

Abstract
Many tumors are associated with deficiency of the tumor suppressor, PTEN, a PIP3 phosphatase that turns off PIP3 signaling. The major site of PTEN action is the plasma membrane, where PIP3 is produced by PI3 kinases. However, the mechanism and functional importance of PTEN membrane recruitment are poorly defined. Using the heterologous expression system in which human PTEN is expressed in Dictyostelium discoideum, we defined the molecular mechanisms that regulate the membrane-binding site through inhibitory interactions with the phosphorylated C-terminal tail. In addition, we potentiated mechanisms that mediate PTEN membrane association and engineered an enhanced PTEN with increased tumor suppressor functions. Moreover, we identified a new class of cancer-associated PTEN mutations that are specifically defective in membrane association. In this review, we summarize recent advances in PTEN-membrane interactions and methods useful in addressing PTEN function.
Keywords: Dictyostelium, PIP3, Membrane, Mutation
1. Introduction
The PIP3 phosphatase, PTEN, converts PIP3 to PIP2. Because PIP3 is a potent signaling lipid that stimulates cell proliferation, survival, and migration, PTEN plays a key negative role in PIP3 signaling (Fig. 1) [1–3]. Defects in PTEN abnormally enhance PIP3 signaling and contribute to many human cancers. PTEN consists of four domains, including the N-terminal lipid-binding domain, which interacts with PIP2 in vitro; the phosphatase domain; the C2 domain, which binds to phosphatidylserine; and the C-terminal tail region (Fig. 2A) [4–9]. It has been suggested that the N-terminal lipid-binding and C2 domains mediate interactions of PTEN with phospholipids in the plasma membrane [6,7,10–14]. However, human PTEN is mainly present in the cytosol, without clear enrichment at the plasma membrane (Fig. 2C) [4,15]. It is poorly understood how PTEN is recruited to the plasma membrane to access its substrate. To address this important question, we have developed an experimental platform using the social amoeba Dictyostelium discoideum [10,11,16]. The domain structure and function of PTEN are conserved in Dictyostelium and human cells (Fig. 2). Interestingly, Dictyostelium PTEN are more stably associated with the plasma membrane compared to human PTEN (Fig. 2C), likely due to the lack of the phosphorylation sites in the inhibitory C-terminal region, as discussed in Section 2.4 – Identification of mutations. Similar to PTEN-defective human cells, PTEN-null Dictyostelium cells dramatically increase PIP3 levels, leading to alterations in cell proliferation, differentiation, and chemotaxis [17–19]. These phenotypes are fully rescued by ectopic expression of human PTEN [10,11,14]. The use of Dictyostelium enabled us to define mechanisms underlying the localization, activity, and function of human PTEN. Here, we will discuss useful methods to study human PTEN in both Dictyostelium and human cells.
Fig. 1.
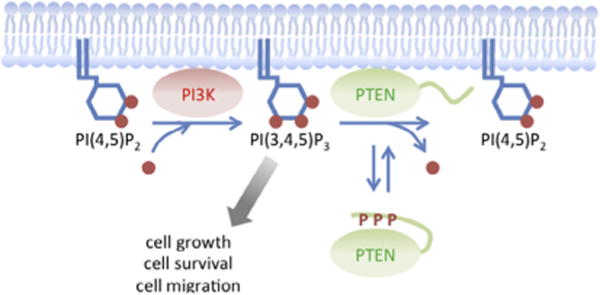
Regulation of PIP3 signaling by PTEN conformational switch. PIP2 is converted to PIP3 by PI3-kinases at the plasma membrane. Dissociating the core region of PTEN from the inhibitory C-terminal tail opens the conformation and promotes the association of PTEN with the plasma membrane. PTEN dephosphorylates PIP3 at the plasma membrane.
Fig. 2.
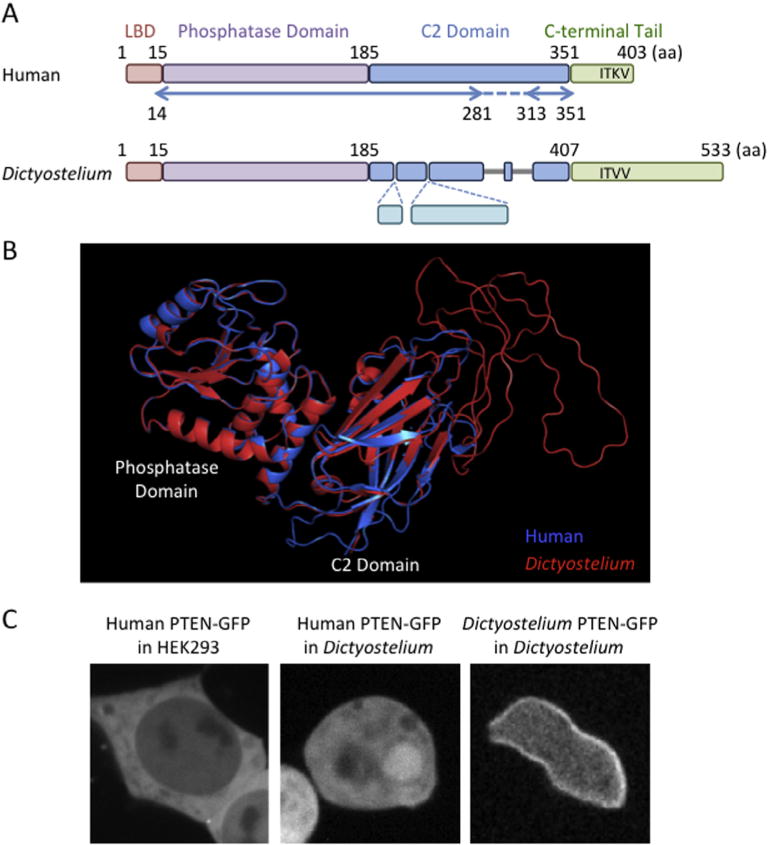
Human and Dictyostelium PTENs. (A) Diagrammatic domain structures of human and Dictyostelium PTENs. The lipid binding domain (LBD) and the phosphatase domain are highly conserved between human and Dictyostelium. Although the C2 domains is also conserved, Dictyostelium PTEN has insertions and deletions. The C-terminus of human PTEN contains the PDZ-binding domain (XTXV) while Dictyostelium PTEN has additional sequences after the domain. Arrows indicate regions of human PTEN that have been visualized in the crystal structure. (B) The 3D structure of Dictyostelium PTEN is predicted by SWISS-MODEL and aligned with human PTEN by PyMol. (C) Localization of human PTEN-GFP in HEK293T cells, human PTEN-GFP in Dictyostelium cells and Dictyostelium PTEN-GFP in Dictyostelium cells is shown.
2. Screening for PTEN mutations that increase membrane association
To define mechanisms for PTEN membrane recruitment, we took an unbiased genetic approach to collect mutations that promote association of human PTEN with the plasma membrane. We expressed a pool of mutant PTEN-green fluorescent protein (GFP) fusion proteins in Dictyostelium cells. As a platform to screen human PTEN localization, the Dictyostelilum system has the following advantages: (1) we can use PTEN-null cells to differentiate potential effects of interactions with endogenous wild-type PTEN on the localization of mutant PTEN; (2) cloning of cells that express individual PTEN mutants is straightforward after visual inspection of PTEN-GFP localization; (3) DNA can be efficiently recovered from small numbers of cells to identify mutations in PTEN; (4) once we identify PTEN mutants, we can test their functions in PTEN-null cells.
2.1. Generation of a plasmid library
To generate a plasmid library containing a large number of human PTEN variants with a wide range of mutations, we randomly mutagenized human PTEN cDNA (UniProtKB P60484) by errorprone PCR using a Diversity PCR random mutagenesis kit (Clontech) [10]. We optimized the PCR conditions to obtain an average of 5.5 mutations per PTEN molecule. The PCR products were cloned into the Dictyostelium expression plasmid pKF3, which creates GFP fusion proteins with mutant PTEN, and electroporated into MegaX DH10B competent cells (Invitrogen). We collected more than 120,000 bacterial colonies. Because PTEN cDNA contains approximately 1200 nucleotides, we estimate the probability that each nucleotide can be mutated ~550 times (120,000 colonies/1200 nucleotides × 5.5 mutations) [10,11]. We confirmed the expected random distribution and diversity of mutations by sequencing ten of the plasmids. Plasmids were extracted from bacteria and pooled to create the PTEN library. We electroporated the library into PTEN-null Dictyostelium cells and selected transformants in HL5 medium (10 g/L glucose, 10 g/L proteose peptone, 5 g/L yeast extract, 5 mM phosphate buffer, pH 6.5) supplemented with 10 μg/mL G418 for 5 days [18,20]. To ensure screening of a large number of mutated-PTEN-GFP plasmids, we collected and pooled more than 20,000 colonies of Dictyostelium cells. These transformants were further cultured, with shaking, in HL5 medium in the presence of 20 μg/mL G418.
2.2. Isolation of PTEN mutants
To visually inspect the localization of PTEN, we plated Dictyostelium cells expressing mutagenized human PTEN-GFP on 96-well optical plates (Thermo Scientific) at a density of 1000 cells/well [10]. After 3 days, we replaced the culture medium with DB (2 mM MgSO4, 0.2 mM CaCl2, 10 mM phosphate buffer, pH 6.5) and examined the localization of PTEN-GFP under a Zeiss HAL100 epifluorescence inverted microscope equipped with a 40 × objective. This step decreased autofluorescence-derived from the HL5 medium. We handpicked cells with increased membrane localization of PTEN-GFP using a P200 pipetman and transferred them to new wells. We repeated this purification process and re-examined the localization of PTEN to increase the homogeneity of cell population. In typical experiments, we repeated this enrichment process three times to obtain more than 50% of cells with increased membrane localization. To further isolate single clones, we plated cells at low density (50–100 cells per 10 cm plate) on SM agar (10 g/L glucose, 10 g/L proteose peptone, 1 g/L yeast extract, 1 g/L MgSO4 7H2O, 20 mM phosphate buffer, pH 6.5, 15 g/L agar) with the bacterium Klebsiella aerogenes. This step allowed each Dictyostelium cell to form a single plaque on the bacterial plate. After 5 days, we transferred cells from individual plaques to the optical plate and confirmed the enhancement of membrane associated PTEN-GFP. From ~20,000 visually inspected Dictyostelium colonies, we isolated 18 clones that have increased robust membrane association of PTEN-GFP [10,11].
2.3. Quantification of PTEN subcellular localization
To quantitatively assess the localization of PTEN, we placed Dictyostelium cells expressing PTEN-GFP on eight-well chambered coverglasses (Lab-TekII, Nunc) and captured fluorescence images under a Leica DMI 6000 inverted microscope equipped with a 63 × objective and a CoolSNAP EZ camera. Fluorescence intensity of PTEN-GFP at the plasma membrane and nucleus was determined relative to that in the cytosol by measuring average fluorescence intensity in 1 pixel area from three different positions in each compartment. Background fluorescence intensity was subtracted from each measurement [10,11]. We used DAPI staining to identify the nucleus. Wild-type human PTEN showed values of approximately 1, whereas mutant PTEN, with increased membrane association, showed 2–5-fold increases. To determine whether the localization of PTEN-GFP observed in Dictyostelium is conserved in human cells, we subcloned PTEN-GFP into the mammalian expression vector pcDNA and transiently transfected human embryonic kidney 293 (HEK293T) cells. HEK293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Invitrogen). Cells were incubated for 24 h before observation. We similarly quantified the distribution of PTEN-GFP in HEK293T cells. Essentially, all mutant PTEN-GFP tested showed similar localization in both Dictyostelium and HEK293T cells, suggesting that mechanisms that control the subcellular distribution of PTEN are highly conserved and that Dictyostelium cells provide a powerful system to study the localization of human PTEN [10,11].
2.4. Identification of mutations
To identify mutations that increased membrane localization, we isolated DNA from Dictyostelium cells grown to confluence in 6-well plates (~1 × 106 cells) [10,11]. We PCR-amplified the human PTEN gene and sequenced the PCR products. Because virtually all mutant PTEN molecules contained multiple mutations, we separated these mutations using subcloning and site-directed mutagenesis. We re-expressed the resulting PTEN-GFP plasmids having fewer or single mutations in PTEN-null Dictyostelium cells. By examining the localization of the mutants, we determined which mutations cause increased membrane association (Fig. 3) [10–12]. The mutations that recruit PTEN to the plasma membrane can be divided into four groups. The first group is located in the catalytic domain, with substitution of Cys124 by either arginine or serine. These mutations have been reported to strengthen interactions of PTEN with PIP3 due to creation of enzyme–substrate intermediates [7]. However, we found that PTENC124R is still bound to the plasma membrane, even after depletion of PIP3, suggesting that this increased membrane interaction is independent of PTEN-PIP3 associations [10]. The second group of mutations is located in the C-terminal phosphorylation sites. Phosphorylation of this cluster increases interaction of the C-terminal and core regions of PTEN, masking the membrane-binding site in the core region. Mutations, blocking phosphorylation, release this inhibitory mechanism and open the conformation of PTEN, allowing PTEN to interact with the plasma membrane. The third group is located in the core region (R41G, E73D, N262Y, and N329H) and inhibits binding to the C-terminal tail [10]. The last mutation, Q17R, is located in the PIP2-binding region and introduces a positive charge [10,11]. Although the exact molecular basis is currently unknown, addition of positive residues may stabilize association with negatively charged head groups of PIP2 through electrostatic interactions.
Fig. 3.
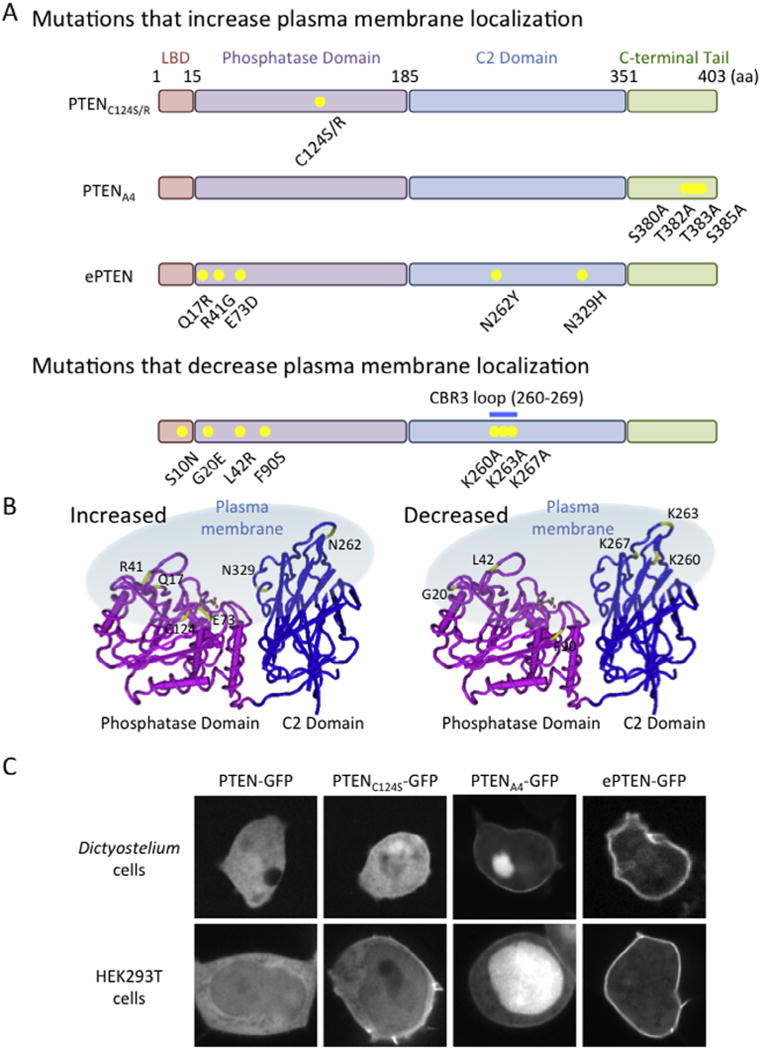
Mutations that change membrane association of PTEN. (A) Mutations that increase and decrease association of human PTEN with the plasma membrane are shown. (B) The positions of the mutations are indicated in the 3D structure of PTEN. (C) Images of Dictyostelium and HEK293T cells expressing PTEN-GFP, PTENC124S-GFP, PTENA4-GFP and ePTEN-GFP are shown.
3. Enhanced PTEN (ePTEN) with increased membrane association
3.1. Generation of ePTEN
Based on the information obtained from the screen described above, we designed PTEN molecules that maximize its membrane association (Fig. 3) [11]. We reasoned that the combination of opening the conformation by dissociating the core and tail regions and introducing the Q17R mutation may synergistically increase membrane association. To open the conformation, we used four mutations (R41G, E73D, N262Y and N329H) located on the surface of the core region. Mutant PTEN carrying those mutations and Q17R showed the strongest association with the plasma membrane observed so far, with an 8-fold increase compared with wild-type PTEN [11]. We designated this PTEN variant ePTEN [11]. The addition of the A4 mutation (C-terminal phosphorylation sites, S380, T382, T383, S385, are mutated to alanines) [6–8,10,11,21,22] to ePTEN did not further increase its membrane association; therefore the conformation may be fully open.
3.2. Function of ePTEN in PIP3 signaling
Demonstrating that increasing the membrane association of PTEN enhances its function, we found that ePTEN has increased activity in suppressing PIP3 signaling [11]. To observe PIP3 levels in cells using live-cell imaging, we expressed ePTEN-GFP together with a PIP3 biosensor, PHAKT-RFP, in HEK293T cells. In the absence of ectopic expression of PTEN, PHAKT-RFP accumulated at the plasma membrane in HEK293T cells, indicating high levels of PIP3. Upon expression, PTEN-GFP significantly lowered PHAKT-RFP signals at the plasma membrane. Remarkably, ePTEN-GFP decreased PHAKT-RFP to undetectable levels at the plasma membrane. Consistent with these results, immunoblotting analysis of phosphorylation of AKT at S473, which is stimulated by PIP3 [23,24], showed that ePTEN-GFP suppressed AKT phosphorylation more effectively than PTEN-GFP. To further test the function of ePTEN as an enhanced tumor suppressor, we expressed ePTEN in a breast cancer cell model, the MCF-10A PIK3CA knockin cell line, which expresses a constitutively active form of PI3K [25,26], and tested cell migration and proliferation. To examine cell migration, we measured serum-stimulated migration behavior, which depends on PIP3 signaling, in a Transwell migration assay [11]. We infected MCF-10A PIK3CA knockin cells with lentiviruses carrying PTEN-GFP or ePTEN-GFP. Five days after infection, cells were harvested, washed twice with PBS and resuspended in serum-free culture medium. 50,000 cells in 100 μl serum-free medium were plated in Transwell inserts of 24-well Transwell plates (Corning). 650 μl of serum-free or serum-containing medium was added to receiver wells. Transwell plates were incubated at 37 °C with 5% CO2. After overnight incubation, we removed the medium in the inserts and gently wiped the inside of each insert using cotton swabs to remove cells that did not transmigrate. We examined the insert under a epifluorescence microscope and counted cells expressing PTEN-GFP that transmigrated. The percentage of cells that migrated was quantified by dividing the total number of migrated cells expressing GFP to the total number of cells expressing GFP plated per insert. Compared with negative controls, ePTEN-GFP significantly suppressed cell migration.
To examine cell proliferation, MCF-10A PIK3CA knockin cells were cultured in DMEM/F12 medium supplemented with 5% horse serum, 10 μg/mL insulin, 0.5 μg/mL hydrocortisone, and 0.1 μg/mL cholera toxin [25,26]. Five days after infection with PTEN-GFP or ePTEN-GFP, cells were plated at the density of 100 cells/well on 96-well plates and cultured for 7–9 days. The number of cells was quantified in individual colonies that expressed PTEN-GFP under a epifluorescence microscope. ePTEN showed stronger inhibition for cell proliferation compared with wild-type PTEN.
4. Assays of human PTEN function
4.1. In vitro phosphatase activity assay
Because PIP3 phosphatase activity is essential for PTEN function, it is important to test whether isolated mutations affect the enzymatic activity of PTEN. Since it is easy to scale up to obtain large amounts of proteins from Dictyostelium cells, we purified PTEN and measured the activity using Dictyostelium cells expressing human PTEN-GFP [10,11,17]. Both Dictyostelium PTEN-GFP and human PTEN-GFP are fully functional. It is important to use the eukaryotic expression system to assess. PTEN’s enzymatic activity since the phosphorylation of the C-terminal tail that regulates the association of PTEN core and C-tail is lacking in prokaryotic systems. We expressed wild-type and mutant forms of PTEN-GFP in Dictyostelium cells (10–50 mL of ~5 × 106 cells/mL in HL5 medium) and lysed cells in 1% Nonidet P-40, 50 mM NaCl, 20 mM Tris–HCl (pH 7.5), 10% glycerol, 0.1 mM EDTA, phosphatase inhibitor cocktail (Sigma) and protease inhibitor cocktail (Roche) [11]. PTEN-GFP was immunopurified using GFP-Trap agarose beads (7 μl) (ChromoTek). In typical experiments, we purified 0.5–2 μg of PTEN-GFP. We incubated PTEN-GFP bound to the beads (0.05–0.1 μg of protein) with a soluble form of PIP3 (3000 pmol in 25 μl), PIP3 diC8 (Echelon), for 30 min at 37 °C. Release of phosphates from PIP3 diC8 was determined using a malachite green phosphatase assay (100 μl malachite green solution to 25 μl sample at room temperature for 20 min) (Echelon), and absorbance at 620 nm was read by a plate reader. We determined protein amounts by quantifying band intensity after SDS–PAGE and Coomassie Brilliant Blue staining. The phosphatase activity was normalized to the level of purified PTEN-GFP. We found that mutant PTENs with increased membrane association, such as PTENA4 and ePTEN, showed higher phosphatase activities [11]. The increased activity is not due to increased accessibility to the plasma membrane, because the assay was performed using a soluble substrate in vitro. Instead, dissociation of the core region from the C-terminal tail exposes the catalytic domain, located in the core domain, to the surface of PTEN, increasing substrate accessibility to the catalytic center.
4.2. Use of PTENA4 mutants to analyze membrane association
The function of PTEN is lost when it fails to associate with the plasma membrane. We, therefore, hypothesized that mutations blocking membrane association are likely tumorigenic. To test our hypothesis, we asked whether there are cancer-associated PTEN mutations that are defective in membrane association [16]. This simple question is difficult to address, because the majority of wild-type human PTEN is present in the cytosol [10,11,16], and this precludes PTEN membrane dissociation of PTEN. To overcome this problem, we used PTENA4. PTENA4 carries mutations in all four phosphorylation sites in the C-terminus and showed increased association with the plasma membrane. Therefore, dissociation from the plasma membrane can be easily observed by fluorescence microscopy. Many cancer-associated mutations are distributed throughout the PTEN molecule, and ~40% are missense mutations [4,27–31]. Most of these missense mutations inhibit PTEN’s lipid phosphatase activity. However, there is a group of PTEN mutants that maintain its phosphatase activity [9,32,33]. We reasoned that these catalytically active mutants are good candidates for the loss of membrane association. To test this idea, we chose four mutations, S10N, G20E, L42R, and F90S, and individually introduced them into PTENA4-GFP [16]. We focused on these four mutations since previous reports have shown that these mutations do not block the PIP3 phosphatase activity and since they are located near the regulatory membrane binding interface we have identified [16]. We first confirmed that these mutations do not block PIP3 phosphatase activity. We then observed their distribution in HEK293T cells. Whereas the membrane localization of PTENA4-GFP can be clearly observed, the additional mutations completely blocked this localization. To test whether tethering these mutants to the plasma membrane reactivates their function, we introduced the myristoylation motif derived from the plasma membrane protein, PKBR1. Addition of membrane tethering rescued the function of the S10N, G20E, L42R, and F90S mutants, demonstrating that detachment from the plasma membrane is the primary defect in these mutants.
4.3. Assays of interactions between PTEN and its C-terminal tail
To analyze interactions between the core region and C-terminal tail of PTEN (PTEN352–403-YFP-FLAG), we set up an in-trans coprecipitation assay (Fig. 4) [7,10,11,16]. We grew HEK293T cells in DMEM supplemented with 10% FBS in a 10 cm dish and transiently transfected the cells with the plasmid carrying PTEN352–403-YFP-FLAG or wild-type and mutant PTEN-GFP. After 24 h, we lysed the cells in 1 mL of lysis buffer containing 1% Nonidet P-40, 50 mM NaCl, 20 mM Tris–HCl (pH 7.5), 10% glycerol, 0.1 mM EDTA, phosphatase inhibitor cocktail (Sigma), and protease inhibitor cocktail (Roche). The lysates were cleared by centrifugation at 13,000 rpm in a tabletop microfuge for 20 min at 4 °C. We also successfully used Dictyostelium to express wild-type and mutant PTEN-GFP. When Dictyostelium cells (1 × 108 cells) were used, we prepared lysates in a solution of 0.5% Nonidet P-40, 150 mM NaCl, 20 mM Tris–HCl (pH 7.5), 1 mM EDTA, phosphatase inhibitor cocktail (Sigma), and protease inhibitor cocktail (Roche). The lysates were then cleared by centrifugation at 13,000 rpm in a tabletop microfuge for 20 min at 4 °C. We mixed the lysates containing PTEN352–403-YFP-FLAG (100 μl) and the lysates containing full length PTEN-GFP (500 μl). Next, we added anti-FLAG M2 beads (15 μl) (Sigma) to the mixtures and incubated for 2 h. The beads were washed twice in the lysis buffer, and the bound fractions were analyzed by SDS–PAGE and immunoblotting using antibodies specific to FLAG and GFP. Wild-type PTEN-GFP does not coprecipitate with PTEN352–403-YFP-FLAG because the core region is tightly bound to its own C-terminal tail [7,10,11,16]. In contrast, PTENA4 -GFP, in which C-terminal phosphorylation sites are mutated, preventing binding of the tail to the core region, strongly binds to PTEN352–403-YFP-FLAG. Thus, this assay allows us to define whether and how mutations affect intramolecular interactions between the core region and the tail of PTEN.
Fig. 4.
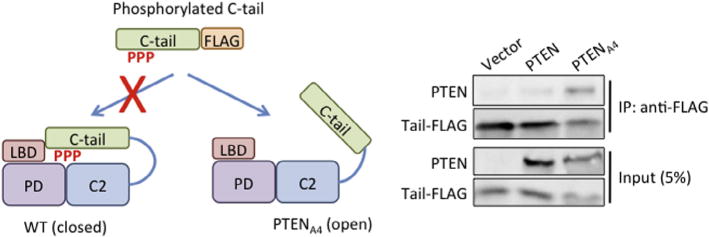
Assay for intramolecular interactions. Cell lysates expressing PTEN-GFP or PTENA4-GFP are incubated with cell lysates expressing PTEN352–403-YFP-FLAG. Introducing the A4 mutation into full length PTEN allowed interactions of the core region of PTENA4 and the FLAG-tagged C-terminal tail. PTEN352–403-YFP-FLAG was immunoprecipitated with beads coupled to anti-FLAG antibodies. Bound fractions (IP) are analyzed with antibodies to GFP and FLAG.
4.4. Evaluation of human PTEN function in Dictyostelium cells
In PTEN-null Dictyostelium cells, PIP3 signaling is overactivated, leading to defects in cell proliferation, differentiation upon starvation, and chemotaxis [18]. These phenotypes can be used to test the function of human PTEN, because ectopic expression of human PTEN can rescue these phenotypes of PTEN-null Dictyostelium cells [10]. For example, we tested whether human PTEN and its mutant forms can rescue the differentiation phenotypes. We transfected PTEN-null Dictyostelium cells with Dictyostelium PTEN-GFP, human PTEN-GFP, and mutant versions of human PTEN-GFP, and starved the cells. Upon starvation, Dictyostelium cells move toward cAMP, aggregate, and differentiate into multicellular structures called fruiting bodies, which consist of spore and stalk cells [34]. PTEN is necessary for chemotactic migration toward cAMP, and PTEN-null cells are defective in aggregation and differentiation [18]. Cells in exponential growth were harvested, washed twice in DB (2 mM MgSO4, 0.2 mM CaCl2, 10 mM phosphate buffer, pH 6.5), and plated at 5 × 105 cells/cm2 on 1% non-nutrient DB agar for 36 h [35,36]. We observed cells under an Olympus SZ-PT dissecting microscope equipped with 6.3 × objective. Wild-type Dictyostelium and human PTEN rescued the differentiation phenotypes in PTEN-null Dictyostelium cells. However, mutant PTEN defective in association with the plasma membrane failed to rescue the phenotypes. Similar to differentiation, cell proliferation phenotypes of PTEN-null Dictyostelium cells were rescued by wild-type Dictyostelium and human PTEN-GFP, but not by mutants defective in membrane association. Therefore, the recruitment of PTEN to the plasma membrane is essential for its function in controlling PIP3 levels in cells.
5. Conclusion and future directions
PTEN-membrane interactions involve assembly of the regulatory membrane-binding interface on the surface of PTEN molecules. This interface is subjected to negative regulation by the inhibitory C-terminal tail. Opening the conformation of PTEN, by releasing this inhibitory mechanism, promotes membrane translocation. The use of D. discoideum made it possible to reveal these key mechanisms and to design ePTEN with increased membrane association and PIP3 phosphatase activity. These new discoveries raise important new questions. For example, although lipid elements, such as PIP2 and PS, are important for PTEN-membrane interactions, we do not know whether and how protein components function in recruitment of PTEN to the plasma membrane. Identifying PTEN receptors will be one of the big challenges to understanding PTEN function. Also, it is not clear how PTEN phosphorylation is controlled under different physiological conditions. Understanding the dynamics of PTEN phosphorylation/dephosphorylation is a crucial next step in understanding PTEN regulation. Finally, it will be very important to understand the entire spectrum of cancer-associated mutations that affect interactions of PTEN with the plasma membrane. Answering these questions will facilitate development of cancer interventions that promote the activation of PTEN.
Acknowledgments
This work was supported by NIH grants to MI (GM084015), PND (GM28007 and GM34933), and HS (GM089853 and NS084154).
References
- 1.Vanhaesebroeck B, Stephens L, Hawkins P. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 2.Sulis ML, Parsons R. Trends Cell Biol. 2003;13:478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 3.Carracedo A, Alimonti A, Pandolfi PP. Cancer Res. 2011;71:629–633. doi: 10.1158/0008-5472.CAN-10-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song MS, Salmena L, Pandolfi PP. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 5.Tamguney T, Stokoe D. J Cell Sci. 2007;120:4071–4079. doi: 10.1242/jcs.015230. [DOI] [PubMed] [Google Scholar]
- 6.Gericke A, Leslie NR, Losche M, Ross AH. Adv Exp Med Biol. 2013;991:85–104. doi: 10.1007/978-94-007-6331-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN. Proc Natl Acad Sci USA. 2009;106:480–485. doi: 10.1073/pnas.0811212106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker SM, Leslie NR, Perera NM, Batty IH, Downes CP. Biochem J. 2004;379:301–307. doi: 10.1042/BJ20031839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denning G, Jean-Joseph B, Prince C, Durden DL. PK Vogt, Oncogene. 2007;26:3930–3940. doi: 10.1038/sj.onc.1210175. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen HN, Afkari Y, Senoo H, Sesaki H, Devreotes PN, Iijima M. Oncogene. 2013 doi: 10.1038/onc.2013.507. Epub ahead. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen HN, Yang JM, Afkari Y, Park BH, Sesaki H, Devreotes PN, Iijima M. Proc Natl Acad Sci USA. 2014;111:E2684–E2693. doi: 10.1073/pnas.1409433111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das S, Dixon JE, Cho W. Proc Natl Acad Sci USA. 2003;100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keniry M, Parsons R. Oncogene. 2008;27:5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 14.Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN. Proc Natl Acad Sci USA. 2006;103:3633–3638. doi: 10.1073/pnas.0510570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker SJ. Cell. 2007;128:25–28. doi: 10.1016/j.cell.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen HN, Yang JM, Rahdar M, Keniry M, Parsons R, Park BH, Sesaki H, Devreotes PN, Iijima M. Oncogene. 2014 doi: 10.1038/onc.2014.293. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iijima M, Huang YE, Luo HR, Vazquez F, Devreotes PN. J Biol Chem. 2004;279:16606–16613. doi: 10.1074/jbc.M312098200. [DOI] [PubMed] [Google Scholar]
- 18.Iijima M, Devreotes P. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- 19.Iijima M, Huang YE, Devreotes P. Dev Cell. 2002;3:469–478. doi: 10.1016/s1534-5807(02)00292-7. [DOI] [PubMed] [Google Scholar]
- 20.Chen CL, Wang Y, Sesaki H, Iijima M. Sci Signal. 2012;5:ra10. doi: 10.1126/scisignal.2002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Odriozola L, Singh G, Hoang T, Chan AM. J Biol Chem. 2007;282:23306–23315. doi: 10.1074/jbc.M611240200. [DOI] [PubMed] [Google Scholar]
- 22.Campbell RB, Liu F, Ross AH. J Biol Chem. 2003;278:33617–33620. doi: 10.1074/jbc.C300296200. [DOI] [PubMed] [Google Scholar]
- 23.Hawkins PT, Anderson KE, Davidson K, Stephens LR. Biochem Soc Trans. 2006;34:647–662. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- 24.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 25.Gustin JP, Karakas B, Weiss MB, Abukhdeir AM, Lauring J, Garay JP, Cosgrove D, Tamaki A, Konishi H, Konishi Y, Mohseni M, Wang G, Rosen DM, Denmeade SR, Higgins MJ, Vitolo MI, Bachman KE, Park BH. Proc Natl Acad Sci USA. 2009;106:2835–2840. doi: 10.1073/pnas.0813351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang GM, Wong HY, Konishi H, Blair BG, Abukhdeir AM, Gustin JP, Rosen DM, Denmeade SR, Rasheed Z, Matsui W, Garay JP, Mohseni M, Higgins MJ, Cidado J, Jelovac D, Croessmann S, Cochran RL, Karnan S, Konishi Y, Ota A, Hosokawa Y, Argani P, Lauring J, Park BH. Cancer Res. 2013;73:3248–3261. doi: 10.1158/0008-5472.CAN-12-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chalhoub N, Baker SJ. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ali IU, Schriml LM, Dean M. J Natl Cancer Inst. 1999;91:1922–1932. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 29.Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Clin Cancer Res. 2012;18:400–407. doi: 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollander MC, Blumenthal GM, Dennis PA. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Oncogene. 2008;27:5464–5476. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- 32.Han SY, Kato H, Kato S, Suzuki T, Shibata H, Ishii S, Shiiba K, Matsuno S, Kanamaru R, Ishioka C. Cancer Res. 2000;60:3147–3151. [PubMed] [Google Scholar]
- 33.Rodriguez-Escudero I, Oliver MD, Andres-Pons A, Molina M, Cid VJ, Pulido R. Hum Mol Genet. 2011;20:4132–4142. doi: 10.1093/hmg/ddr337. [DOI] [PubMed] [Google Scholar]
- 34.Fey P, Kowal AS, Gaudet P, Pilcher KE, Chisholm RL. Nat Protoc. 2007;2:1307–1316. doi: 10.1038/nprot.2007.178. [DOI] [PubMed] [Google Scholar]
- 35.Cai H, Huang CH, Devreotes PN, Iijima M. Methods Mol Biol. 2012;757:451–468. doi: 10.1007/978-1-61779-166-6_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Steimle PA, Ren Y, Ross CA, Robinson DN, Egelhoff TT, Sesaki H, Iijima M. Mol Biol Cell. 2011;22:2270–2281. doi: 10.1091/mbc.E10-11-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]