2013;19: E79–80. 10.1097/mib.0b013e318280e790 [DOI] [PubMed] [Google Scholar]
- 79. Faubion WA Jr., Fletcher JG, O'Byrne S, Feagan BG, de Villiers WJ, Salzberg B, et al. EMerging BiomARKers in Inflammatory Bowel Disease (EMBARK) study identifies fecal calprotectin, serum MMP9, and serum IL-22 as a novel combination of biomarkers for Crohn's disease activity: role of cross-sectional imaging. Am J Gastroenterol. 2013;108: 1891–1900. 10.1038/ajg.2013.354 [DOI] [PubMed] [Google Scholar]
- 80. Raelson JV, Little RD, Ruether A, Fournier H, Paquin B, Van Eerdewegh P, et al. Genome-wide association study for Crohn's disease in the Quebec Founder Population identifies multiple validated disease loci. Proc Natl Acad Sci U S A. 2007;104: 14747–14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu C, Batliwalla F, Li W, Lee A, Roubenoff R, Beckman E, et al. Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis. Mol Med. 2008;14: 575–581. 10.2119/2008-00056.Liu [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Plant D, Bowes J, Potter C, Hyrich KL, Morgan AW, Wilson AG, et al. Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum. 2011;63: 645–653. 10.1002/art.30130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kremer JM. The CORRONA database. Autoimmun Rev. 2006;5: 46–54. [DOI] [PubMed] [Google Scholar]
- 85. Kurreeman FA, Padyukov L, Marques RB, Schrodi SJ, Seddighzadeh M, Stoeken-Rijsbergen G, et al. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med. 2007;4: e278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Miceli-Richard C, Comets E, Verstuyft C, Tamouza R, Loiseau P, Ravaud P, et al. A single tumour necrosis factor haplotype influences the response to adalimumab in rheumatoid arthritis. Ann Rheum Dis. 2008;67: 478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Canhao H, Faustino A, Martins F, Fonseca JE. Reuma.pt—the rheumatic diseases portuguese register. Acta Reumatol Port. 2011;36: 45–56. [PubMed] [Google Scholar]
- 88. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22: 568–576. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38: e164–e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kurreeman F, Liao K, Chibnik L, Hickey B, Stahl E, Gainer V, et al. Genetic basis of autoantibody positive and negative rheumatoid arthritis risk in a multi-ethnic cohort derived from electronic health records. Am J Hum Genet. 2011;88: 57–69. 10.1016/j.ajhg.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liao KP, Diogo D, Cui J, Cai T, Okada Y, Gainer VS, et al. Association between low density lipoprotein and rheumatoid arthritis genetic factors with low density lipoprotein levels in rheumatoid arthritis and non-rheumatoid arthritis controls. Ann Rheum Dis. 2014;73: 1170–1175. 10.1136/annrheumdis-2012-203202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Carroll MD, Kit BK, Lacher DA, Shero ST, Mussolino ME. Trends in lipids and lipoproteins in US adults, 1988–2010. JAMA. 2012;308: 1545–1554. 10.1001/jama.2012.13260 [DOI] [PubMed] [Google Scholar]
- 95. Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19: 149–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
An accumulation of true rare missense variants (MAF<0.5%) predicted to be damaging was observed in the Protein kinase 1 domain of TYK2. Association results from 500 bp sliding window tests in SKAT-O restricted to nonsense variants (pink) and missense variants predicted to be damaging (red) are shown. Variants with MAF>1% (indicated by a star) were excluded in the test. In TYK2, we further excluded the A928V and A53T variants with 0.5%<MAF<1% (indicated by a star) that were independently investigated using Exomechip data. The light blue background highlights the coding sequence region with P<0.05.
(PDF)
Only the 3 variants with MAF>0.5% confirmed to be associated to RA in our study reached P<0.05, in either the disease-specific analyses or the diseases-combined analysis.
(PDF)
(A) We first tested association of the A928V variant to 502 PheWAS phenotypes with frequency>1% in two independent EMR collections. Pvalues of each PheWAS phenotype in meta-analysis of the two EMR collections are shown. We also tested association of the A928V variant with LDL levels (B), and white blood cell counts (WBC) (C). Effect sizes and confidence intervals in each EMR collection are shown.
(PDF)
We estimated the power to detect an association at P<1x10-4 for a variant with MAF = 3.4%, based on phenotype frequency in the EMR and estimated OR. (A) Power estimations for a sample size of 3,005 subjects. (B) Power estimations for a sample size of 26,372 subjects. The left panel shows results for the minor allele associated with increased risk. The right panel shows results for the minor allele with a protective effect. The dashed red line indicated a phenotype frequency of 1%. The barplots highlight the number of cases per phenotype in the EMR collections.
(PDF)
We estimated the power to detect an association at P<1x10-4 for a variant with MAF = 8.5%, based on phenotype frequency in the EMR and estimated OR. (A) Power estimations for a sample size of 3,005 subjects. (B) Power estimations for a sample size of 26,372 subjects. The left panel shows results for the minor allele associated with increased risk. The right panel shows results for the minor allele with a protective effect. The dashed red line indicated a phenotype frequency of 1%. The barplots highlight the number of cases per phenotype in the EMR collections.
(PDF)
We estimated the power to detect an association at P<1x10-4 for a variant with MAF = 0.7%, based on phenotype frequency in the EMR and estimated OR. (A) Power estimations for a sample size of 3,005 subjects. (B) Power estimations for a sample size of 26,372 subjects. The left panel shows results for the minor allele associated with increased risk. The right panel shows results for the minor allele with a protective effect. The dashed red line indicated a phenotype frequency of 1%. The barplots highlight the number of cases per phenotype in the EMR collections.
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(XLS)
(XLS)
(PDF)
Data Availability Statement
Detailed summary statistic levels data from our analyses are available in the manuscript and in supplementary data. De-identified genotype level data at the TYK2 locus cannot be made publicly available due to restrictions in the consent forms signed by participants. These data are available upon request. RA sequencing data should be requested to Dr Soumya Raychaudhuri (soumya@broadinstitute.org). RA Immunochip data should be requested to Dr Jane Worthington (Jane.Worthington@manchester.ac.uk), RA, SLE and IBD TYK2 Exomechip data should be requested to Dr Robert Graham (graham.robert@gene.com). i2b2 EMR-linked TYK2 genetic data should be requested to Dr Katherine Liao (kliao@partners.org). BioVU EMR-linked TYK2 genetic data should be requested to Dr Joshua Denny (josh.denny@Vanderbilt.Edu).