

Abstract
Background
Skin barrier integrity requires a highly coordinated molecular system involving the structural protein filaggrin. Mutational loss of the skin barrier protein filaggrin predisposes individuals to the development of atopic dermatitis (AD).
Objective
to determine the role of SIRT1 in skin barrier function, filaggrin expression, and the development of AD.
Methods
Skin histology of mice with skin-specific SIRT1 deletion and wild-type controls was examined by Hematoxyline and Eosin (H&E). Protein and mRNA abundance was analyzed by immunoblot, immunohistochemistry, immunofluorescence, and RT-PCR. Serum antibody levels were assessed by ELISA.
Results
Here we show that filaggrin is regulated by the protein deacetylase SIRT1, and that SIRT1 is critical for skin barrier integrity. Epidermis-specific SIRT1 ablation causes AD-like skin lesions in mice, and mice with epidermal SIRT1 deletion are sensitive to percutaneous challenge by the protein allergen ovalbumin. In normal human keratinocytes and mouse skin, SIRT1 knockdown or genetic deletion down-regulates filaggrin, and regulation of filaggrin expression by SIRT1 requires the deacetylase activity of SIRT1. SIRT1 also promotes the activation of the aryl hydrocarbon receptor (AhR), and the AhR ligand restores filaggrin expression in SIRT1-inhibited cells. As compared with normal human skin, SIRT1 is down-regulated in the lesions of atopic dermatitis as well as non-atopic dermatitis.
Conclusion
Our findings demonstrate a critical role of SIRT1 in skin barrier maintenance, open up new opportunities to use SIRT1 as a pharmacological target, and may facilitate the development of mechanism-based agents for AD prevention and therapy.
Keywords: SIRT1, skin barrier, filaggrin
Introduction
Skin is the essential barrier protecting against environmental insults including infectious pathogens, chemicals and UV radiation, and minimizing the water loss from the body. As the most abundant cells forming the epidermis, keratinocytes proliferate and differentiate to form an impermeable barrier. Defects in the skin barrier have an active role in the pathogenesis of several chronic inflammatory skin diseases, including atopic dermatitis (1–4).
Atopic dermatitis (AD) is an increasingly common pruritic inflammatory disease affecting 10–20% of children and 3% of adults in the US and other developed countries (5, 6). The clinical picture evolves in stages and is frequently associated with an increased serum immunoglobulin E (IgE) concentration and a number of cutaneous, ocular, and other atopic disorders such as allergic rhinitis and asthma. There is no cure for AD and its molecular pathogenesis is still poorly understood. Although AD was, for many years, considered to be primarily an immunologically-driven disease with a secondary barrier defect (the so-called inside-outside hypothesis), investigators hypothesized that the primary defect was in the skin barrier (the outside-inside hypothesis) (4, 6). Abnormal barrier function of the skin has long been noted in ichthyosis vulgaris, a common skin condition characterized by postnatal appearance of dry, flaking skin, even in the absence of AD. Recent studies have disclosed a strong association between a defect in the skin barrier and the pathogenesis of AD (6). This defect is due to a genetic loss-of-function mutation of the gene FLG, encoding the skin barrier protein profilaggrin/filaggrin (7, 8). These mutations, which are carried by up to 10% of Europeans, represent a strong genetic predisposing factor for AD, asthma, and allergies (9).
Although FLG mutation carriers have a greatly increased risk of AD, AD develops in only approximately 42% of all mutation carriers (10). This outcome implies that genetic modifiers and environmental factors are both important. Impaired barrier function increases transepidermal penetration of environmental allergens. This is supported by the findings that FLG deficiency in mice facilitates and permits increased percutaneous sensitization with protein allergens, irritants, and haptens (11–14).
Sirtuin 1 (SIRT1), a mammalian counterpart of the yeast silent information regulator 2 (Sir2) and a proto member of the sirtuin family, is an NAD-dependent protein deacetylase crucial for cell survival, metabolism, senescence, and stress response in several cell types and tissues (15–19). Both histone and non-histone targets of SIRT1 have been identified, including FOXO, p53, PGC-1α, NF-κB, and PPARs (16, 18, 20). SIRT1 also deacetylates XPA to regulate UV-induced DNA damage repair (21). We have demonstrated previously that SIRT1 positively regulates UV-induced DNA damage repair by promoting XPC expression, and that it has a critical role in skin tumorigenesis and homeostasis (22, 23). In addition, SIRT1 has been found to promote differentiation of normal human keratinocytes in vitro (24), suggesting a possible role in barrier function and thus the development of AD.
Although genetic FLG mutations are strongly associated with the risk of AD, two independent studies have shown that both carriers and non-carriers of FLG mutations develop childhood AD. In one study of AD patients, 90% of whom were Caucasian, only 26.7% of AD patients carried FLG mutations (8). In Irish childhood AD cases, ~ 47% of individuals carried at least one null FLG mutation (7). These findings indicate that more than half of all AD patients do not carry FLG mutations. One implication is that non-genetic loss of profilaggrin/filaggrin, including down-regulation of FLG expression, may play an important role. FLG can be down-regulated by disrupting the fine balance of the complex regulation of FLG promoter activity during epidermal differentiation (9). However, the mechanisms that regulate FLG expression remain poorly understood.
Here we show that epidermal SIRT1 deletion down-regulates filaggrin and sensitizes mouse skin to epicutaneous allergen-induced response. SIRT1 regulates filaggrin expression through AhR. Our findings demonstrate a new role of SIRT1 in skin barrier function and shed light on the molecular mechanisms for maintaining the skin barrier and preventing inflammatory skin diseases.
METHODS
Human normal skin, atopic dermatitis and non-atopic dermatitis samples
All human specimens were studied after approval by the University of Chicago Institutional Review Board. Formalin-fixed paraffin-embedded tissue blocks were obtained from the archives in the tissue bank of Section of Dermatology, Department of Medicine, University of Chicago. Normal skin (sun-protected), atopic dermatitis, and non-atopic dermatitis (spongiotic dermatitis without a history of atopy) were used for immunohistochemical analysis of SIRT1 protein levels. The SIRT1 staining intensity was scored blindly by two independent investigators as 0 (negative), 1 (low), 2 (medium), or 3 (high) as in our previous studies (22, 25).
Animal treatments
All animal resources were approved by the University of Chicago Institutional Animal Care and Use Committee. Floxed mice carrying the Sirt1 allele (Jackson Laboratory, Bar Harbor, Maine) were bred with mice expressing Cre recombinase driven by the K14 promoter (Jackson Laboratory) to generate skin keratinocyte-specific heterozygous SIRT1 deletion (cHet) and homozygous SIRT1 knockout (cKO) mice with a B6 background as described previously (23). To generate WT and cKO mice in the SKH1 hairless background, mice were backcrossed with SKH1 females at least five times (23). Mice were housed five animals per cage, and there was no evidence of dorsal wounds caused by fighting. Female mice (n=15 for each group) were kept for 24 months for observation of survival and aging-related phenotypes.
Cutaneous treatment with ovalbumin (OVA)
SIRT1 WT and cKO mice with an SKH-1 background were treated with OVA (Sigma Chemicals, Saint Louis, MO) by three cycles of daily cutaneous exposure to OVA for 5 consecutive days as described previously (11). OVA (fraction V, Sigma) was prepared in PBS (Dulbecco’s PBS, Sigma) at 1 mg/ml. Mice were restrained, and 50 μl of OVA solution, or 50 μl PBS, was applied to the dorsal skin and allowed to air dry.
Measurement of trans-epidermal water loss (TEWL)
TEWL was measured on the dorsal skin of shaved mice by using a DPM9003 device (Nova Technology) as described previously (26). Measurements were performed at room temperature and results were recorded. Three readings from dorsal skin were taken on each mouse and averaged.
Microarray gene expression
RNA was extracted from Normal human epidermal keratinocytes (NHEK) cells transfected with NC/siSIRT1 according to the manufacturer’s instructions (RNeasy mini kit, Qiagen). Total RNA concentration and purity were determined with NanoDrop and total RNA integrity was confirmed with Agilent Bioanalyzer. The RNA samples were processed with an Affymetrix GeneChip microarray at the Functional Genomics Core Facility at the University of Chicago.
Cell culture and siRNA/plasmid transfection
SIRT1 wild-type (WT) and knockout (KO) mouse embryonic fibroblast (MEF) cells (a gift from Dr. Xiaoling Li, NIEHS/NIH) were maintained in a monolayer culture in 95% air/5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units per mL penicillin, and 100 mg per mL streptomycin (Invitrogen, Carlsbad, California). MEF cells were cultured for fewer than 20 passages. Normal human epidermal keratinocytes (NHEK) were obtained from Clonetics (Lonza, Inc., Allendale, NJ) and cultured in KGM Gold BulletKit medium (Clonetics, Lonza) according to the manufacturer’s instructions. NHEK cells were cultured for fewer than 4 passages. Cells were transfected with negative control (NC) or siRNA (ON-TARGETplus SMARTpool, Dharmacon, Inc., Pittsburgh, PA) targeting SIRT1, using Amaxa Nucleofector according to the manufacturer’s instructions as described previously (22, 23).
Immunohistochemistry and immunofluorescence
Immunohistochemical staining of SIRT1, K10, loricrin, and filaggrin were performed in human skin and dermatitis specimens. SIRT1 levels were determined using the alkaline phosphatase anti-alkaline phosphatase (APAAP) method in which the substrate staining (red) is easily distinguishable from melanin (brown). Immunofluorescence analysis of SIRT and CYP1B1 was performed as described in our recent studies (27, 28).
Western blotting
Protein concentrations were determined using the BCA assay (Pierce, Inc., Rockford, IL, USA). Equal amounts of protein were subjected to electrophoresis. Western blotting was performed as described previously (22). Antibodies used included SIRT1, CYP1B1, AKT, p53, p21, GAPDH (Santa Cruz Biotechnology, Inc., Dallas, TX), p-AKT at serine 473 (Cell Signaling, Inc., Danvers, MA), filaggrin, and acetylated p53 (ac-p53) at K382 (Abcam, Inc., Cambridge, MA).
Real-time PCR
Quantitative real-time PCR assays were performed using ABI7300 (Applied Biosystems, Foster City, CA). Real time RT-PCR fluorescence detection was performed in 96-well plates with the SYBR Green PCR Master Mix (Applied Biosystems). Amplification primers for filaggrin, CYP1B1, were purchased from Origene. 5′-ACTGGAACGGTGAAGGTGACA-3′ (forward) and 5′-ATGGCAAGGGACTTCCTGTAAC-3′ (reverse) were used for β-actin. The threshold cycle number (Ct) for each sample was determined in triplicate. In all RT-PCR data, mRNA abundance of specific targets was relative expression (fold) to the control group (taken as 1) after normalization to β-actin (22).
Luciferase Reporter Assays
The plasmid mixtures, containing 1μg of XRE promoter luciferase construct (XRE-luc in pGL3 vector, kindly provided by Dr. Michael S. Denison from University of California) and 0.025 μg of pRL-TK (Promega, used as a transfection efficiency control), were used for transfection with electroporation according to the manufacturer’s protocol. The luciferase activity was measured as described previously (22).
Toluidine blue staining for mast cells
Toluidine blue staining for mast cells was performed according to the manufacturer’s protocol (NovaUltra, Waltham, MA, IW-3013). Briefly, the sections were deparaffinized and hydrated, then stained in toluidine blue working solution. After washing, the sections were dehydrated and then coverslipped with resinous mounting medium. The numbers of mast cells were calculated as the average from four or seven different fields of each sample (×40 magnification).
Serum IgE levels
IgE concentrations were measured using a mouse IgE ELISA kit (BioLegend, Inc., San Diego, CA) according to the manufacturer’s protocol.
Electron microscopy
Transmission electron microscopy was carried out on dorsal skin of three female WT or SIRT1 cKO mice at 6 weeks of age. Sections were embedded in Spurr resin, and examined in a FEI Tecnai G2 F30 by our Electron Microscopy core facility on campus.
Statistical analyses
Statistical analyses were performed using Prism 5 (GraphPad software, San Diego, CA). Kaplan-Meier survival estimates and log-rank tests were used to evaluate the skin lesion onset in mice. Data were expressed as the mean of three independent experiments and analyzed by Student’s t-test or two-way ANOVA. A P value of less than 0.05 was considered statistically significant.
RESULTS
Development of AD-like skin lesions in mice with epidermal-specific SIRT1 deletion
We have demonstrated recently the essential role of epidermal SIRT1 in UV-induced skin carcinogenesis and skin injury using mice with keratinocyte-specific SIRT1 deletion (23) (Fig. S1). Since SIRT1 is critical for preventing age-related pathologies (15), we asked whether SIRT1 has an active role in skin homeostasis during aging. We monitored wild-type (WT) mice and mice with epidermal SIRT1 heterozygous (cHet) and homozygous deletion (cKO) housed in our clean barrier facility for 24 months. At 51 weeks of age, cKO mice spontaneously developed AD-like skin lesions (Fig. 1A–B). In comparison, at 64 weeks of age, cHet mice started to developed similar AD-like skin lesions (Fig. 1A–B). The clinical severities of skin lesions included erythema, edema, erosion, and scaling. In contrast, no cutaneous manifestation was observed in SIRT1 WT mice. To further elucidate AD-associated immunological status of SIRT1 cHet and cKO mice prior to development of AD-like lesions, we measured serum IgE concentrations, histology, and numbers of cutaneous eosinophils and mast cells in mice without visible AD-like lesions, as IgE antibodies, eosinophils and mast cells have been linked to the pathophysiology of allergic disorders including AD (29). IgE concentrations were significantly higher in SIRT1 cHet and cKO mice than in age-matched 18-month-old SIRT1 WT mice (Fig. 1C). At 6 months of age, most of the dorsal skin examined in cHet and cKO did not show significant acanthosis or alterations to the epidermis (Fig. 1D). However, we did observe significant increase in the number of eosinophils in the SIRT1 cHet and cKO mouse skin (Fig. 1E). In addition, the numbers of mast cells increased significantly in the dermis of SIRT1 cHet and cKO mice as compared with WT mice (Fig. 1F–G). To determine the effect of SIRT1 deletion on the integrity of the skin barrier prior to formation of AD-like lesions, we measured transepidermal water loss (TEWL). SIRT1 cHet and cKO mice showed increased TEWL, as compared with WT mice (Fig. 1H). These results indicate that SIRT1 loss causes age- and SIRT1-dose-dependent spontaneous development of AD-like lesions.
Figure 1. Development of AD-like skin lesions in mice with epidermal SIRT1 deletion (cKO).

A, Gross phenotype of 51–64 week-old SIRT1 cKO mice. B, percent (%) of skin lesion-free mice in SIRT1 WT, cHet, and cKO mice (N=15). C, mouse serum IgE level in SIRT1 WT, cHet, and cKO mice at 18 months of age. D, representative photomicrographs of HE skin sections from 6-month-old SIRT1 WT, cHet, and cKO mice. Scale bar=50μM. E, Numbers of eosinophils detected per high-power field (HPF, 400× magnification). F, Toluidine Blue staining of mast cells in the skin of SIRT1 WT, cHet, and cKO mice at 6 months of age. Scale bar=200μM. G, Scatter chart for numbers of mast cells per low power field (LPF, 100× magnification) in SIRT1 WT, cHet, and cKO mice skin (n=3). H, TEWL in 18-month old SIRT1 WT, cHet and cKO mice. For C, data are shown as mean ± SE for n = 3. *, P < 0.05; t-test, compared with the WT group. For E, G and H, data are shown as means ± SD for n = 3. *, P < 0.05; t-test, compared with the WT group.
Reduced filaggrin expression in mice with epidermal SIRT1 deletion
To determine the molecular role of SIRT1 in skin barrier function, we carried out a microarray analysis of gene expression in undifferentiated NHEK cells transfected with siRNA targeting SIRT1 (siSIRT1) or negative control siRNA (NC). Among the genes that were up- or down-regulated by SIRT1 knockdown by greater than two fold, we found that SIRT1 inhibition decreased the expression of filaggrin (Supplemental Table S1), while it had less or little effect on other epidermal differentiation genes, including involucrin, keratin 14, keratin 5, desmogleins, KLKs, martriptase, or SPINK 5 (Supplemental Fig. S2). Further analysis showed that filaggrin mRNA expression and profilaggrin (proFLG) protein abundance both were significantly reduced in undifferentiated normal human epidermal keratinocytes (NHEK) cells transfected with siSIRT1, as compared with NC (Fig. 2A–B). Furthermore, the mRNA level of filaggrin and the protein levels of profilaggrin (proFLG) and filaggrin monomer (FLG) were decreased in the skin of SIRT1 cKO mice as compared with WT mice (Fig. 2C–E). However, epidermal SIRT1 deletion did not affect the protein abundance of K10 or loricrin (Fig. 2E). Electron microscopy showed that the granular layer of the SIRT1 cKO epidermis lacked the large, irregular shaped keratohyalin (F-granules) (30) that contains profilaggrin as compared with SIRT1 WT epidermis, while the small round loricrin-containing keratohyalin (L-granules) were present in the epidermal granular cells in both WT and SIRT1 cKO mice (Fig. 2F–G), similar to the phenotype detected in flaky tail mice that are deficient in filaggrin (31). These findings indicated that SIRT1 loss inhibits filaggrin expression in vitro and in vivo. Further in vitro and in vivo investigation will elucidate the role of other epidermal differentiation genes in the function of SIRT1 in skin barrier integrity.
Figure 2. Reduced filaggrin expression in keratinocytes and mouse skin with SIRT1 loss.

A, Filaggrin mRNA level in undifferentiated NHEK cells transfected with negative control (NC) or siRNA targeting SIRT1 (siSIRT1). Data are shown as mean ± SE from three independent experiments. *, P < 0.05; t-test, compared with the NC group. B, Filaggrin protein level in NHEK cells transfected with NC or siSIRT1 as in A. C, Filaggrin mRNA level in 6-month-old SIRT1 WT and cKO mouse skin. D, Profilaggrin (proFLG) and filaggrin monomer (FLG) protein level in the skin from 6-month-old SIRT1 WT and cKO mouse skin. E, Immunohistochemical staining of filaggrin, K10 (keratin 10), and loricrin in the skin of 6-month-old WT and SIRT1 cKO mice. Scale bar=50μM. F and G, Electron microscopic analysis of WT (F) and SIRT1 cKO (G) epidermis from 6-week-old mice. Red arrow indicates F-granule (F), while blue arrow indicates L-granule (L). Scale bar, 2 μm.
SIRT1 deficiency in keratinocytes sensitizes mouse skin to OVA-induced histological and immunological response
To determine whether loss of SIRT1 impairs the skin barrier, we tested the hypothesis that skin with SIRT1 loss is permissive to epicutaneous challenges by protein antigens normally excluded from the skin. To do this, we treated mice with ovalbumin (OVA) as described previously (11) (Fig. 3A). Allergen challenge induced epidermal acanthosis in SIRT1 cKO mice but not in WT mice (Fig. 3B). In SIRT1 cKO mice, OVA significantly increased the serum IgE level, but it did not affect IgE levels in WT mice (Fig. 3C). To determine the effect of OVA on the integrity of the skin barrier, we measured transepidermal water loss (TEWL). At the site of the allergen challenge at 6 weeks of allergen challenge treatments, OVA significantly increased TEWL in SIRT1 cKO mice, but not in WT mice (Fig. 3D). These findings demonstrate that SIRT1 deficiency in the epidermis impairs skin barrier integrity.
Figure 3. Response to ovalbumin (OVA) in WT and SIRT1 cKO mice.
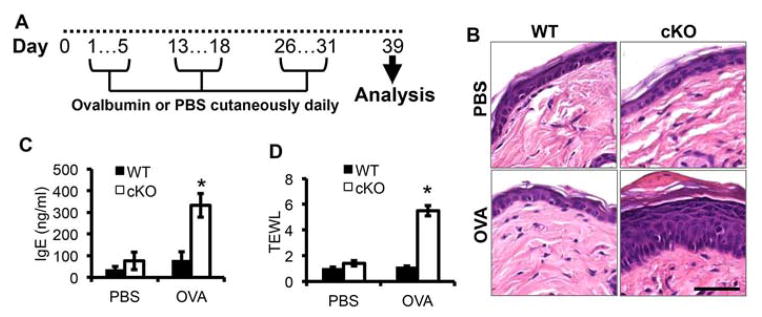
A, schematic for ovalbumin treatment. B, Histology of mouse skin following PBS and OVA treatment as in Fig A. Scale bar: 50 μM. C, ELISA assay of the serum IgE levels in SIRT1 WT and cKO mice following PBS and OVA treatment. D, TEWL in SIRT1 WT and cKO mice following PBS and OVA treatment. For all of the graphs, data are shown as mean ± SE for n = 4. *, P < 0.05; t-test, compared with the WT group. The mice were 6–8 weeks old.
SIRT1 regulates filaggrin expression in a deacetylase-dependent manner
To determine the role of SIRT1 deacetylase activity in filaggrin expression, we compared the difference in filaggrin expression in NHEK and 293T cells transfected with vector (Vec), wild-type SIRT1 (WT), and inactive mutant SIRT1 H355Y (Mut) (32). As compared with vector-transfected NHEK/293T cells, WT SIRT1 transfection increased profilaggrin (proFLG) protein abundance, whereas mutant SIRT1 had no effect (Fig. 4A–B). WT SIRT1 reduced the level of acetylated p53 (Ac-p53), whereas Mut SIRT1 had no effect, confirming loss of the deacetylase activity of Mut SIRT1 (Fig. 4A–B). Filaggrin mRNA concentration was significantly increased in SIRT1 WT-transfected 293T cells but unaltered in mutant SIRT1-transfected cells (Fig. 4C). These findings indicate that the deacetylase activity of SIRT1 is required for its regulation of filaggrin. Since SIRT1 is known to deacetylate p53 and thus inhibit its transcriptional function of preventing apoptosis induced by DNA damage (33), next we investigated whether p53 plays a role in filaggrin regulation. Profilaggrin protein abundance remained unaltered in NHEK cells transfected with siRNA targeting p53 (Fig. 4D). Taken together, these observations indicate that SIRT1 promotes filaggrin expression through its deacetylase activity independent of p53.
Figure 4. Role of SIRT1 deacetylase activity in its regulation of filaggrin.
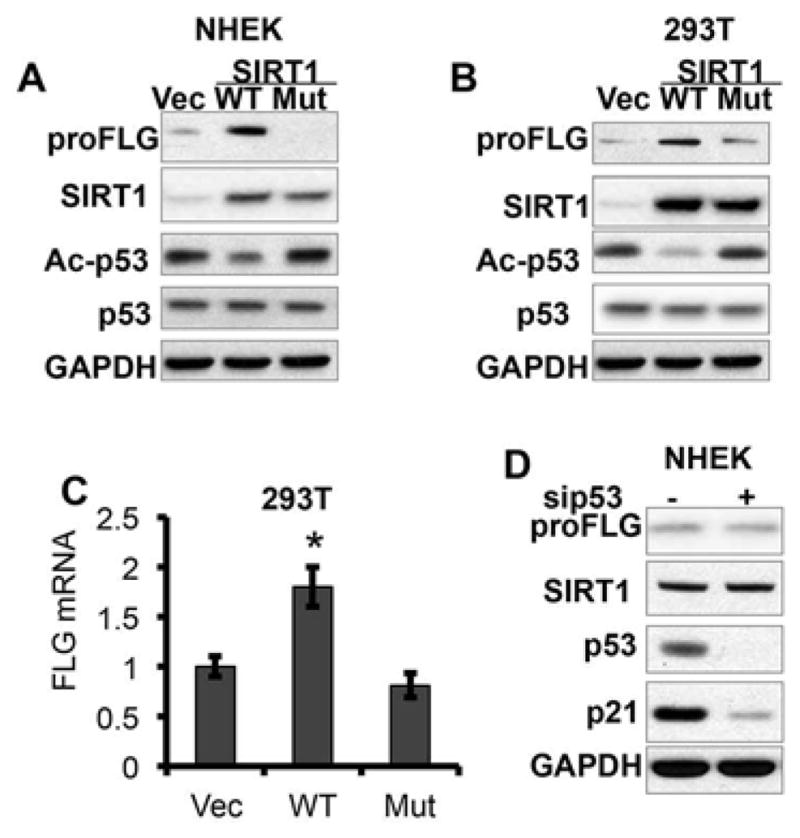
A, Immunoblot analysis of profilaggrin (proFLG), SIRT1, ac-p53, p53, and GAPDH protein levels in NHEK cells transfected with SIRT1 vector (Vec), wild-type (WT), or inactive mutant (Mut) SIRT1 plasmids. B, Immunoblot analysis of profilaggrin (proFLG), SIRT1, ac-p53, p53, and GAPDH protein levels in 293T cells transfected with Vec, WT or Mut SIRT1 plasmids. C, Filaggrin mRNA levels in 293T cells transfected with Vec, WT or Mut SIRT1 plasmids. Data are shown as mean ± SE from three independent experiments. *, P < 0.05; t-test, compared with the vector (Vec) group. D, immunoblot analysis of profilaggrin (proFLG), SIRT1, p53, p21 and GAPDH in NHEK cells transfected with negative control or siRNA targeting p53 (sip53).
SIRT1 loss inhibits the activation of AhR
Previous reports indicated that coal-tar-medicated activation of the AhR signaling pathway leads to enhanced epidermal differentiation and epidermal barrier function (9). To determine the molecular function of SIRT1, we examined whether SIRT1 regulates AhR target genes obtained from our microarray analysis. Among the genes that were up- or down-regulated by SIRT1 knockdown by greater than two fold, we found that SIRT1 inhibition decreased the expression of CYP1B1 (Supplemental Table S1), a known AhR target gene, but had no effect on AhR expression (Fig. 5A). Real-time PCR analysis indicated that SIRT1 knockdown significantly decreased CYP1B1 mRNA expression (Fig. 5B). Epidermal SIRT1 deletion significantly reduced the CYP1B1 mRNA expression in mouse skin (Fig. 5C). Next we assessed the effect of SIRT1 inhibition on AhR activation, using the AhR reporter assay with a luciferase plasmid driven by a xenobiotic response element (XRE-Luc). The transcriptional activity of the AhR reporter was significantly lower in SIRT1 KO cells than in WT cells (Fig. 5D). Immunofluorescence analysis showed that SIRT1 knockdown decreased CYP1B1 abundance (Fig. 5E). These findings demonstrate that SIRT1 is required for basal AhR activity in keratinocytes.
Figure 5. Role of AhR in SIRT1 regulation of filaggrin.

A, Gene array analysis of mRNA levels of CYP1B1 and AhR in NHEK cells transfected with NC or siSIRT1. B, Real-time PCR analysis of CYP1B1 mRNA expression in NHEK cells transfected with NC or siSIRT1. C, Real-time PCR analysis of CYP1B1 mRNA expression in SIRT1 WT and cKO mouse skin (n=3). D, Luciferase reporter assay of AhR reporter activity (XRE-luc) in SIRT1 WT and KO MEF cells. E, Immunofluorescence analysis of SIRT1 and CYP1B1 in NHEK cells transfected with NC or siSIRT1. DAPI staining (blue) was used as a nuclear counterstain. Scale bar: 50 μm. F, Immunoblot analysis of filaggrin, SIRT1, and GAPDH protein levels in NHEK cells transfected with NC or siSIRT1 and then treated with Bap (10μM) for 48h. G, Real-time PCR analysis of filaggrin mRNA level in NHEK cells transfected with NC or siSIRT1 and treated with BaP (10 μM) for 48h. H, Real-time PCR analysis of CYP1B1 mRNA levels in NHEK cells transfected with NC or siSIRT1 and treated with BaP (10 μM) for 48h. I, Immunoblot analysis of filaggrin monomer (FLG) and GAPDH protein levels in SIRT1 cKO mice treated with OVA for 6 weeks followed by treatment with BaP for 3 or 7 days. J, representative pictures of H&E staining for SIRT1 cKO mice treated with OVA for 6 weeks followed by treatment with Bap for 3 or 7 days. K, mouse serum IgE level in SIRT1 cKO mice treated with OVA for 6 weeks followed by treatment with Bap for 7 days. Scale bar: 50 μM. For all of the graphs, data are shown as mean ± SE from three independent experiments. *, P < 0.05; t-test, compared with the control (NC or WT) group for A–D, or vehicle (Veh) group in G–H.
To determine the role of AhR in filaggrin suppression in SIRT1-inhibited cells, we investigated whether AhR activation in SIRT1-inhibited cells restored filaggrin expression. NHEK cells were transfected with NC/siRNA targeting SIRT1, and then treated with the AhR ligand benzo(a)pyrene (BaP). In both NC- and siSIRT1-transfected cells, BaP increased the protein abundance and mRNA expression of both filaggrin (Fig. 5F–G) and CYP1B1 (Fig. 5F–H). However, SIRT1 knockdown reduced the basal and BaP-induced protein and mRNA levels of filaggrin and CYP1B1 (Fig. 5F–H). To further confirm the role of BaP-induced filaggrin expression in vivo, we treated SIRT1 cKO mice with OVA for 6 weeks and then with BaP for 3 or 7 days. BaP increased mature filaggrin (FLG) abundance in SIRT1 cKO mouse skin (Fig. 5I), and reduced OVA-induced acanthosis (Fig. 5J) and the IgE level (Fig. 5K). These findings indicate that SIRT1 promotes AhR activity in the regulation of filaggrin.
AKT activation is involved in AhR regulation of filaggrin
It had been reported that AhR regulates the PI3K/AKT pathway (34). To further determine whether AKT activation plays a role in the AhR regulation of filaggrin, we measured the effect of BaP on AKT activation and the effect of AKT knockdown on BaP-induced filaggrin protein concentrations in NHEK cells. BaP increased filaggrin abundance and AKT phosphorylation (Fig. 6A), while SIRT1 knockdown reduced the basal and BaP-induced AKT phosphorylation (Fig. 6A). Blocking the AKT pathway with siRNA targeting the major AKT isoform AKT1 (siAKT1) decreased BaP-induced filaggrin abundance, while it did not affect the CYP1B1 protein abundance (Fig. 6B), implying a distinct regulation of filaggrin and CYP1B1 by the SIRT1/AhR axis. Similar effect of AKT inhibition on filaggrin protein levels was detected with siAKT1 or the PI3K/AKT inhibitor LY294002 (LY) in the keratinocyte line HaCaT cells (Fig. S3A–B). These findings indicate that SRIT1-modulated AhR activation regulates filaggrin through AKT (Fig. 6C), suggesting an indirect mechanism in filaggrin regulation by the SIRT1/AhR pathway.
Figure 6. Role of AKT in AhR regulation of filaggrin.
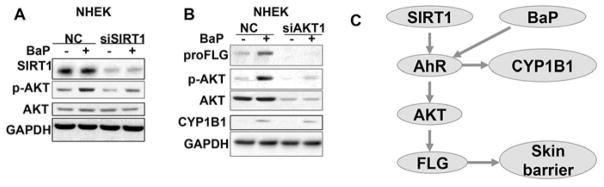
A, Immunoblot analysis of filaggrin, CYP1B1, p-AKT, AKT, and GAPDH in NHEK cells treated with BaP (10 μM) for 48h. B. Immunoblot analysis of filaggrin, p-AKT, AKT, CYP1B1, and GAPDH in NHEK cells transfected with siRNA targeting AKT1 (siAKT1) or negative control (NC) and then treated with vehicle (V) or BaP (10 μM).
SIRT1 protein level is down-regulated in human atopic dermatitis and non-atopic dermatitis
To further investigate the specific function of SIRT1 in human dermatitis, we used immunohistochemical analysis to evaluate SIRT1 protein levels in 7 atopic dermatitis and 13 non-atopic dermatitis specimens (spongiotic dermatitis without a history of atopy), as compared with 10 sun-protected normal skin specimens. Although some SIRT1 was localized in the cytoplasm, the majority of the protein was localized to the nucleus in epidermal keratinocytes of normal skin (Fig. 7A). The SIRT1 levels were reduced (score 0 or 1) in all 7 atopic dermatitis and all 13 non-atopic dermatitis samples, as compared with 10% of normal skin samples (1/10) (Fig. 7B). This reduction was statistically significant as analyzed by the Mann-Whitney Test (P<0.005 between normal skin and atopic dermatitis, and between normal skin and non-atopic dermatitis).
Figure 7. SIRT1 protein levels in human normal skin, atopic dermatitis, and non-atopic dermatitis.
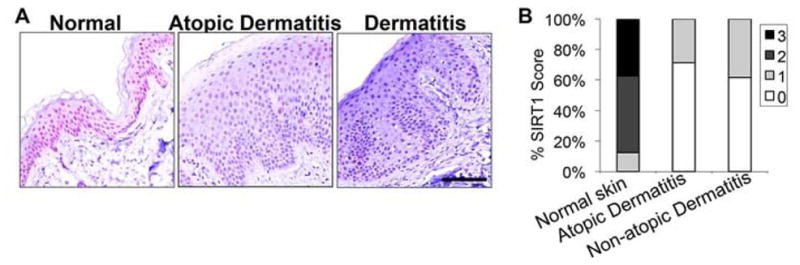
A. APAAP red staining for SIRT1 protein levels in human normal skin, atopic dermatitis, and non-atopic dermatitis. Scale bar: 100 μM. B, Percent (in stacked column format) for each score of SIRT1 expression.
DISCUSSION
In this study we have shown that SIRT1 regulates skin barrier integrity and promotes filaggrin expression. Epidermal SIRT1 deletion causes AD-like skin lesions in older mice and sensitizes mice to epicutaneous allergen challenge. Filaggrin regulation by SIRT1 requires the deacetylase activity of SIRT1 and the AhR/AKT pathway by SIRT1 (Fig. 6C). As compared with normal human skin, SIRT1 is down-regulated in human atopic dermatitis and non-atopic dermatitis. Our results demonstrate the critical role of SIRT1 in skin barrier function and protecting the skin against allergen challenge.
SIRT1 plays vital roles in multiple physiological and pathological contexts, in aging, metabolism, and cancer (15, 19). The role of SIRT1 in skin diseases and function is just now beginning to be understood. Our recent studies have delineated the critical role of SIRT1 in UV-induced skin cancer and injury (23). Recent microarray studies have shown that SIRT1 promotes keratinocyte differentiation in vitro and suggested the role of several structural proteins and enzymes including filaggrin (35). Our findings in the current study indicate that SIRT1 regulates filaggrin expression, but not K10 or loricrin, in its deacetylase-dependent mechanism.
At the molecular level, our findings indicate that SIRT1 modulates AhR activity, which regulates AKT activation and thus promotes filaggrin expression (Fig. 6C). As one of the critical regulators of the skin barrier, AhR is a cytoplasmic transcription factor in xenobiotic detoxification; upon ligand binding, AhR translocates to the nucleus (36) and binds to the DNA motifs known as xenobiotic response elements (XREs) (37). AhR activation by coal tar induces filaggrin expression and skin barrier repair (38). As one of the oldest treatments for AD, coal tar is found to promote epidermal differentiation by activating AhR (38). We found that SIRT1 positively regulates AhR activation. SIRT1 loss in keratinocytes and mouse skin inhibited basal and ligand-induced AhR activity. AhR ligand increased filaggrin expression in control and SIRT1-inhibited keratinocytes, indicating that SIRT1 fine-tunes AhR activation. SIRT1 utilizes its deacetylase function to regulate filaggrin. However, we failed to detect the acetylation of either AhR or its partner ARNT (aryl hydrocarbon receptor nuclear translocator, data not shown). It is possible that histone deacetylation (19) mediates the effect of SIRT1 on AhR activity and filaggrin expression. Further investigation is needed to elucidate the mechanism of AhR regulation by SIRT1. Nevertheless, our results imply that the combination of SIRT1 activation and AhR/AKT activation may maximize skin barrier repair and the therapeutic effect on AD.
Both AhR and ARNT have been shown to regulate keratinocyte differentiation and barrier function (38, 39). AhR activation by coal tar promotes keratinocyte differentiation and the skin barrier (38). ARNT promotes the activation of EGFR/ERK to induce the expression of filaggrin and other epidermal structure proteins (39). Our study showed that AKT activation is required for AhR ligand-induced expression of filaggrin but not the expression of CYP1B1 (Fig. 6C). Given that AKT is the downstream pathway of EGFR, it is possible that non-canonical AhR activation activates the EGFR/AKT pathway to regulate filaggrin expression, further underscoring the critical role of AhR in skin barrier function.
In addition to regulating filaggrin expression, other functions of SIRT1 may play a role in the development of AD-like skin lesions. For example, SIRT1 inhibits activation of the pro-inflammatory transcription factor NF-κB by acetylating its subunit p65 (40, 41), and topical inhibition of NF-κB has been shown to improve AD in mice (42–44). SIRT1 activators and inhibitors have been shown to modulate airway inflammation in an ovalbumin-challenged mouse model (45, 46). However, it remains unclear whether SIRT1 regulation of NF-κB has a role in either AD or in airway inflammation. It is possible that the epithelial barrier is involved in the effect of SIRT1 modulators in airway inflammation in the mouse model of asthma as it is in AD. Indeed, we note that mice with epidermal SIRT1 deletion developed AD-like lesions only at an older age, suggesting a critical role of SIRT1 loss in skin inflammation. On the other hand, NF-κB may play a role in filaggrin regulation by SIRT1, since TNFα has been shown to suppress filaggrin expression (47). Further studies are needed to define the molecular mechanisms in the role of SIRT1 in both the skin barrier and the airway epithelial barrier.
In human dermatitis tissue, SIRT1 down-regulation may involve multiple mechanisms. For example, in flaky tail mice harboring a homozygous frameshift filaggrin mutation (11–14, 31), loss of filaggrin reduced SIRT1 expression (48). SIRT1 and filaggrin may form a positive feedback loop in maintaining the availability of both proteins and thus skin barrier function. Loss of one leads to the suppression of the other. In addition, AD-associated cytokines may also contribute to SIRT1 down-regulation. Future investigation can elucidate the potential regulation of SIRT1 expression by critical cytokines involved in AD pathogenesis and filaggrin regulation, including but not limited to TNFα, interleukin 4 (IL-4), or IL-13 (47, 49–53).
In summary, we have demonstrated that SIRT1 promotes filaggrin expression and is required for skin barrier function. Loss of SIRT1 predisposes mice to susceptibility to epicutaneous allergen challenge and causes late-onset AD-like skin inflammation. SIRT1 enhances the activation of AhR and AhR/AKT-induced filaggrin expression. Our findings may provide new molecular insights into the regulation of skin barrier integrity and the pathogenesis of AD, and open up new opportunities for developing targeted mechanism-based strategies for AD prevention and therapy.
Supplementary Material
Clinical Implications.
SIRT1 deficiency inhibits filaggrin expression, predisposes mice to spontaneous development of AD-like lesion, sensitizes mouse skin to epicutaneous allergen challenge, and is associated with human dermatitis.
Acknowledgments
We thank Dr. Lawrence S. Chan, Julian Solway, and Sarah Stein for helpful discussion, Dr. Xiaoling Li for providing the WT and SIRT1 KO MEF cells and the wild-type and mutant SIRT1 plasmids, and Dr. Michael S. Denison for providing the XRE-Luc plasmid. This work was supported by the NIH/NIEHS grant ES016936 (YYH), the American Cancer Society (ACS) grant RSG-13-078-01 (YYH), the University of Chicago Cancer Research Center (P30 CA014599), the CTSA (NIH UL1RR024999), and the University of Chicago Friends of Dermatology Endowment Fund. We thank Terri Li for immunohistochemical analysis, Yimei Chen for electron microscopy, and Dr. Ann Motten for critical reading of the manuscript.
Abbreviations
- Ac-p53
acetylated p53
- AD
atopic dermatitis
- AhR
aryl hydrocarbon receptor
- AKT
protein kinase B
- ARNT
Aryl hydrocarbon receptor nuclear translocator
- BaP
benzo(a)pyrene
- cHet
epidermis-specific heterozygous SIRT1 deletion
- cKO
epidermis-specific homozygous SIRT1 deletion
- CYP1B1
cytochrome P450 1B1, An AhR target gene
- ELISA
the enzyme-linked immunosorbent assay
- FLG
filaggrin
- H&E
Hematoxyline and Eosin
- HPF
high power field
- IgE
immunoglobin E
- K10
keratin 10
- KO
SIRT1 null cells
- LPF
low power field
- MEF
mouse embryonic fibroblast cells
- NC
negative control
- NHEK
normal human epidermal keratinocytes
- OVA
ovalbumin
- p-AKT
phosphorylated AKT
- PBS
phosphate buffered saline
- proFLG
profilaggrin
- SIRT1
sirtuin 1
- siAKT1
siRNA targeting AKT1
- sip53
siRNA targeting p53
- siSIRT1
siRNA targeting SIRT1
- TEWL
trans-epidermal water loss
- WT
wild type
- XRE
xenobiotic response element
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elias PM. Skin barrier function. Curr Allergy Asthma Rep. 2008 Jul;8(4):299–305. doi: 10.1007/s11882-008-0048-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elias PM, Schmuth M. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Opin Allergy Clin Immunol. 2009 Oct;9(5):437–46. doi: 10.1097/ACI.0b013e32832e7d36. Epub 2009/06/25. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elias PM, Wakefield JS. Therapeutic implications of a barrier-based pathogenesis of atopic dermatitis. Clin Rev Allergy Immunol. 2011 Dec;41(3):282–95. doi: 10.1007/s12016-010-8231-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin H, He R, Oyoshi M, Geha RS. Animal models of atopic dermatitis. J Invest Dermatol. 2009 Jan;129(1):31–40. doi: 10.1038/jid.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams H, Flohr C. How epidemiology has challenged 3 prevailing concepts about atopic dermatitis. J Allergy Clin Immunol. 2006 Jul;118(1):209–13. doi: 10.1016/j.jaci.2006.04.043. Epub 2006/07/04. eng. [DOI] [PubMed] [Google Scholar]
- 6.Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. The New England journal of medicine. 2011 Oct 6;365(14):1315–27. doi: 10.1056/NEJMra1011040. [DOI] [PubMed] [Google Scholar]
- 7.Sandilands A, Terron-Kwiatkowski A, Hull PR, O’Regan GM, Clayton TH, Watson RM, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007 May;39(5):650–4. doi: 10.1038/ng2020. Epub 2007/04/10. eng. [DOI] [PubMed] [Google Scholar]
- 8.Morar N, Cookson WO, Harper JI, Moffatt MF. Filaggrin mutations in children with severe atopic dermatitis. J Invest Dermatol. 2007 Jul;127(7):1667–72. doi: 10.1038/sj.jid.5700739. Epub 2007/02/16. eng. [DOI] [PubMed] [Google Scholar]
- 9.Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009 May 1;122(Pt 9):1285–94. doi: 10.1242/jcs.033969. Epub 2009/04/24. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henderson J, Northstone K, Lee SP, Liao H, Zhao Y, Pembrey M, et al. The burden of disease associated with filaggrin mutations: a population-based, longitudinal birth cohort study. J Allergy Clin Immunol. 2008 Apr;121(4):872–7. e9. doi: 10.1016/j.jaci.2008.01.026. Epub 2008/03/08. eng. [DOI] [PubMed] [Google Scholar]
- 11.Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009 May;41(5):602–8. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oyoshi MK, Murphy GF, Geha RS. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol. 2009 Sep;124(3):485–93. 93 e1. doi: 10.1016/j.jaci.2009.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scharschmidt TC, Man MQ, Hatano Y, Crumrine D, Gunathilake R, Sundberg JP, et al. Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. J Allergy Clin Immunol. 2009 Sep;124(3):496–506. e1–6. doi: 10.1016/j.jaci.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawasaki H, Nagao K, Kubo A, Hata T, Shimizu A, Mizuno H, et al. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J Allergy Clin Immunol. 2012 Jun;129(6):1538–46. e6. doi: 10.1016/j.jaci.2012.01.068. [DOI] [PubMed] [Google Scholar]
- 15.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes & development. 2006 Nov 1;20(21):2913–21. doi: 10.1101/gad.1467506. eng. [DOI] [PubMed] [Google Scholar]
- 16.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annual review of biochemistry. 2004;73:417–35. doi: 10.1146/annurev.biochem.73.011303.073651. eng. [DOI] [PubMed] [Google Scholar]
- 17.Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes & development. 2000 May 1;14(9):1021–6. eng. [PubMed] [Google Scholar]
- 18.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. The Biochemical journal. 2007 May 15;404(1):1–13. doi: 10.1042/BJ20070140. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annual review of pathology. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009 Feb;9(2):123–8. doi: 10.1038/nrc2562. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Molecular cell. 2010 Jul 30;39(2):247–58. doi: 10.1016/j.molcel.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Ming M, Shea CR, Guo X, Li X, Soltani K, Han W, et al. Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proceedings of the National Academy of Sciences of the United States of America. 2010 Dec 28;107(52):22623–8. doi: 10.1073/pnas.1010377108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ming M, Soltani K, Shea CR, Li X, He YY. Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene. 2014 Jan 20; doi: 10.1038/onc.2013.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blander G, Bhimavarapu A, Mammone T, Maes D, Elliston K, Reich C, et al. SIRT1 promotes differentiation of normal human keratinocytes. J Invest Dermatol. 2009 Jan;129(1):41–9. doi: 10.1038/jid.2008.179. Epub 2008/06/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ming M, Feng L, Shea CR, Soltani K, Zhao B, Han W, et al. PTEN positively regulates UVB-induced DNA damage repair. Cancer research. 2011 Aug 1;71(15):5287–95. doi: 10.1158/0008-5472.CAN-10-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C, Lasse S, Lee P, Nakasaki M, Chen SW, Yamasaki K, et al. Development of atopic dermatitis-like skin disease from the chronic loss of epidermal caspase-8. Proc Natl Acad Sci U S A. 2010 Dec 21;107(51):22249–54. doi: 10.1073/pnas.1009751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiang L, Wu C, Ming M, Viollet B, He YY. Autophagy controls p38 activation to promote cell survival under genotoxic stress. The Journal of biological chemistry. 2013 Jan 18;288(3):1603–11. doi: 10.1074/jbc.M112.415224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiang L, Zhao BZ, Ming M, Wang N, He TC, Hwang S, et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:9241–6. doi: 10.1073/pnas.1322913111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nature medicine. 2012 May;18(5):693–704. doi: 10.1038/nm.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steven AC, Bisher ME, Roop DR, Steinert PM. Biosynthetic pathways of filaggrin and loricrin--two major proteins expressed by terminally differentiated epidermal keratinocytes. Journal of structural biology. 1990 Jul-Sep;104(1–3):150–62. doi: 10.1016/1047-8477(90)90071-j. [DOI] [PubMed] [Google Scholar]
- 31.Presland RB, Boggess D, Lewis SP, Hull C, Fleckman P, Sundberg JP. Loss of normal profilaggrin and filaggrin in flaky tail (ft/ft) mice: an animal model for the filaggrin-deficient skin disease ichthyosis vulgaris. The Journal of investigative dermatology. 2000 Dec;115(6):1072–81. doi: 10.1046/j.1523-1747.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- 32.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. The Journal of biological chemistry. 2008 Oct 10;283(41):27628–35. doi: 10.1074/jbc.M805711200. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001 Oct 19;107(2):137–48. doi: 10.1016/s0092-8674(01)00524-4. eng. [DOI] [PubMed] [Google Scholar]
- 34.Wu R, Zhang L, Hoagland MS, Swanson HI. Lack of the aryl hydrocarbon receptor leads to impaired activation of AKT/protein kinase B and enhanced sensitivity to apoptosis induced via the intrinsic pathway. The Journal of pharmacology and experimental therapeutics. 2007 Jan;320(1):448–57. doi: 10.1124/jpet.106.111773. [DOI] [PubMed] [Google Scholar]
- 35.Blander G, Bhimavarapu A, Mammone T, Maes D, Elliston K, Reich C, et al. SIRT1 Promotes Differentiation of Normal Human Keratinocytes. The Journal of investigative dermatology. 2009 Jun 19; doi: 10.1038/jid.2008.179. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A. 1992 Sep 1;89(17):8185–9. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denison MS, Fisher JM, Whitlock JP., Jr Inducible, receptor-dependent protein-DNA interactions at a dioxin-responsive transcriptional enhancer. Proc Natl Acad Sci U S A. 1988 Apr;85(8):2528–32. doi: 10.1073/pnas.85.8.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van den Bogaard EH, Bergboer JG, Vonk-Bergers M, van Vlijmen-Willems IM, Hato SV, van der Valk PG, et al. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest. 2013 Feb 1;123(2):917–27. doi: 10.1172/JCI65642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robertson ED, Weir L, Romanowska M, Leigh IM, Panteleyev AA. ARNT controls the expression of epidermal differentiation genes through HDAC- and EGFR-dependent pathways. Journal of cell science. 2012 Jul 15;125(Pt 14):3320–32. doi: 10.1242/jcs.095125. [DOI] [PubMed] [Google Scholar]
- 40.Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M, et al. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell host & microbe. 2008 Mar 13;3(3):158–67. doi: 10.1016/j.chom.2008.02.002. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schug TT, Xu Q, Gao H, Peres-da-Silva A, Draper DW, Fessler MB, et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol. 2010 Oct;30(19):4712–21. doi: 10.1128/MCB.00657-10. Epub 2010/07/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamura H, Aoki M, Tamai K, Oishi M, Ogihara T, Kaneda Y, et al. Prevention and regression of atopic dermatitis by ointment containing NF-kB decoy oligodeoxynucleotides in NC/Nga atopic mouse model. Gene Ther. 2002 Sep;9(18):1221–9. doi: 10.1038/sj.gt.3301724. Epub 2002/09/07. eng. [DOI] [PubMed] [Google Scholar]
- 43.Dajee M, Muchamuel T, Schryver B, Oo A, Alleman-Sposeto J, De Vry CG, et al. Blockade of experimental atopic dermatitis via topical NF-kappaB decoy oligonucleotide. J Invest Dermatol. 2006 Aug;126(8):1792–803. doi: 10.1038/sj.jid.5700307. Epub 2006/04/22. eng. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka A, Muto S, Jung K, Itai A, Matsuda H. Topical application with a new NF-kappaB inhibitor improves atopic dermatitis in NC/NgaTnd mice. J Invest Dermatol. 2007 Apr;127(4):855–63. doi: 10.1038/sj.jid.5700603. Epub 2006/10/28. eng. [DOI] [PubMed] [Google Scholar]
- 45.Kim SR, Lee KS, Park SJ, Min KH, Choe YH, Moon H, et al. Involvement of sirtuin 1 in airway inflammation and hyperresponsiveness of allergic airway disease. The Journal of allergy and clinical immunology. 2010 Feb;125(2):449–60. e14. doi: 10.1016/j.jaci.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Ichikawa T, Hayashi R, Suzuki K, Imanishi S, Kambara K, Okazawa S, et al. Sirtuin 1 activator SRT1720 suppresses inflammation in an ovalbumin-induced mouse model of asthma. Respirology. 2013 Feb;18(2):332–9. doi: 10.1111/j.1440-1843.2012.02284.x. [DOI] [PubMed] [Google Scholar]
- 47.Kim BE, Howell MD, Guttman-Yassky E, Gilleaudeau PM, Cardinale IR, Boguniewicz M, et al. TNF-alpha downregulates filaggrin and loricrin through c-Jun N-terminal kinase: role for TNF-alpha antagonists to improve skin barrier. J Invest Dermatol. 2011 Jun;131(6):1272–9. doi: 10.1038/jid.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakai K, Yoneda K, Hosokawa Y, Moriue T, Presland RB, Fallon PG, et al. Reduced expression of epidermal growth factor receptor, E-cadherin, and occludin in the skin of flaky tail mice is due to filaggrin and loricrin deficiencies. Am J Pathol. 2012 Sep;181(3):969–77. doi: 10.1016/j.ajpath.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. The Journal of allergy and clinical immunology. 2007 Jul;120(1):150–5. doi: 10.1016/j.jaci.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. The New England journal of medicine. 2002 Oct 10;347(15):1151–60. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 51.Fiset PO, Leung DY, Hamid Q. Immunopathology of atopic dermatitis. The Journal of allergy and clinical immunology. 2006 Jul;118(1):287–90. doi: 10.1016/j.jaci.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 52.Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006 Mar;24(3):341–8. doi: 10.1016/j.immuni.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 53.Chan LS, Robinson N, Xu L. Expression of interleukin-4 in the epidermis of transgenic mice results in a pruritic inflammatory skin disease: an experimental animal model to study atopic dermatitis. The Journal of investigative dermatology. 2001 Oct;117(4):977–83. doi: 10.1046/j.0022-202x.2001.01484.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.