

Abstract
PTEN, a tumor suppressor protein that dephosphorylates phosphoinositides at the 3-position of the inositol ring, is a cytosolic protein that needs to associate with the plasma membrane or other subcellular membranes to exert its lipid phosphatase function. Upon membrane association PTEN interacts with at least three different lipid entities: An anionic lipid that is present in sufficiently high concentration to create a negative potential that allows PTEN to interact electrostatically with the membrane, phosphatidylinositol-4,5-bisphosphate, which interacts with PTEN's N-terminal end and the substrate, usually phosphatidylinositol-3,4,5-trisphosphate. Many parameters influence PTEN's interaction with the lipid bilayer, for example, the lateral organization of the lipids or the presence of other chemical species like cholesterol or other lipids. To investigate systematically the different steps of PTEN's complex binding mechanism and to explore its dynamic behavior in the membrane bound state, in vitro methods need to be employed that allow for a systematic variation of the experimental conditions. In this review we survey a variety of methods that can be used to assess PTEN lipid binding affinity, the dynamics of its membrane association as well as its dynamic behavior in the membrane bound state.
Keywords: phosphatase, PTEN, peripheral membrane protein, conformational change, spectroscopy, single-particle tracking
1 Introduction
The subcellular distribution of PTEN creates a fundamental problem to understanding its regulation and action. The most important substrate for PTEN is diacylphosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3), which is localized in the inner leaflet of the plasma membrane [1]. In contrast, PTEN is primarily localized in the cytoplasm where it freely diffuses [2]. Hence, in order to hydrolyze PI(3,4,5)P3, PTEN must bind to the membrane and be activated. The first step is dephosphorylation of the PTEN tail (particularly sites S385, S380, T383 and T382). Sellers and coworkers proposed this influential model [3, 4], positing that the phosphorylated tail folds back onto the PTEN core. This closed conformation blocks binding of PTEN to the membrane [5]. Dephosphorylation of the PTEN tail allows the second step, binding of PTEN to phospholipid membranes. Bound PTEN shows a shallow penetration of the membrane [6]; binding is dominated by interactions with the headgroups of negatively charged lipids [7, 8]. An N-terminus domain preferentially binds diacylphosphatidylinositol-4,5-bisphosphate PI(4,5)P2 [9, 10]. The C2 domain binds negatively charged lipids, including phosphatidylserine (PS) [8]. The third step is a PTEN conformational change induced by PI(4,5)P2. This conformation change activates the phosphatase domain [11] and was verified by infrared spectroscopy [9]. The fourth step is to diffuse laterally until it binds PI(3,4,5)P3. The fifth step is hydrolysis of the phosphate in the 3-position of PI(3,4,5)P3. Careful analysis of membrane-bound PTEN reveals that the lateral diffusion allows hydrolysis of multiple PI(3,4,5)P3 molecules prior to the sixth step, release of PTEN from the membrane [12].
The goal of this review is to discuss biophysical methods for analyzing the kinetic and thermodynamic parameters for each step of PTEN activation. It should be emphasized that PTEN function is highly quantitative. Tumor formation can be enhanced by PTEN haploinsufficiency [13]. Hence, a quantitative understanding of PTEN regulation and membrane binding is essential to understand wild type and mutated PTEN proteins [14].
2. Biomembrane mimics for the characterization of membrane-associated PTEN
PTEN membrane association involves binding to multiple lipid entities. The C2 domain and probably other positively charged parts of the protein interact electrostatically with anionic lipids that generate a negatively charged environment at the water/membrane interface. Typically this role has been assigned to phosphatidylserine (PS) [9, 15]. PTEN's N-terminal end interacts selectively with PI(4,5)P2, which leads to enhanced binding and allosteric activation of the phosphatase [9]. Finally, the substrate PI(3,4,5)P3 binds to the phosphatase domain active site. While PI(3,4,5)P3 is in all likelihood the most important substrate in vivo, PTEN shows also in vitro activity towards the other 3-phosphorylated lipids. Interactions of other anionic lipids with the active site may contribute to PTEN membrane binding in the absence of 3-phosphorylated phosphoinositides. Considering PTEN's multifaceted lipid-binding properties, special consideration has to be given to the choice of the appropriate membrane model system and its composition. For a recent review on model membrane systems for the characterization of peripheral membrane proteins, please see Czogalla et al. [16].
2.1 Choosing the lipid composition
Phosphoinositides (PIPs) occur in nature almost exclusively with a stearoyl-arachidonoyl (SAPIPs) acyl chain composition. Since the acyl chain composition has a major impact on lipid phase behavior and hence lipid mobility as well as headgroup spacing, studies aimed at investigating phosphoinositide/PTEN interaction should utilize lipids with these acyl chains whenever possible. Phosphoinositides are also commercially available with di-oleoyl (DOPIPs) or di-palmitoyl acyl (di-C16 PIPs) chains. DOPIPs will form fluid phases, however, the phase behavior in the presence of other membrane components like cholesterol deviates slightly from what is observed for SAPIPs [17]. DPPIPs form ordered (gel) phases at room temperature [18], resulting in a reduced mobility and lateral spacing of di-C16PIPs in comparison to SAPIPs and even though PTEN penetrates the bilayer only to a limited extent [19, 20], bilayer model systems involving saturated chain phosphoinositides are less suitable to study PTEN membrane association. Short-chain PIPs (di-butanoyl or di-octanoyl PIPs are commercially available from a variety of vendors, see Table 1) are widely used to determine the activity of PTEN against soluble substrates. In some instances this test is used as a “quality control” measure to ensure that an expressed protein is active, while in some other cases the comparison of PTEN activity against soluble or vesicle resident PI(3,4,5)P3 substrate highlights differences in interfacial vs. lipid bilayer independent activation. The critical micelle concentrations (CMCs) for short chain PIPs is around 0.5 – 1 mM [21] and typically experiments involving short chain PIPs are carried out at a concentration well below the CMC. However, others [21] have found some evidence that PTEN may promote PIP clustering through its various anionic lipid interaction sites, resulting in a significant lowering of the CMC like it was observed for other protein/amphiphile complexes. When interpreting data involving data obtained with soluble PIP lipids, this potential caveat should be kept in mind.
Table 1. Acyl chain composition of commercially available PI(4,5)P2 and PI(3,4,5)P3 lipids.
| Lipid | di-C4 | di-C8 | di-C16 | di-C18:1 | C18:0/C 20:4 | natural |
|---|---|---|---|---|---|---|
| PI(4,5)P2 | various* | various* | various* | Avanti | Avanti | Avanti (brain) |
| PI(3,4,5)P3 | various* | various* | various* | Avanti | Avanti | - |
several vendors offer phosphoinositides with saturated acyl chains, including Ag Scientific, Alexis, Avanti Polar Lipids, Echelon, Matreya, Mobitech, Sichem, Sigma Aldrich
Phosphoinositides alone do not form stable vesicles and therefore, need to be mixed with other membrane constituents. To study the interaction of PTEN or PTEN derivatives with phosphoinositides (e.g., PI(4,5)P2 or PI(3,4,5)P3), phosphatidylcholine (PC) can be used as a “matrix” lipid to form vesicles since PTEN has a very small binding affinity for PC [9]. Alternatively, we have found that cholesterol stabilizes phosphoinositide vesicles if at least 10 mol% cholesterol is present [17]. While PTEN's interaction with PI(4,5)P2 presumably involves hydrogen bonds between N-terminal amino acids and the inositol ring phosphomonoester groups, other parts of the protein have been found to interact non-specifically, electrostatically with anionic lipids [9]. To investigate synergistic lipid binding of PTEN, PI(4,5)P2 can be mixed with PS or phosphatidylinositol, where the latter two will bind electrostatically, non-specifically to PTEN. The presence of cholesterol in PI(4,5)P2 containing vesicles enhances PTEN binding, presumably due to cholesterol-induced PI(4,5)P2 clustering [17].
The choice of the lipid composition is generally driven by the experimental goals. For example, experiments aimed at comparing the binding properties of wt and mutant PTEN may employ mixtures like brain PI(4,5)P2/POPS/POPC (1:20:79). PI(3,4,5)P3 hydrolysis can be measured by using binary PC/PI(3,4,5)P3 with different lipid ratios (see below). To mimic plasma membrane inner leaflet lipid compositions, one might use lipid mixtures such as PE (33%)/PC (10%)/PS (21%)/PI (4.5%)/SM (4.5%)/cholesterol (25%)/PIPs (2%) [22], which is a frequently used model system. PI(4,5)P2 concentration in the plasma membrane is about 1 mol%, while the other PIPs are found at lower concentrations [23].
2.2 Fluorescently and isotopically labeled phosphoinositides
Several types of experiments require the use of fluorescently labeled phospho-inositides, which are available with different excitation/emission wavelengths from either Echelon or Avanti Polar Lipids (see Table 2). All of these phosphoinositide derivatives are labeled at the acyl chain, i.e., the PIP headgroup is non-derivatized. It should be noted that the fluorophore group for all of these PIPs is quite bulky, which might affect their organization in lipid bilayers. In general, chain labeled phospholipids will prefer fluid, disordered phases over liquid-ordered or gel phases. Furthermore, weak mutual phosphoinositide interaction, e.g., via hydrogen bond formation, cannot be effectively monitored with these labeled PIPs [17, 24]. However, fluorescently labeled PIPs can be used very effectively to monitor protein induced PIP clustering. Photobleaching is a general problem in fluorescence experiments and we found TopFluor PIPs to be slightly more photostable than the regular BODIPY fluorophores. As noted above, PTEN interacts with anionic lipids like phosphatidylserine, which can be obtained fluorescently labeled from Avanti Polar Lipids.
Table 2. Commercially available fluorescently labeled phosphoinositides.
| Fluorophore | Type | Excitation/Emission | Vendor |
|---|---|---|---|
| BODIPY FL | PI & all PIPs | 505/513 nm | Echelon |
| BODIPY TMR | PI & all PIPs | 542/574 nm | Echelon |
| TopFluor | PI(4)P, PI(3,5)P2 PI(4,5)P2 | 495/503 nm | Avanti Polar Lipids |
Radioactively labeled 3-[33P] PI(3,4,5)P3, which is often used to determine the interfacial kinetics of PTEN action, can be obtained by using PI(4,5)P2 as the substrate for PI 3-kinase. The detailed procedure for this reaction is given elsewhere in this issue [25].
2.3 Preparation of multilamellar mixed lipid vesicles
Most procedures aimed at fabricating model membrane systems start with the formation of multilamellar vesicles (for a recent review on vesicle preparation procedures see [26]). There are a variety of ways to make multilamellar vesicles and we describe here the method employed in our lab, which is suitable for phosphoinositide-containing lipid mixtures [9].
Lipids dissolved in appropriate organic solvents are combined in a sample vial. Phosphoinositides need to be dissolved in a quite polar solvent mixture (e.g., CHCl3/CH3OH/H2O 20:9:1) and less polar lipids (e.g., phosphatidylcholine) need to be dissolved in solvents that are miscible with this mixture. To minimize fractional precipitation of lipids, which may translate into lateral demixing in the vesicles, the organic solvent is rapidly evaporated at an elevated temperature with a stream of nitrogen gas. To remove any trace of remaining solvent, the samples are kept for a couple of hours in a vacuum oven at an elevated temperature (∼ 80°C). Multilamellar vesicles are obtained by re-suspending the dried lipid mixture in a buffer solution, which is followed by “vortexing” the lipid mixture at a temperature above the highest gel/liquid-crystalline phase transition temperature of the lipid components in the mixture.
2.4 Preparation of unilamellar vesicles
Large unilamellar vesicles (LUVs; ≥ 100 nm) are obtained by extrusion of the multilamellar vesicles at a temperature above the gel/liquid-crystalline phase transition temperature through a polycarbonate filter with a pore size equal to the desired diameter of the unilamellar vesicles. Extrusion kits are available from a variety of commercial vendors (e.g., Avanti Polar Lipids). To obtain a narrow size distribution of the unilamellar vesicles, the vesicle suspension is moved multiple times through the filter. Preferably, the vesicle size distribution should be checked using a dynamic light scattering instrument. Since the vesicles elongate as they are moved through the extrusion filter, vesicle are slightly larger than the pore size of the filter (e.g., for a 100 nm pore size filter, vesicles with a 110 – 125 nm diameter are typically obtained). A quality extrusion should yield a narrow size distribution.
Small unilamellar vesicles (SUVs) can be obtained either also by extrusion using a 30 – 50 nm pore size filter or high power sonication (e.g., tip sonicator or high power bath sonicator). For LUVs one can assume that 50% of the total lipid amount make up the inner and outer leaflet of the vesicles, respectively, this is not the case for SUVs, where about 60% of the total lipid is found in the outer leaflet.
A potential pitfall for all methods for vesicle preparations is the recent observation that within a single sample preparation the lipid ratios may vary between single vesicles [27].
2.5 Preparation of giant unilamellar vesicles
Giant unilamellar vesicles (GUVs) are typically prepared using the electroformation or gentle hydration method. Phosphoinositide phase behavior is highly dependent on the ionization state of the phosphomonoester groups, which in turn depends on the ionic strength of the buffer solution [17]. Therefore, it is important to conduct experiments at physiological salt concentrations, which might preclude the conventional electroformation method [28], which requires either buffer solutions with low salt concentrations or >10% of anionic lipid. Recently developed electroformation methods that can be utilized at physiological salt concentrations are discussed in detail by Patil and Jadhav [26].
The “gentle hydration method” [28] is often the method of choice for the preparation of phosphoinositide containing GUVs [17]. Lipid stock solutions are prepared as described above to yield a concentration of about 10 mM. 0.2 μmol of stock lipid solution is mixed with 0.1 mol% of a labeled lipid (e.g., TopFluor PI(4,5)P2 from Avanti Polar Lipids) and diluted with 300 μL organic solvent. Subsequently, the sample is placed in a jointed test tube (∼1.5 cm diameter) and the solvent is evaporated at an elevated temperature (the exact temperature is solvent dependent) with a slow moving rotary evaporator. The thin film that is formed at the lower portion of the tube is dried for about 4 hrs in a vacuum oven to remove any traces of solvent. The dried lipid film is prehydrated for about 45 minutes at 50°C with water-saturated nitrogen gas. Subsequently, 2 mL of nitrogen-purged buffer solution are gently added to the test tube. The test tube is sealed under nitrogen, wrapped with aluminum foil and incubated at 50°C for about 18 hrs. The GUVs slowly develop during this incubation. Finally, the GUV suspension is very slowly cooled to room temperature.
GUVs have been used to image PTEN bilayer interactions and lipid reorganization due to PTEN [17].
2.6 Langmuir Films
Monolayers at the air/water interface have been used extensively to study the interaction of interfacial proteins (including enzymes) with lipids. The typical experiment involves the spreading of an appropriate organic solution of a lipid or lipid mixture at the air/water interface, compression of the resulting lipid monolayer with movable barriers to a particular surface pressure (which governs the monolayer phase state and morphology) and injection of the protein of interest underneath the monolayer. Alternatively, the lipid solution can be spread at the air/water interface to yield directly the desired surface pressure. This technique is often favored when only small amounts of the lipid and/or protein are available since smaller troughs can be used compared to those used for monolayer compression experiments. Several techniques are available to probe changes of the thermodynamic, morphological or structural properties of the monolayer upon interaction with the protein. Thermodynamic information is obtained from surface pressure changes that can be used as a semi-quantitative measure of protein penetration into the lipid monolayer or protein-induced lipid condensation [29, 30]. In combination with optical techniques like fluorescence microscopy, information about the localization of labeled protein relative to the lateral distribution of lipids and/or the lipid phase state (fluid vs. ordered) can be obtained [31, 32], infrared reflection-absorption spectroscopy (IRRAS) provides information about the secondary structure of the protein in the lipid bound state [33-35] and x-ray techniques can be applied to obtain structural information for the lipid/protein complex. Monolayer techniques have not been utilized to date for studies related to PTEN, however, Langmuir film measurements have been extensively used to characterize phosphoinositide monolayers and to investigate the interaction of proteins with phosphoinositides [36-48]. Monolayers obviously represent only one leaflet of the bilayer, and experiments are limited to proteins that insert only into one leaflet of the bilayer. The advantage of the monolayer technique is that the lipid composition in the monolayer is the same as that in the organic solution used for the spreading of the lipids.
2.7 Preparation of solid-supported phosphoinositide containing lipid bilayers
Several different approaches are used to prepare solid supported bilayers [49, 50]. For non-tethered solid supported phosphoinositide-containing lipid bilayers, we adopted a method described by the Steinem group [51]: SUVs in a 50 mM KCl, 20 mM citrate buffer (pH 4.8) are prepared by the steps described above. This SUV suspension is mixed 1:1 with a 1 M NaCl-containing citrate buffer and placed on a glass support (the type of glass support depends on the nature of the experiment; e.g., for TIRF experiments we use acid- and plasma-treated clean glass cover slips). After ½ hour incubation time, the sample is washed with distilled water followed by the final buffer solution (e.g., 140 mM KCl, 15 mM NaCl, 25 mM Hepes, pH 7.5). Using a lower pH during the bilayer formation results in a reduced number of defects (holes). To ensure that no glass is exposed, a bovine serum albumin solution (100 μg/mL) can be used to block any defects and also improve bilayer fluidity [52].
In some instances it might be preferable to use tethered lipid bilayers [53, 54]. The Lösche group described the use of sparsely tethered lipid bilayers for the characterization of PTEN membrane association [6].
2.8 Choosing the most appropriate model system for physiological questions
While striving to mimic the properties of cellular membranes, one needs to be aware of the limits of lipid model systems. A range of lipid bilayer properties affect the functions of interfacial enzymes and the choice of the most appropriate model system will depend on the physiological context of the enzyme function one wishes to investigate, the technique used for the experiment and the biophysical question one aims to answer. These questions will be illustrated in the following two general examples, membrane curvature and bilayer fluidity in laterally heterogeneous systems.
Membrane curvature is well-known to affect the activity of interfacial enzymes [55]. In the context of PTEN function, membrane curvature is a relevant topic because a range of PI(3,4,5)P3 mediated cellular functions are linked to membrane regions exhibiting high curvature, for example, filipodia during chemotaxis [56] or synapses [57-59]. For the model systems described above, only SUVs exhibit a curvature that is relevant on the length scale of a PTEN protein. When setting up the experiment in a way that the PTEN protein interacts with the outside of the SUVs (which is a typical setup), one needs to be aware that SUVs exhibit a positive curvature, which may or may not be appropriate for the physiological condition one wishes to mimic. A powerful approach to study the effect of negative or positive curvature on the function of interfacial proteins is the use of optical tweezers to pull tube like structure and to investigate the distribution of fluorescently labeled proteins relative to the curved regions using fluorescence microscopy [60-62]. It is worth mentioning that the effect of curvature on PTEN activity has not been addressed in detail at this point.
We have already discussed in section 2.1 the relationship between lipid bilayer fluidity and PTEN bilayer association. Lipid demixing (domain formation), whether inherent to the respective lipid system or due to the interaction with bivalent cations or positively charged proteins like PTEN, complicates the experimental analysis for all techniques that do not generate spatially resolved data. A range of papers have discussed the influence of bivalent cations [38, 63-65], lipids with hydrogen bonding capabilities [24] and/or cholesterol [17, 63, 66] on phosphoinositide domain formation. In cases where domain formation is observed, one may be advised to conduct a series of experiments, each mimicking the composition and physical properties of one spatial component of the complex system. For example, a PC/PI(4,5)P2/cholesterol system could be characterized by investigating the interaction of PTEN with PC as well as PI(4,5)P2/cholesterol systems. Equally, mixing cholesterol and saturated (gel phase) phosphoinositide lipids can potentially mimic a liquid-ordered phosphoinositide-enriched environment. In these cases, one needs to first prove that the compositionally reduced lipid system is an accurate representation of the spatial region in the compositionally complex system. For experiments that provide spatially resolved information (e.g., microscopy), monolayers at the air/water interface, solid supported lipid bilayers (tethered or untethered) as well as GUVs are appropriate model systems.
3. PTEN binding studies
PTEN activity towards lipids requires its association with the membrane, and it is therefore important to determine the lipid-binding properties of PTEN and its derivatives. The available methods can be divided into qualitative, semi-quantitative and quantitative assays.
3.1 Qualitative PTEN binding methods
PIP Strips (Echelon Biosciences Inc., Salt Lake City, USA) and Inositol Snoopers (Avanti Polar Lipids, Alabaster, USA) [67] are hydrophobic membranes spotted with certain amounts (100 pmol and 1 μg, respectively) of physiologically relevant lipids, including PIPs. Using an appropriate PTEN antibody, these strips can be used to obtain qualitative information about the binding of PTEN and its derivatives to anionic lipids. While these strips are useful as initial screening tools, the results must be verified with other experimental methods using lipids in a more native environment.
3.2 Semi-quantitative PTEN binding methods
The concept of lipid strips for the characterization of phosphoinositide binding proteins has been extended to array strips that have spots or well plates with different lipid compositions. These model systems have been successfully used to identify relative binding affinities for protein binding to different phosphoinositide derivatives and other anionic lipids [68, 69]. Busse et al. [70] describe a flotation assay where SUVs and the protein of interest were mixed, overlaid a non-continuous Nycodenz gradient and a buffer layer on top. The sample is spun at 55,000 rpm (275,000 × g) during which the liposomes with the bound protein migrate to the top buffer layer. Subsequently, samples are analyzed with immunoblotting.
In our lab we have used a quenching assay of fluorescently labeled phosphoinositides to evaluate the relative binding preferences of PTEN to naturally occurring phosphoinositide derivatives [9]. For this assay, phosphatidylcholine LUVs are doped with a small amount of BODIPY-phosphoinositide (∼ 0.1 mol%). Upon interaction with PTEN, the respective BODIPY-phosphoinositide clusters, which leads to a reduction of the fluorescence due to self-quenching. A standard fluorescence spectrometer with a temperature-controlled cuvette holder is sufficient to conduct these experiments. BODIPY TMR labeled phosphoinositide lipids are excited at 542 nm and the fluorescence intensity at 574 nm is monitored to investigate the interaction of PTEN with the lipid vesicles [9]. We initially utilized this assay because at the time some phosphoinositide derivatives were not available with an unsaturated acyl chain composition, thereby limiting our ability to investigate PTEN phosphoinositide binding preferences using the Tryptophan (TRP) quenching assay described below. However, it is worth mentioning that the PTEN phosphoinositide binding preferences established with this assay withstood the test of time and have been largely confirmed using other methods. However, we believe that now better binding assays are available (see below).
3.3 Determination of PTEN binding constants
Lipid binding constants of PTEN and PTEN derivatives have been determined using tryptophan quenching experiments [9, 14] and surface plasmon resonance [6, 15].
3.3.1 Tryptophan quenching experiments
Determining PTEN lipid-binding constants using TRP quenching involves titration of PTEN or a PTEN mutant with LUVs, which are labeled with a small amount of a fluorescently labeled lipid. The TRP and the lipid fluorophore (pyrene or dansyl) form a FRET pair, with TRP as the donor. Titration of the protein results in a reduction of the TRP and an increase of the pyrene or dansyl fluorescence. In our experiments [9, 14] (carried out in a standard fluorescence spectrometer equipped with a temperature-controlled cuvette holder) we found a protein concentration of 1 μM to be ideal for the experiment. Lipid vesicles are titrated into the protein solution in about 15 – 20 steps, reaching a final lipid concentration of ∼1 mM. The TRP fluorophores are excited at 290 nm and fluorescence spectra in the range 300 – 380 nm are obtained. The maximum fluorescence intensity values for each titration step are corrected for dilution. The environment of the TRP residues and hence its fluorescence properties may change upon membrane association. To account for these changes, the fluorescence spectra for each titration step should be corrected by titrating PTEN with vesicles lacking the labeled lipid [9]. In the last step, the fluorescence values are normalized to the fluorescence intensity obtained prior to the addition of any lipids. Typically, the data can be fitted with a simple binding model (y = Fmax[lipid]/(Kd + [lipid]; Fmax is the maximum fluorescence intensity).
A potential caveat of this method is vesicle and/or PTEN aggregation, which impacts the amount of free protein or vesicles (lipid) as well as the fluorescence intensity (either directly by altering the fluorophore environment or through enhanced light scattering). It is therefore important to check by DLS prior to the experiment the size distribution of the LUVs as well as whether the protein is partially aggregated. If a fraction of protein is aggregated it should be removed by centrifugation and the protein concentration be re-determined. Upon completion of the experiment, the sample should be checked again for the presence of large particles due protein or vesicle aggregation.
3.3.2 Surface Plasmon Resonance Measurements
Surface Plasmon Resonance (SPR) is a powerful method for determining the lipid binding constants of peripheral membrane proteins. In SPR, the lipids of interest are immobilized on a biosensor chip and the association/dissociation of the protein is monitored by following the temporal evolution of the surface plasmon resonance signal, which depends on the refractive index and hence on the amount of material deposited on the chip surface.
SPR has been used to obtain some initial PTEN/lipid binding constants [15]. More recent experiments have shown that these earlier data [15] over estimated the binding avidity of PTEN [6, 9, 14]. There are a couple of potential reasons for this overestimate: (1) Dextran, which is used as a coating in a variety of lipid-capturing SPR chips, can bind proteins. If the lipid coverage of the dextran layer shows some defects after blocking with BSA, the test protein will bind strongly to the dextran. (2) To obtain binding constants from SPR experiments two general approaches can be employed: In the kinetic analysis a protein concentration dependent global fit of the time-dependent protein association and dissociation SPR signal response is used to obtain kon and koff. Assuming a simple 1:1 stoichiometry, the binding constant can be obtained by the ratio of these two rate constants. For some PIP-binding proteins (e.g., PLCδ1 PH domain), this method furnishes higher apparent affinities than the affinities obtained by other methods [71]. An alternative approach is a “saturation analysis”, where the steady state values for the concentration-dependent protein association curves are taken as the equilibrium binding for the respective protein concentration. Using tethered lipid bilayers as the substrate, this approach yielded PTEN binding constants consistent with other methods (e.g., TRP quenching described above) [6].
3.3.3 Potential caveats of binding studies
PTEN establishes multiple contacts with lipids as it associates with the plasma membrane. The C2 domain and positively charged amino acids within the phosphatase domain have non-specific, electrostatic interactions with anionic lipids in the membrane at high enough concentrations to generate negative interfacial potentials. The N-terminal end binds presumably via electrostatic interactions and hydrogen bond formation to PI(4,5)P2. As a result, PTEN membrane affinity will be affected by the local concentration of these lipids, which is tightly linked to the membrane morphology (lateral distribution of the respective lipids). Agents that promote clustering of either or both lipid-binding partners, alter PTEN binding affinities. For example, the addition of cholesterol to phosphoinositide-containing vesicles resulted in enhanced PTEN binding [17] even though the PI(4,5)P2 concentration was constant. When comparing binding affinities for PTEN and PTEN mutants, it is therefore important that exactly the same lipid mixtures are employed to ensure the same lateral distribution of the PTEN binding partners. This poses a serious challenge to compare binding affinities across different studies. It is therefore recommended that each study includes wt PTEN as reference for each lipid system. Equally, studies that compare binding affinities to different lipids need to ensure that the (local) lateral density of the investigated lipids is the same for all experiments. One might argue that binding constants for laterally heterogeneous lipid bilayer systems have only limited value since they represent the binding properties of the protein and the lipid organization.
4. Structural characterization of PTEN in the membrane bound state
Only few methods are available to obtain structural information about PTEN in the membrane-bound state. Neutron reflectivity measurements in combination with molecular dynamics calculations have been successfully used to obtain structural information for PTEN and mutant PTEN bound to phosphatidylserine-containing lipid bilayers [6, 7]. For a detailed description of this method the reader is referred to a review in this issue [72]. A new promising method for the characterization of PIP-binding proteins is hydrogen/deuterium exchange mass spectrometry. While this method has not been applied to PTEN yet, it provided a map of dynamic interactions for a variety PI 3-kinases in the membrane bound state [73-77].
Infrared spectroscopy is in structural terms a low-resolution method that can monitor secondary structure changes of peripherally bound proteins upon interaction with lipid bilayers [78]. While for proteins in solution circular dichroism (CD) and IR spectroscopy provide a similar level of detail, IR spectroscopy is the superior technique when investigating proteins in the presence of lipid vesicles. In contrast to CD spectroscopy, light scattering from vesicles does not negatively impact the quality of IR spectra. Protein IR spectra can be obtained in two principal modes: In the transmission experiment, the protein/lipid vesicle sample is sandwiched between two IR transparent windows (typically BaF2 or CaF2) using a thin spacer. The secondary structure analysis relies on the protein amide I band, which is conformationally sensitive and is found between ∼ 1620 – 1700 cm-1. Since the H2O scissoring mode is also found in this region, either thin spacers (∼ 6 μm) need to be used to limit the pathlength and hence the intensity of the water absorption band, or the protein/vesicle suspension needs to be transferred into a D2O buffer. The second general method to obtain IR spectra of lipid-bound proteins is attenuated total reflection (ATR) IR spectroscopy. In ATR IR spectroscopy, the IR light impinges on the ATR crystal at an angle above the critical angle, and therefore, experiences total reflection. An evanescent wave penetrates a few micrometers into the medium above the crystal and since the presence of protein and lipids alters the refractive index of the interface relative to the bare substrate, the IR beam gets attenuated, which manifests itself in a spectrum similar to the one obtained in transmission mode. For a detailed description of Fourier transform infrared (FTIR) ATR methods and how they can be used to obtain orientational information, the reader may consult reference [79].
We have used a micro ATR FTIR unit to study PTEN/lipid vesicle interactions [9]. For this experiment, PTEN samples are concentrated using 10,000 molecular weight cutoff (MWCO) Centricon tubes, to reach a concentration between 8 – 9 mg/ml. To avoid overlap of the protein amide I band with the H2O scissoring mode, the protein samples are exchanged against D2O buffer using dialysis cartridges (in comparison to H2O, the D2O scissoring mode is shifted to lower wavenumbers). To ensure that the final buffer solution contains a minimum of H2O, this buffer exchange is repeated three times. The lipid free PTEN spectrum is obtained by placing ∼ 12 μL of PTEN/D2O buffer solution in the sample cell of the BioATRCell II (Bruker, Billerica, MA). FTIR interferograms are collected at 2 cm-1 spectral resolution (500 – 1000 scans), apodized with a Blackman-Harris function and Fourier transformed with one level of zero filling to yield spectra encoded at 1 cm-1 intervals. As reference spectrum the bare ATR crystal is used and the absorbance spectrum is computed using the instrument software. Spectra of lipid bound PTEN are obtained by resuspending the appropriate amount of dried lipid in PTEN/D2O buffer solution (see preparation of multilamellar vesicles described above). The use of multilamellar vesicles instead of unilamellar vesicles is advantageous for IR experiments since the protein is present in all aqueous compartments between the various lamellar layers. The final protein or protein/lipid IR spectrum is obtained by subtracting a D2O buffer spectrum. The D2O buffer samples are adjusted with respect to their H2O (HOD) content so that the intensities of the H2O and HOD bands match the respective bands in the protein spectra.
5. PTEN membrane binding kinetics, lateral dynamics and interfacial kinetics
The kinetics of PTEN membrane association, its dynamics when bound to the membrane and the kinetics that govern its membrane dissociation, are important factors for PTEN's targeting to distinct membrane environment, its activation and substrate turnover.
5.1 Stopped-flow fluorescence measurements of PTEN membrane association and dissociation kinetics
Stopped-flow spectrophotometry experiments have been widely used to study the transient kinetics of protein processes [80, 81], including the binding of proteins to phosphoinositides [82-86].
For lipid-binding proteins, the typical stopped-flow experiment is based on the quenching of TRP fluorescence and involves rapid mixing of the protein of interest dissolved in buffer solution and a lipid vesicle suspension. The lipid vesicles contain a small amount (∼ 2 mol%) of a labeled lipid that quenches TRP fluorescence (typically dansyl- or pyrene-labeled phosphatidylethanolamine). TRP (donor) and dansyl or pyrene (acceptors) form a FRET pair, and the binding kinetics can therefore be followed either based upon the reduction of the TRP fluorescence or the increase of dansyl or pyrene fluorescence. For the principle of the stopped-flow experiment, see Figure 1.
Figure 1.

Principle of stopped-flow fluorescence experiment. Top: The kinetics of PTEN binding is investigated by mixing PTEN with dansyl-PE labeled lipid vesicles. TRP and Dansyl form a FRET pair and the PTEN bilayer association can be followed by monitoring either the decrease of the TRP or the increase of the dansyl fluorescence (shown here). Bottom: The kinetics of PTEN bilayer dissociation is followed by mixing PTEN bound to labeled lipid vesicles with an excess of unlabeled lipid vesicles (except for the labeled lipid, both types of lipid vesicles have the same lipid composition).
Figure 2 shows the time evolution of dansyl fluorescence for the mixing of wt PTEN with PS (98%)/dansyl-PE (2%) vesicles for different lipid concentrations. The TRP fluorophore is excited at 280 nm (mercury xenon lamp in combination with a monochromator). Our instrument (Applied Photophysics SX20) allows for the simultaneous observation of the decrease in TRP fluorescence intensity (40 nm bandpass filter centered at 320 nm) and the increase in dansyl fluorescence intensity (395 nm long pass filter). We found that the S/N ratio is better for the channel that monitors the change in dansyl fluorescence intensity and therefore, we base our analysis of PTEN lipid binding kinetics on the increase in dansyl fluorescence.
Figure 2.



In a typical binding experiment, a 0.5 μM PTEN solution is placed in one of the stopped-flow syringes, while a unilamellar (100 nm), dansyl labeled (2%) lipid vesicle suspension is placed in the second syringe. One should obtain the association kinetics for at least 5 - 7 lipid concentrations (in our case ranging from 5 to 320 μM outer leaflet lipid concentration). For each lipid concentration, a minimum of eight mixing drives are carried out and the time dependent fluorescence intensity data are averaged. To avoid vesicle shearing during the driving of the syringe, the drive ram pressure should not exceed 32 psi (∼ 2.2 bar). The readout of the instrument are time-dependent fluorescence intensity curves (Figure 2A). For simple binding processes, such as the binding of PTEN to PS vesicles, these data can be fit with a simple single exponential fit [87]:
F: fluorescence intensity; Fmax and Fmin are the maximum and minimum fluorescence intensities
kobs: observed rate constant
Plotting of the observed rate constant kobs as a function of surface lipid concentration yields a straight line, where the slope is kon and the y-intercept is a first estimate of koff (Figure 2B). To obtain accurate koff values, however, a dissociation experiment should be performed. In this case, one syringe is loaded with PTEN bound to dansyl-labeled lipid vesicles, while the other syringe is filled with unlabeled lipid vesicles. The concentration of unlabeled lipid vesicle solution should be at least 15 times high than the concentration of the labeled lipid vesicles in the other syringe. To ensure a strong response, the protein concentration should be higher than in the previous experiment. In our case we used 1 uM (see Figure 2C). After mixing, the concentration of unlabeled lipid vesicles is significantly higher than the corresponding concentration of the labeled vesicles with bound PTEN and as a result, PTEN dissociation from the labeled vesicles and binding to unlabeled vesicles is more probable. This process leads to a FRET reduction and hence to an increase in TRP fluorescence intensity and a decrease of the dansyl emission intensity. Since a first order rate law characterizes the dissociation step, it can be fit with a simple exponential fit to yield koff (Figure 2C).
Binding of PTEN to PS vesicles is from a kinetic point of view a rather simple process, consistent with the simple binding model described above. However, for more complex lipid environments, this simple binding model does not describe satisfactorily the binding mechanism. From a mathematical point of view, this manifests itself in a non-linear behavior in the concentration dependent plot of kobs obtained using the procedure described above. In these cases, a more complicated binding model needs to be developed that accurately accounts for the different lipid environments the PTEN protein is binding to. This usually requires a global fit of several PTEN association experiments carried out for several lipid concentrations [88-90], which yields the rate constants characteristic for the respective lipid environments.
5.2 Single molecule dynamics of model membrane associated PTEN
Single molecule techniques have emerged in recent years as tools for the characterization of transient interactions between biomolecules. One such sensitive single molecule imaging technique is total internal reflection fluorescence (TIRF) microscopy [91, 92]. Many signaling events occur at the plasma membrane and involve the recruitment of intracellular proteins to membrane surfaces. TIRF microscopy has the advantage that plasma membrane-associated proteins can be easily studied. The results are independent from proteins distant to the bilayer since only fluorophores within ∼100 nm of the substrate-sample interface are illuminated. This eliminates background noise, enhances the signal-to-noise ratio and makes single molecule observation of interfacial processes possible. Single molecule TIRF microscopy has been widely used to investigate the interaction of labeled proteins with cell membranes in vivo [93-98] as well as to investigate the dynamics of proteins to membrane mimics [99, 100]. Most importantly, PTEN binding to membranes has been observed in living cells, such as Dictyostelium discoideum, at the single molecule level [101-103].
The capacity of TIRF to preferentially detect bound proteins can also be applied to artificial supported lipid bilayers in vitro [100, 104]. Our group has applied TIRF microscopy to study the interaction of labeled PTEN with membranes. This technique has the distinct advantage of following the events of single protein molecules at the membrane, such as binding and release, lateral diffusion along the membrane as well as membrane-binding lifetimes. The supported membranes provide a completely controlled system in which membrane components, protein concentration and buffer compositions can be varied, allowing study of the underlying mechanisms of PTEN-membrane interaction. In order to achieve this plasma membrane biomimic, a planar supported lipid bilayer (SLB) with different composition can be prepared either using vesicle fusion or Langmuir-Blodgett (LB) technique or Langmuir-Schaefer (LS) deposition or combination of both LB/LS method [50, 105, 106]. The SLB is a transparent film that is flat, stable, and displays the lateral diffusion of native plasma membrane making it a perfect system for TIRF microscopy. However, preparing a defect-free and stable SLB that mimics the complexity of native cell membrane has always been a challenge. In recent years, the simple and versatile vesicle fusion technique has been widely used for SLB preparation and researchers have optimized parameters such as temperature, pH, ionic strength, ion type to form a stable and complete SLBs [49, 51, 107]. In addition to the formation of stable SLBs, labeling the protein with the optimum fluorophore decides the success of the single molecule experiment. For instance, an efficient single-molecule fluorophore should have high fluorescence quantum yield (close to unity), be sufficiently stable against bleaching and have the protein of interest tagged with a single molecule of fluorophore. Figure 3A shows an example of a single molecule TIRF image of wtPTEN bound to a DOPC/DOPS (70:30) supported bilayer membrane, and the single molecule trajectories show the lateral mobility of PTEN along the membrane (Figure 3B). Here the SLB is prepared using the vesicle fusion method on glass coverslip as mentioned in section 2.5 and the protein is labeled with Alexa fluor 647 using Sfp phosphopantetheinyl transferase method [108], where a single fluorophore is site specifically tagged to the protein such that the fluorescence intensity from individual protein molecules is uniform. Also, the lipids used for preparing SLBs are monounsaturated, dioleyl acyl chains to ensure that the membrane is uniformly fluid.
Figure 3.
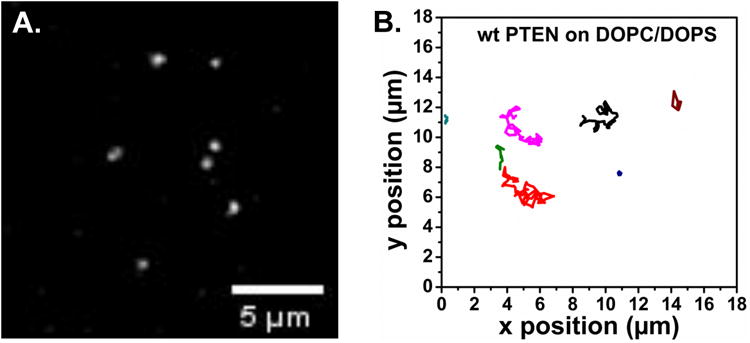
TIRF single molecule images. A. TIRF image showing 200 pM of wt PTEN-AF647 molecules bound to the supported lipid bilayer composed of DOPC/DOPS (70:30). B. Single molecule trajectories of wt PTEN-AF647 on DOPC/DOPS membrane acquired with an exposure of 30 ms/frame. Measurements were performed at room temperature in a near physiological buffer (20 mM HEPES, 140 mM KCl, 15 mM NaCl, pH 7.5).
5.2.1 Single particle tracking and data analysis
The single molecule trajectories of protein molecules in movies can be generated using tracking software tools such as Mtrack2, SpotTracker, Speckle Tracker, ImarisTrack, ICY, TrackMate, uTrack, Particle Tracker, and many more. An exhaustive list of particle tracker software tools can be found in [109]. In this study we have used the Particle Tracker plugin for ImageJ to generate the trajectories [110] and then the data were imported into MATLAB for further analysis. Briefly, the Particle Tracker determines the center position and the intensity of each particle using an intensity threshold set by the user, which is followed by linking them together in the successive frames to form trajectories. The particle intensity threshold and the linking parameters were set so as to avoid spurious trajectories. Further, the trajectories were subjected to some of the exclusion tests as described in Knight et al 2009, [100]. The dim and bright contaminants were excluded by setting minimum and maximum particle intensity cutoff that is determined empirically by calculating the mean intensity of individual trajectory. Also, we eliminated trajectories shorter than 10 frames, removing trajectories resulting from contaminants as well as trajectories that are not properly linked. Furthermore, to eliminate the immobile and slow moving particles the diffusion coefficient D was calculated for individual particles and particles with D below a threshold (0.1 μm2/s) were excluded. These exclusion criteria gave us the trajectories that more-or-less belong to a single mobile fraction.
All the trajectories that passed the exclusion tests were exported to MATLAB (The Mathworks, Inc.) and subjected to ‘msdanalyzer’ package that is specifically developed for mean square displacement (MSD) analysis [111]. Using this package, the MSD function was calculated and fitted for individual trajectories. The fits for which the R2 coefficient, reflecting the quality of the fit, was <0.8 were excluded. The mean squared displacement values 〈r2〉, were generated as well the diffusion coefficient D for individual trajectories was calculated. These data were exported to GraphPad Prism 6 (GraphPad Software, Inc.) for further analysis. Two different approaches were used to determine the average diffusion coefficient of the particles. In the first approach, Figure 4A, the MSD plot 〈r2〉 versus time interval (8 frames, 0.24 s) was generated and the D value for the particle was determined by least-squares fitting to a straight line through the origin. In the second approach, Figure 4B, the D-value calculated for individual trajectories was converted to probability distribution histograms with 30 equally spaced bins. The histogram was best fit to a log Gaussian distribution. The diffusion constants generated from these two approaches are summarized in Table 3. The D value of 0.49 μm2/s reported here is an overall average diffusion coefficient of the PTEN molecule on DOPC/DOPS membrane and is about five times slower than the diffusion coefficient of the lipids, ∼2.7 μm2/s (data not shown). This suggests that the protein is strongly bound to the membrane either by binding to multiple lipid molecules or by increased bilayer contact [104, 112].
Figure 4.
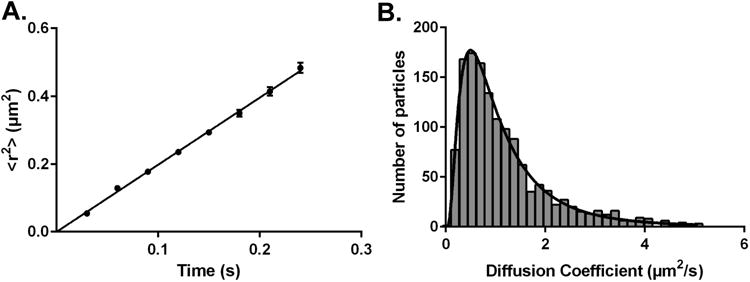
Diffusion analysis of wt PTEN bound to a DOPC/DOPS (70:30) solid supported lipid bilayer. (A) Plot of the mean square displacement 〈r2〉 versus time interval (8 frames, 0.24s) (B) Probability distribution histogram of diffusion coefficients generated from individual trajectories.
Table 3. Diffusion coefficients of wt PTEN on DOPC/DOPS (70:30) membrane.
| Method | D (μm2/s) | 95% confidence interval | trajectories |
|---|---|---|---|
| MSD plot | 0.49 ± 0.02 | 0.48 to 0.50 | 2223 |
| Histogram | 0.49 ± 0.01 | 0.48 to 0.51 | 1374 |
Multi-domain proteins such as PTEN often show changes in the diffusion states within the trajectories, likely due to the difference in the number of lipid head group bound to the protein and its bilayer contact. The increase in the protein-lipid, protein-bilayer contact as well as interaction with other molecules (complex formation) lowers the mobility of the protein, thereby decreasing its diffusion coefficient. The authors suggest a few of the references [99, 102, 104, 112-115] for analyzing the different membrane bound states that occur within the trajectories. Overall, the diffusion coefficient of a protein molecule can be helpful and provide essential information about the functional state, mechanistic insight, membrane interaction, and to a certain extent determine the lipid binding stoichiometry of the protein.
5.3 In terfacial kinetics of PTEN substrate turnover
The analysis of PTEN catalytic activity against soluble, short-chain PI(3,4,5)P3 is carried out in many labs to study PTEN function and involves the quantification of released phosphate using a malachite green reaction [116]. The analysis of PTEN activity in lipid vesicle systems, which is a physiologically more relevant substrate environment, is more challenging. While initially also in this case the malachite green reaction was used to determine the amount of released inorganic phosphate [15], most more recent studies analyze the release of phosphate using (natural chain) 3-[32P] PI(3,4,5)P3 embedded in lipid vesicles [5, 117]. The experimental conditions for both techniques have been discussed in detail in another article in this issue [25]. For a general discussion of interfacial kinetics and models used to analyze the data, the reader is referred to a short paper by Stahelin [118]
Like for PTEN binding experiments, the choice of the lipid composition may significantly impact the outcomes of PTEN activity experiments, since local accumulation of PI(4,5)P2 and/or PI(3,4,5)P3 will result in increased PTEN binding and hence substrate turnover. The kinetic analysis of PTEN lipid turnover is carried out by following a formalism introduced by Dennis and co-workers, which yields the initial velocity of PTEN substrate turnover [119-122]
where V0 and Vmax are the initial and maximal velocities of the enzymatic reaction, respectively, KS is the dissociation constant of PTEN from the vesicle surface, XS is the surface concentration (molar fraction) of PI(3,4,5)P3, [So] is the bulk PI(3,4,5)P3 concentration and iKm is the interfacial value of the apparent Michaelis constant Km (mole fraction, unitless) [122]. To evaluate this equation two types of experiments are performed: The first type of experiment is a bulk dilution (BD) experiment where for a fixed surface concentration XS (fixed mole fraction) of PI(3,4,5)P3 in mixed PC/PI(3,4,5)P3 vesicles PTEN activity is measured for different PI(3,4,5)P3 bulk concentrations. Different PI(3,4,5)P3 bulk concentrations are obtained by varying the total lipid (total vesicle) concentration [117]. The second type of experiment is a surface dilution (SD) experiment, where the PI(3,4,5)P3 surface concentration XS is varied by varying the concentration of PC in the mixed PC/PI(3,4,5)P3 vesicles [117]. Apparent Vmax and apparent Km values are fitted to the following equations to obtain the kinetic parameters for PTEN [5]
This formalism will work well when the goal is to compare the activities of, for example, wtPTEN and mutant PTEN when interacting with binary PC/PI(3,4,5)P3 mixed lipid vesicles. However, the use of more complex lipid mixtures may pose a problem. Similar to what was described when discussing PTEN binding to complex lipid systems, lipid demixing (domain formation) will have a profound effect on the kinetics of the enzymatic reaction since the local surface concentration will change dramatically and the dissociation constant will change (see above). The effect of a varying lipid ratio across the lipid vesicle population [27] is a caveat that cannot be avoided, but should be kept in mind when discussing the experimental data.
5.4 Combining several kinetic experiments - a new way to tackle PTEN kinetics in complex membrane environments?
We have presented three experimental approaches that furnish kinetic data. While some quantities can be obtained from all three methods, for example, dissociation constants, other quantities are unique to a particular method. While interfacial kinetics measurements provide information about the PI(3,4,5)P3 hydrolysis, single molecule experiments can provide spatially resolved information about kon and koff rates as well as lateral diffusion. In combination the three methods, stopped-flow experiments, single molecule measurements and interfacial kinetic measurements, should be able to provide a description of PTEN dynamics that reveals new important insights about PTEN membrane association, activation, substrate hydrolysis and dissociation. Unfortunately, a comprehensive analysis of data from all three experimental modalities based upon existing literature data is currently not possible because deviations in the respective experimental setups and lipid compositions.
6. Concluding remarks
PTEN function is characterized by a complex series of steps that leads to its membrane association, subsequent activation by PI(4,5)P2, PI(3,4,5)P3 dephosphorylation and PTEN dissociation. The membrane binding properties can be determined by a combination of binding experiments discussed in section 3.3 and kinetic experiments detailed in sections 5.1 and 5.2. The substrate hydrolysis can be investigated using interfacial kinetics techniques described in section 5.3, while the dissociation step can be determined using a combination of binding and kinetic methods (section 3.3 and 5.1/5.2). While each of these methods provides valuable information, none of these methods alone can provide exhaustive information about PTEN function in a particular experimental scenario. In order to be able to combine the various methods, one should strive to use the same or similar lipid model system across all experimental platforms, so that a global analysis can be performed (see Table 4 for a summary of methods).
Table 4. Steps of PTEN function and corresponding characterization methods in lipid model systems.
| Equilibrium | Kinetics | Dynamics | Conformation | |
|---|---|---|---|---|
| Association | TRP quenching/SPR | Stopped-Flow/SM TIRF | Neutron Diffraction | |
| Lateral Distribution | Fluorescence Microscopy (Langmuir Films/GUVs) | SM TIRF | ||
| Activation | Interfacial kinetics | IR Spectroscopy | ||
| Hydrolysis | Interfacial kinetics | |||
| Dissociation | Stopped-Flow SM TIRF |
Highlights.
Peripheral binding to phospholipid membranes is a critical step for PTEN action.
PTEN binding to membranes is dominated by interactions with anionic lipids.
Biophysical methods provide insight by quantifying binding affinities and kinetics.
Acknowledgments
We gratefully acknowledge financial support from NSF through grant CHE 1216827 and the NIH grant NS021716.
Footnotes
attenuated total reflection ATR; circular dichroism CD; diacylphosphatidylinositol-3,4,5-trisphosphate PI(3,4,5)P3; diacylphosphatidylinositol-4,5-bisphosphate PI(4,5)P2; di-oleoyl DOPIP; di-palmitoyl di-C16; fluorescence resonance energy transfer FRET; Fourier transform infrared FTIR; giant unilamellar vesicle GUV; infrared IR; large unilamellar vesicle LUV; molecular weight cutoff MWCO; phosphatidylcholine PC; phosphatidylinositol PI; phosphatidylserine PS; phosphoinositide PIP; small unilamellar vesicle SUV; stearoyl-arachidonoyl SAPIPs; surface plasmon resonance SPR
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Worby CA, Dixon JE. Annu Rev Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 2.Liu F, Wagner S, Campbell RB, Nickerson JA, Schiffer CA, Ross AH. J Cell Biochem. 2005;96:221–234. doi: 10.1002/jcb.20525. [DOI] [PubMed] [Google Scholar]
- 3.Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. J Biol Chem. 2001;276:48627–48630. doi: 10.1074/jbc.C100556200. [DOI] [PubMed] [Google Scholar]
- 4.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolduc D, Rahdar M, Tu-Sekine B, Sivakumaren SC, Raben D, Amzel LM, Devreotes P, Gabelli SB, Cole P. eLife. 2013;2:e00691. doi: 10.7554/eLife.00691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shenoy S, Shekhar P, Heinrich F, Daou MC, Gericke A, Ross AH, Losche M. PLoS One. 2012;7:e32591. doi: 10.1371/journal.pone.0032591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shenoy SS, Nanda H, Losche M. J Struct Biol. 2012;180:394–408. doi: 10.1016/j.jsb.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 9.Redfern RE, Redfern DA, Furgason MLM, Muonson M, Ross AH, Gericke A. Biochemistry. 2008;47:2162–2171. doi: 10.1021/bi702114w. [DOI] [PubMed] [Google Scholar]
- 10.Iijima M, Huang YE, Luo HR, Vazquez F, Devreotes PN. J Biol Chem. 2004;279:16606–16613. doi: 10.1074/jbc.M312098200. [DOI] [PubMed] [Google Scholar]
- 11.Campbell RB, Liu FH, Ross AH. J Biol Chem. 2003;278:33617–33620. doi: 10.1074/jbc.C300296200. [DOI] [PubMed] [Google Scholar]
- 12.Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN. Proc Natl Acad Sci U S A. 2009;106:480–485. doi: 10.1073/pnas.0811212106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redfern RE, Daou MC, Li L, Munson M, Gericke A, Ross AH. Protein Sci. 2010;19:1948–1956. doi: 10.1002/pro.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das S, Dixon JE, Cho W. Proc Natl Acad Sci U S A. 2003;100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czogalla A, Grzybek M, Jones W, Coskun U. Biochim Biophys Acta. 2014;1841:1049–1059. doi: 10.1016/j.bbalip.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Jiang Z, Redfern RE, Isler Y, Ross AH, Gericke A. Chem Phys Lipids. 2014;182:52–61. doi: 10.1016/j.chemphyslip.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Redfern DA, Gericke A. J Lip Res. 2005;46:504–515. doi: 10.1194/jlr.M400367-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Nanda H, Heinrich F, Losche M. Methods. 2014 doi: 10.1016/j.ymeth.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gericke A, Leslie NR, Losche M, Ross AH. Adv Exp Med Biol. 2013;991:85–104. doi: 10.1007/978-94-007-6331-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei Y, Stec B, Redfield AG, Weerapana E, Roberts MF. J Biol Chem. 2014 doi: 10.1074/jbc.M114.588590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landgraf KE, Pilling C, Falke JJ. Biochemistry. 2008;47:12260–12269. doi: 10.1021/bi801683k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaughlin S, Murray D. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 24.Graber ZT, Jiang Z, Gericke A, Kooijman EE. Chem Phys Lipids. 2012;165:696–704. doi: 10.1016/j.chemphyslip.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Spinelli L, Leslie NR. Methods. 2014 [Google Scholar]
- 26.Patil YP, Jadhav S. Chem Phys Lipids. 2014;177:8–18. doi: 10.1016/j.chemphyslip.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 27.Larsen J, Hatzakis NS, Stamou D. J Am Chem Soc. 2011;133:10685–10687. doi: 10.1021/ja203984j. [DOI] [PubMed] [Google Scholar]
- 28.Akashi K, Miyata H, Itoh H, Kinosita K., Jr Biophys J. 1996;71:3242–3250. doi: 10.1016/S0006-3495(96)79517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calvez P, Demers E, Boisselier E, Salesse C. Langmuir. 2011;27:1373–1379. doi: 10.1021/la104097n. [DOI] [PubMed] [Google Scholar]
- 30.Calvez P, Bussieres S, Eric D, Salesse C. Biochimie. 2009;91:718–733. doi: 10.1016/j.biochi.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 31.Stottrup BL, Nguyen AH, Tuzel E. Biochim Biophys Acta. 2010;1798:1289–1300. doi: 10.1016/j.bbamem.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Fanani ML, Hartel S, Maggio B, De Tullio L, Jara J, Olmos F, Oliveira RG. Biochim Biophys Acta. 2010;1798:1309–1323. doi: 10.1016/j.bbamem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Blume A, Kerth A. Biochim Biophys Acta. 2013;1828:2294–2305. doi: 10.1016/j.bbamem.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Mendelsohn R, Mao G, Flach CR. Biochim Biophys Acta. 2010;1798:788–800. doi: 10.1016/j.bbamem.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendelsohn R, Brauner JW, Gericke A. Annu Rev Phys Chem. 1995;46:305–334. doi: 10.1146/annurev.pc.46.100195.001513. [DOI] [PubMed] [Google Scholar]
- 36.Sarmento MJ, Coutinho A, Fedorov A, Prieto M, Fernandes F. Biochim Biophys Acta. 2014;1838:822–830. doi: 10.1016/j.bbamem.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 37.Rebaud S, Wang CK, Sarkis J, Mason L, Simon A, Blum LJ, Hofmann A, Girard-Egrot AP. Biochim Biophys Acta. 2014;1838:2698–2707. doi: 10.1016/j.bbamem.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 38.Ellenbroek WG, Wang YH, Christian DA, Discher DE, Janmey PA, Liu AJ. Biophys J. 2011;101:2178–2184. doi: 10.1016/j.bpj.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh SK, Castorph S, Konovalov O, Jahn R, Holt M, Salditt T. New Journal of Physics. 2010;12 [Google Scholar]
- 40.Hermelink A, Kirsch C, Klinger R, Reiter G, Brezesinski G. Biophys J. 2009;96:1016–1025. doi: 10.1016/j.bpj.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorenz CD, Faraudo J, Travesset A. Langmuir. 2008;24:1654–1658. doi: 10.1021/la703550t. [DOI] [PubMed] [Google Scholar]
- 42.He J, Haney RM, Vora M, Verkhusha VV, Stahelin RV, Kutateladze TG. J Lipid Res. 2008;49:1807–1815. doi: 10.1194/jlr.M800150-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manna D, Albanese A, Park WS, Cho W. J Biol Chem. 2007;282:32093–32105. doi: 10.1074/jbc.M703517200. [DOI] [PubMed] [Google Scholar]
- 44.Stahelin RV, Karathanassis D, Bruzik KS, Waterfield MD, Bravo J, Williams RL, Cho W. J Biol Chem. 2006;281:39396–39406. doi: 10.1074/jbc.M607079200. [DOI] [PubMed] [Google Scholar]
- 45.Malkova S, Stahelin RV, Pingali SV, Cho W, Schlossman ML. Biochemistry. 2006;45:13566–13575. doi: 10.1021/bi061133l. [DOI] [PubMed] [Google Scholar]
- 46.Flesch FM, Yu JW, Lemmon MA, Burger KNJ. Biochem J. 2005;389:435–441. doi: 10.1042/BJ20041721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stahelin RV, Long F, Diraviyam K, Bruzik KS, Murray D, Cho WH. J Biol Chem. 2002;277:26379–26388. doi: 10.1074/jbc.M201106200. [DOI] [PubMed] [Google Scholar]
- 48.Hirasawa K, Irvine RF, Dawson RM. Biochem J. 1981;193:607–614. doi: 10.1042/bj1930607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hardy GJ, Nayak R, Zauscher S. Current opinion in colloid & interface science. 2013;18:448–458. doi: 10.1016/j.cocis.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cremer PS, Boxer SG. Journal of Physical Chemistry B. 1999;103:2554–2559. [Google Scholar]
- 51.Braunger JA, Kramer C, Morick D, Steinem C. Langmuir. 2013;29:14204–14213. doi: 10.1021/la402646k. [DOI] [PubMed] [Google Scholar]
- 52.Nair PM, Salaita K, Petit RS, Groves JT. Nat Protoc. 2011;6:523–539. doi: 10.1038/nprot.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner ML, Tamm LK. Biophys J. 2000;79:1400–1414. doi: 10.1016/S0006-3495(00)76392-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knoll W, Naumann R, Friedrich M, Robertson JW, Losche M, Heinrich F, McGillivray DJ, Schuster B, Gufler PC, Pum D, Sleytr UB. Biointerphases. 2008;3:FA125. doi: 10.1116/1.2913612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Epand RM, D'Souza K, Berno B, Schlame M. Biochim Biophys Acta. 2015;1848:220–228. doi: 10.1016/j.bbamem.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 56.Artemenko Y, Lampert TJ, Devreotes PN. Cell Mol Life Sci. 2014;71:3711–3747. doi: 10.1007/s00018-014-1638-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garcia-Junco-Clemente P, Golshani P. Commun Integr Biol. 2014;7:e28358. doi: 10.4161/cib.28358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kreis P, Leondaritis G, Lieberam I, Eickholt BJ. Front Mol Neurosci. 2014;7:23. doi: 10.3389/fnmol.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haws ME, Jaramillo TC, Espinosa F, Widman AJ, Stuber GD, Sparta DR, Tye KM, Russo SJ, Parada LF, Stavarache M, Kaplitt M, Bonci A, Powell CM. J Comp Neurol. 2014;522:1171–1190. doi: 10.1002/cne.23488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campillo C, Sens P, Koster D, Pontani LL, Levy D, Bassereau P, Nassoy P, Sykes C. Biophys J. 2013;104:1248–1256. doi: 10.1016/j.bpj.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roux A, Koster G, Lenz M, Sorre B, Manneville JB, Nassoy P, Bassereau P. Proc Natl Acad Sci U S A. 2010;107:4141–4146. doi: 10.1073/pnas.0913734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sorre B, Callan-Jones A, Manneville JB, Nassoy P, Joanny JF, Prost J, Goud B, Bassereau P. Proc Natl Acad Sci U S A. 2009;106:5622–5626. doi: 10.1073/pnas.0811243106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levental I, Christian DA, Wang YH, Madara JJ, Discher DE, Janmey PA. Biochemistry. 2009;48:8241–8248. doi: 10.1021/bi9007879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Slochower DR, Wang YH, Tourdot RW, Radhakrishnan R, Janmey PA. Adv Colloid Interface Sci. 2014;208:177–188. doi: 10.1016/j.cis.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang YH, Slochower DR, Janmey PA. Chem Phys Lipids. 2014;182:38–51. doi: 10.1016/j.chemphyslip.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Graber ZT, Gericke A, Kooijman EE. Chem Phys Lipids. 2014;182:62–72. doi: 10.1016/j.chemphyslip.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 67.Sims KH, Tytler EM, Tipton J, Hill KL, Burgess SW, Shaw WA. Biochim Biophys Acta. 2014;1841:1038–1048. doi: 10.1016/j.bbalip.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Bruntz RC, Taylor HE, Lindsley CW, Brown HA. J Biol Chem. 2014;289:600–616. doi: 10.1074/jbc.M113.532978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guittard G, Gerard A, Dupuis-Coronas S, Tronchere H, Mortier E, Favre C, Olive D, Zimmermann P, Payrastre B, Nunes JA. J Immunol. 2009;182:3974–3978. doi: 10.4049/jimmunol.0804172. [DOI] [PubMed] [Google Scholar]
- 70.Busse RAM, Scacioc A, Hernandez JMM, Krick R, Stephan M, Janshoff A, Thumm M, Kuhnel K. Autophagy. 2013;9:770–777. doi: 10.4161/auto.23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Narayan K, Lemmon MA. Methods. 2006;39:122–133. doi: 10.1016/j.ymeth.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nanda H, Heinrich F, Losche M. Methods. 2014 doi: 10.1016/j.ymeth.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walser R, Burke JE, Gogvadze E, Bohnacker T, Zhang X, Hess D, Kuenzi P, Leitges M, Hirsch E, Williams RL, Laffargue M, Wymann MP. PLoS Biol. 2013;11:e1001587. doi: 10.1371/journal.pbio.1001587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vadas O, Dbouk HA, Shymanets A, Perisic O, Burke JE, Abi Saab WF, Khalil BD, Harteneck C, Bresnick AR, Nurnberg B, Backer JM, Williams RL. Proc Natl Acad Sci U S A. 2013;110:18862–18867. doi: 10.1073/pnas.1304801110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burke JE, Williams RL. Advances in biological regulation. 2013;53:97–110. doi: 10.1016/j.jbior.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burke JE, Perisic O, Masson GR, Vadas O, Williams RL. Proc Natl Acad Sci U S A. 2012;109:15259–15264. doi: 10.1073/pnas.1205508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burke JE, Vadas O, Berndt A, Finegan T, Perisic O, Williams RL. Structure. 2011;19:1127–1137. doi: 10.1016/j.str.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barth A. Biochim Biophys Acta. 2007;1767:1073–1101. doi: 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 79.Glassford SE, Byrne B, Kazarian SG. Biochim Biophys Acta. 2013;1834:2849–2858. doi: 10.1016/j.bbapap.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 80.Johnson KA. Curr Opin Biotechnol. 1998;9:87–89. doi: 10.1016/s0958-1669(98)80089-x. [DOI] [PubMed] [Google Scholar]
- 81.Johnson KA. FEBS Lett. 2013;587:2753–2766. doi: 10.1016/j.febslet.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perez-Lara A, Egea-Jimenez AL, Ausili A, Corbalan-Garcia S, Gomez-Fernandez JC. Biochim Biophys Acta. 2012;1821:1434–1442. doi: 10.1016/j.bbalip.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 83.Feeser EA, Ignacio CM, Krendel M, Ostap EM. Biochemistry. 2010;49:9353–9360. doi: 10.1021/bi1012657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McKenna JM, Ostap EM. J Biol Chem. 2009;284:28650–28659. doi: 10.1074/jbc.M109.049791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Corbin JA, Evans JH, Landgraf KE, Falke JJ. Biochemistry. 2007;46:4322–4336. doi: 10.1021/bi062140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Corbin JA, Dirkx RA, Falke JJ. Biochemistry. 2004;43:16161–16173. doi: 10.1021/bi049017a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goodrich JA, Kugel JF. Binding and kinetics for molecular biologists. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2007. [Google Scholar]
- 88.Johnson KA, Simpson ZB, Blom T. Anal Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 89.Johnson KA, Simpson ZB, Blom T. Anal Biochem. 2009;387:30–41. doi: 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 90.Johnson KA. Method Enzymol. 2009;467:601–+. doi: 10.1016/S0076-6879(09)67023-3. [DOI] [PubMed] [Google Scholar]
- 91.Reck-Peterson SL, Derr ND, Stuurman N. Cold Spring Harbor protocols. 2010;2010 doi: 10.1101/pdb.prot5399. pdb top73. [DOI] [PubMed] [Google Scholar]
- 92.Trache A, Meininger GA. Current protocols in microbiology. 2008;Chapter 2:Unit 2A 2 1–2A 2 22. doi: 10.1002/9780471729259.mc02a02s10. [DOI] [PubMed] [Google Scholar]
- 93.Iino R, Koyama I, Kusumi A. Biophys J. 2001;80:2667–2677. doi: 10.1016/S0006-3495(01)76236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Axelrod D. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 95.Sako Y, Minoghchi S, Yanagida T. Nat Cell Biol. 2000;2:168–172. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- 96.Belyy V, Yildiz A. FEBS Lett. 2014;588:3520–3525. doi: 10.1016/j.febslet.2014.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martin-Fernandez ML, Tynan CJ, Webb SE. J Microsc. 2013;252:16–22. doi: 10.1111/jmi.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Coelho M, Maghelli N, Tolic-Norrelykke IM. Integr Biol (Camb) 2013;5:748–758. doi: 10.1039/c3ib40018b. [DOI] [PubMed] [Google Scholar]
- 99.Ziemba BP, Li J, Landgraf KE, Knight JD, Voth GA, Falke JJ. Biochemistry. 2014;53:1697–1713. doi: 10.1021/bi4016082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knight JD, Falke JJ. Biophys J. 2009;96:566–582. doi: 10.1016/j.bpj.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yasui M, Matsuoka S, Ueda M. PLoS Comput Biol. 2014;10:e1003817. doi: 10.1371/journal.pcbi.1003817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsuoka S, Shibata T, Ueda M. PLoS Comput Biol. 2013;9:e1002862. doi: 10.1371/journal.pcbi.1002862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN. Proc Natl Acad Sci U S A. 2006;103:3633–3638. doi: 10.1073/pnas.0510570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Knight JD, Lerner MG, Marcano-Velazquez JG, Pastor RW, Falke JJ. Biophys J. 2010;99:2879–2887. doi: 10.1016/j.bpj.2010.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Picas L, Suarez-Germa C, Teresa Montero M, Hernandez-Borrell J. J Phys Chem B. 2010;114:3543–3549. doi: 10.1021/jp910882e. [DOI] [PubMed] [Google Scholar]
- 106.Liu J, Conboy JC. Langmuir. 2005;21:9091–9097. doi: 10.1021/la051500e. [DOI] [PubMed] [Google Scholar]
- 107.Jackman JA, Choi JH, Zhdanov VP, Cho NJ. Langmuir. 2013;29:11375–11384. doi: 10.1021/la4017992. [DOI] [PubMed] [Google Scholar]
- 108.Yin J, Lin AJ, Golan DE, Walsh CT. Nat Protoc. 2006;1:280–285. doi: 10.1038/nprot.2006.43. [DOI] [PubMed] [Google Scholar]
- 109.Meijering E, Dzyubachyk O, Smal I. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- 110.Sbalzarini IF, Koumoutsakos P. J Struct Biol. 2005;151:182–195. doi: 10.1016/j.jsb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 111.Tarantino N, Tinevez JY, Crowell EF, Boisson B, Henriques R, Mhlanga M, Agou F, Israel A, Laplantine E. J Cell Biol. 2014;204:231–245. doi: 10.1083/jcb.201307172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ziemba BP, Falke JJ. Chem Phys Lipids. 2013;172-173:67–77. doi: 10.1016/j.chemphyslip.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schutz GJ, Schindler H, Schmidt T. Biophys J. 1997;73:1073–1080. doi: 10.1016/S0006-3495(97)78139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ziemba BP, Knight JD, Falke JJ. Biochemistry. 2012;51:1638–1647. doi: 10.1021/bi201743a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bosch PJ, Kanger JS, Subramaniam V. Biophys J. 2014;107:588–598. doi: 10.1016/j.bpj.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Van Veldhoven PP, Mannaerts GP. Anal Biochem. 1987;161:45–48. doi: 10.1016/0003-2697(87)90649-x. [DOI] [PubMed] [Google Scholar]
- 117.McConnachie G, Pass I, Walker SM, Downes CP. Biochem J. 2003;371:947–955. doi: 10.1042/BJ20021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stahelin RV. Biophys J. 2013;105:1–2. doi: 10.1016/j.bpj.2013.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Deems RA, Dennis EA. J Biol Chem. 1975;250:9008–9012. [PubMed] [Google Scholar]
- 120.Deems RA, Eaton BR, Dennis EA. J Biol Chem. 1975;250:9013–9020. [PubMed] [Google Scholar]
- 121.Hendrickson HS, Dennis EA. J Biol Chem. 1984;259:5740–5744. [PubMed] [Google Scholar]
- 122.Hendrickson HS, Dennis EA. J Biol Chem. 1984;259:5734–5739. [PubMed] [Google Scholar]