

Abstract
The Ca2+ channelopathies caused by mutations of the CACNA1A gene that encodes the pore-forming subunit of the human Cav2.1 (P/Q-type) voltage-gated Ca2+ channel include episodic ataxia type 2 (EA2). Although, in EA2 the emphasis has been on cerebellar dysfunction, patients also exhibit episodic, nonmotoric abnormalities involving the cerebral cortex. This study demonstrates episodic, low-frequency oscillations (LFOs) throughout the cerebral cortex of tottering (tg/tg) mice, a widely used model of EA2. Ranging between 0.035 and 0.11 Hz, the LFOs in tg/tg mice can spontaneously develop very high power, referred to as a high-power state. The LFOs in tg/tg mice are mediated in part by neuronal activity as tetrodotoxin decreases the oscillations and cortical neuron discharge contain the same low frequencies. The high-power state involves compensatory mechanisms because acutely decreasing P/Q-type Ca2+ channel function in either wild-type (WT) or tg/tg mice does not induce the high-power state. In contrast, blocking l-type Ca2+ channels, known to be upregulated in tg/tg mice, reduces the high-power state. Intriguingly, basal excitatory glutamatergic neurotransmission constrains the high-power state because blocking ionotropic or metabotropic glutamate receptors results in high-power LFOs in tg/tg but not WT mice. The high-power LFOs are decreased markedly by acetazolamide and 4-aminopyridine, the primary treatments for EA2, suggesting disease relevance. Together, these results demonstrate that the high-power LFOs in the tg/tg cerebral cortex represent a highly abnormal excitability state that may underlie noncerebellar symptoms that characterize CACNA1A mutations.
Keywords: calcium channelopathy, cerebral cortex, episodic ataxia type 2, episodic nervous system dysfunction, flavoprotein optical imaging, tottering mouse
Introduction
A hallmark of channelopathies is that a permanent alteration in a gene encoding an ion channel can lead to transient abnormalities in the excitability state of neural circuits and episodic neurological dysfunction (for reviews, see Kullmann, 2010; Ryan and Ptácek, 2010). One family of Ca2+ channelopathies is caused by mutations of the CACNA1A gene that encodes the α1A, pore-forming subunit of the Cav2.1 (P/Q-type) voltage-gated Ca2+ channel (Kramer et al., 1995; Ophoff et al., 1996). Non-CAG expansion mutations of the CACNA1A gene include episodic ataxia type 2 (EA2) and familial hemiplegic migraine type 1 (FHM1) (Baloh et al., 1997; Jen et al., 2004; Jen et al., 2007; Rajakulendran et al., 2010). Typically, EA2 involves a decrease and FMH1 an increase of Cav2.1 function (Tottene et al., 2002; Wappl et al., 2002; Jen et al., 2004; Spacey et al., 2004).
Motor dysfunction is a prominent feature of EA2 with episodes of cerebellar dysfunction that include limb and gait ataxia and oscillopsia (Rajakulendran et al., 2012). Interictally, patients may exhibit nystagmus and progressive cerebellar dysfunction (Baloh et al., 1997; Denier et al., 1999; Jen et al., 2007). The most widely studied model of EA2 is the tottering (tg/tg) mouse that has a recessive mutation in the pore-forming region of the P/Q-type Ca2+ channel gene, Cacna1a, ortholog of the human CACNA1A gene (Fletcher et al., 1996). Similarly, the tg/tg motor phenotype includes mild ataxia and a dramatic paroxysmal dystonia that are linked to cerebellar dysfunction (Campbell et al., 1999; Hoebeek et al., 2005; Walter et al., 2006; Neychev et al., 2008; Raike et al., 2013).
In addition to the motor symptoms, EA2 patients have a high incidence of migraine headaches and the associated transient visual aura, as well as somatosensory and motor disturbances (Jen et al., 2004). These patients also can exhibit epilepsy, cognitive impairment, abnormal EEG, and elevated cortical excitability (Jouvenceau et al., 2001; Helmich et al., 2010; Baloh, 2012; Rajakulendran et al., 2012). This complex set of findings is not unexpected because P/Q-type Ca2+ channels are distributed widely throughout the CNS, with high expression levels in the cerebral cortex, hippocampus, and cerebellum (Mintz et al., 1992; Westenbroek et al., 1995; Fletcher et al., 1996). Further, P/Q-type Ca2+ channels are central to neurotransmitter release (Catterall, 1998; Pietrobon, 2010). Similarly, tg/tg mice have noncerebellar dysfunction, including absence seizures (Noebels and Sidman, 1979), decreased glutamatergic and GABAergic signaling (Tehrani et al., 1997; Ayata et al., 2000), increased threshold for cortical spreading depression (CSD) (Ayata et al., 2000), and increased norepinephrine innervation (Levitt and Noebels, 1981). Except for absence seizures (Noebels and Sidman, 1979; Helekar and Noebels, 1991; Hewitt et al., 2011), few studies have investigated the excitability changes occurring in the cerebral cortex of tg/tg mice in vivo.
Previously, we described episodic, low-frequency oscillations (LFOs) in the cerebellar cortex of tg/tg mice that are coupled to the dystonic attacks (Chen et al., 2009). We hypothesized that similar oscillations may occur in the cerebral cortex. This study demonstrates episodic LFOs of very high power throughout the cerebral cortex of anesthetized tg/tg mice that represent a highly abnormal excitability state.
Materials and Methods
Animal preparation.
All of the animal studies were approved by and conducted in conformity with the Institutional Animal Care and Use Committee of the University of Minnesota. Male and female tg/tg mice on a C57BL/6 background and male and female C57BL/6 control mice (WT) were used in this study. Homozygous tg/tg mice were obtained by crossing a mouse line containing the tg allele and the semidominant allele Os, which causes oligosyndactalism. The first step in identifying tg/tg homozygotes was by the absence of oligosyndactalism at birth. Putative tg/tg homozygotes were then challenged with caffeine (15 mg/kg, i.p.) or psychological stress (i.e., placement in a novel environment) to verify the expression of episodic dystonia before experimentation (Fureman and Hess, 2005).
Detailed descriptions of the anesthetized mouse preparation for optical imaging of the cerebellar cortex have been described previously (Reinert et al., 2004; Gao et al., 2006). The approach used here is similar except for the exposure and imaging of the cerebral cortex instead of the cerebellar cortex. Therefore, the preparation is only described briefly. Adult mice 3–8 months of age were anesthetized by an initial intramuscular injection of 2.0 mg/kg acepromazine followed by an intraperitoneal injection of 2.0 mg/kg urethane and supplemented with 1.5 mg/kg urethane as needed. Animals were placed in a stereotaxic frame, mechanically ventilated, and their body temperature feedback regulated. Depth of anesthesia was monitored via electrocardiogram and testing for responses to somatosensory stimuli. A craniotomy (∼7.00 × 6.75 mm) was performed to expose the cerebral cortex and the dura carefully removed. A watertight acrylic chamber was constructed around the exposed cortex and filled with Ringer's solution gassed with 95% O2 and 5% CO2.
Optical imaging and neural activity acquisition and analysis.
The anesthetized animal in the stereotaxic frame was placed on an x–y stage mounted under a high-speed, cooled CCD macroscope (Nikon AZ-100) with a 1× (Fig. 12) or 2× (all other figures) objective with a 512 × 512 CCD chip at 16 bit digitization. Binning on the CCD chip was done to achieve a pixel resolution of ∼35 × 35 μm (1×) or ∼28 × 28 μm (2×).
Figure 12.

Cortical responses to direct electrical stimulation and sensory input are reduced in the tg/tg mouse. A, Example power maps of evoked response to air-puff stimulation of the left, C2 whisker in WT (left) and tg/tg (right) mice. The air-puff stimulus consisted of train of 5 pulses of 9 PSI, 120 ms at 5 Hz delivered at random intervals of 6–8 s. These maps were generated at the frequencies of the stimulation (0.125–0.167 Hz) and not at the LFO frequency. Note that a 1× objective was used, resulting in a larger imaged area compared with other figures. B, Population summary of the power of the cortical response evoked by whisker stimulation in the WT and tg/tg mouse (n = 4 each). C, Similar power maps of the response to direct cortical stimulation of the motor cortex using trains of 5 pulses of 200 μA, 100 μs at 5 Hz in the WT mouse (left) and the tg/tg mouse (right). Stimulation electrode is evident on left. D, Summary of the power evoked by cortical stimulation in the ipsilateral (i) cortex (i.e., the side with the stimulating electrode) and the contralateral (c) response in WT and tg/tg mice (n = 4 each).
Flavoprotein (autofluorescence) imaging was used to monitor activity over a large area of the cerebral cortex. Flavoproteins fluoresce when oxidized and the changes in fluorescence are highly correlated with neural activity (Chance et al., 1967; Shibuki et al., 2003; Reinert et al., 2004; Gao et al., 2006; Reinert et al., 2011). Importantly, flavoprotein fluorescence is not dependent on changes in cerebral blood flow (Shibuki et al., 2003; Reinert et al., 2004; Husson et al., 2007; Kitaura et al., 2007; Reinert et al., 2011). An imaging acquisition period consisted of a series of flavoprotein images (625 frames, 200 ms). In a few experiments, 1500 frames were acquired, as noted in the Results. Difference images were then generated by subtracting the average of nine control frames (control average) from each control and experimental frame. These difference images were then divided by the control average, yielding images in which the intensity of each pixel reflects the change in fluorescence (ΔF/F) relative to the control period (Reinert et al., 2004; Gao et al., 2006). Images of the ΔF/F were used to illustrate the spontaneous oscillations in tg/tg versus WT mice by selecting time points in the image series (see Fig. 1). At each time point, 10 frames (five frames before and five frames after the time point) were averaged. Resultant images were scaled to ± 5% ΔF/F for pseudocolor display using MetaMorph (Molecular Devices). In general, imaging sessions lasted 2–4 h.
Figure 1.

Spontaneous LFOs in the cerebral cortex of the tg/tg mouse. A, Background fluorescence image of the cerebral cortex with color-coded ROIs used to quantify the fluorescence change over time. The red star indicates the location of bregma in this and all subsequent figures. The pseudocolored images show the spontaneous change in flavoprotein fluorescence (ΔF/F) in the cerebral cortex of a tg/tg mouse at 2 s intervals over a 16 s period. Note the large cyclical fluctuations at this periodicity throughout the exposed cortex. B, Traces of the ΔF/F over time for the ROIs depicted in the background image in A demonstrate large-amplitude LFOs in both hemispheres. C, Background image and pseudocolored images of the spontaneous change in flavoprotein activity in a WT mouse. D, Traces of the ΔF/F over time for the ROIs depicted in the background image in C show only small-amplitude fluctuations at higher frequencies.
The spectral content of the optical data was obtained as described previously (Chen et al., 2009) and is therefore described only briefly. Images were low-pass filtered using a 13 × 13 pixel mean filter and then the optical signal for each pixel was linearly detrended and transformed into the frequency domain using a 1024-point fast Fourier transform (FFT) algorithm. The power spectrum was computed using a 50% Hanning window with 94% overlap with a final frequency resolution of 0.0049 Hz. The initial analysis quantified the global spectrum by averaging the power spectra at the pixel level across two visually defined regions of interest (ROIs) that included the exposed regions of the left and right cerebral hemispheres. The ROIs were chosen to exclude non-neuronal structures within the imaged field (i.e., the superior sagittal sinus, cranium, and acrylic chamber). This procedure was applied to each image acquisition period. As described in the Results, the average power spectrum from WT and tg/tg mice differed markedly between 0.035 and 0.11 Hz (see Fig. 2A), which was defined as the frequency band of interest. The average of the global spectrum across the frequency band of interest determined the global power of each acquisition period. Subsequent analyses of the optical signals focused on this frequency band. Also as described in the Results, tg/tg mice exhibited very high global power at these LFOs and values >3 SDs above the mean WT power were defined as a high-power state. In the population summaries of experimental conditions and genotype, the power within the ROIs with the frequency band of interest were averaged across multiple imaging periods.
Figure 2.
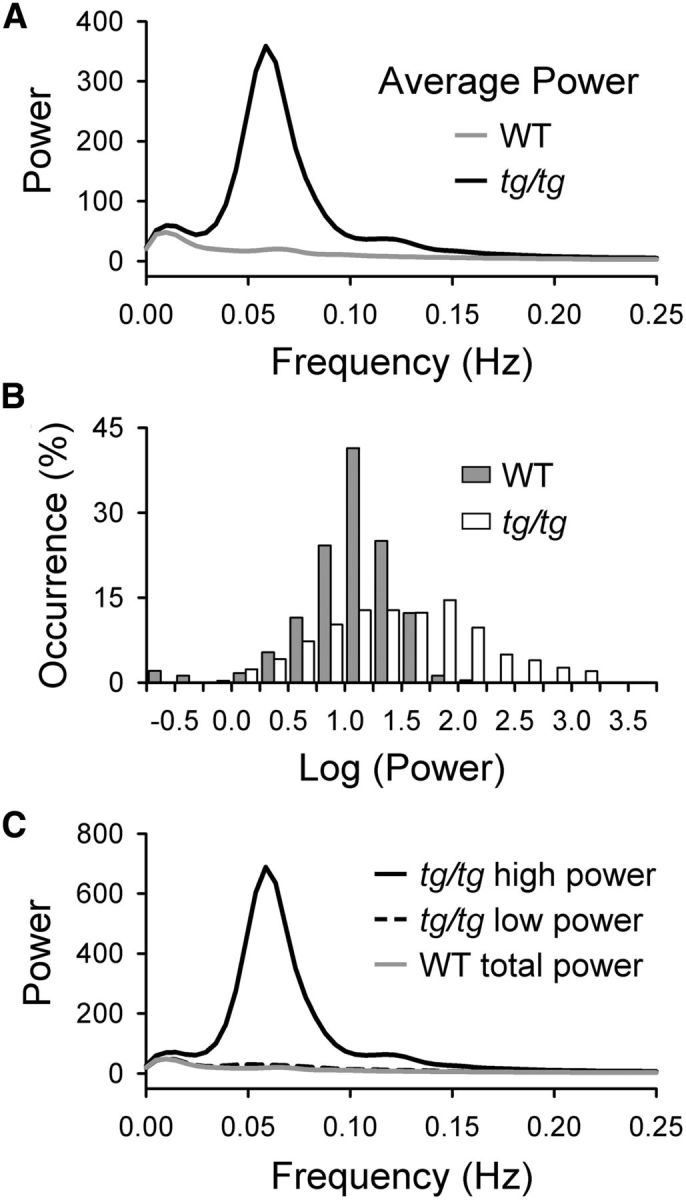
Transient, high-power LFOs in the tg/tg mouse cerebral cortex. A, Power versus frequency plot demonstrates the average power in the cerebral cortex of tg/tg (black) and WT mice (gray) obtained from 947 imaging periods in tg/tg mice (n = 34) and 308 imaging periods in WT mice (n = 20). Spectral analysis demonstrates that the greatest power in the tg/tg mouse cerebral cortex is within a frequency band of 0.035–0.11 Hz. Therefore, we focused on this frequency range for the remaining analysis. B, Occurrence rate (as a percentage of total number of imaging periods) is plotted versus the log10 of the average power histogram of 947 imaging periods in the tg/tg mouse (white bars) and 308 imaging periods in the WT mouse (gray bars). An imaging period was defined as high power if the average power was > 69.7, a level corresponding to the mean power exhibited by WT mice (gray trace, 16.3 ± 17.8) plus 3 SDs. C, Power versus frequency plots of the high-power state (black trace) and baseline (black dashes) averaged across all imaging periods from tg/tg mice. Average power versus frequency pots across all imaging periods from WT mice (gray trace).
Based on the spectral analysis and the definition of a high-power state, maps of the frequency, power, and phase shifts were generated as described previously (Chen et al., 2009). At each pixel, the frequency with the largest power was defined as the dominant frequency. For pixels in which the power exceeded the high-power threshold, the frequency maps show the dominant frequency superimposed on a background image of the cerebral cortex and power maps show the corresponding power at those pixels. The phase maps indicate the phases of the oscillations in the high-power state across the exposed cortex.
A regional analysis was undertaken to determine whether there are differences in the amplitude or propensity to develop high-power LFOs in different cortical regions. Four regions of equal area (∼1.25 mm2) were analyzed, including the primary motor (+0.84 mm, 1.54 mm relative to bregma), barrel (−1.85 mm, 3.00 mm), parietal association (−1.30 mm, 1.00 mm), and visual cortices (−3.5 mm, 1.90 mm; see Fig. 4A), based on previously defined stereotaxic coordinates (Lim et al., 2012).
Figure 4.

Regional occurrence of high-power states. A, Background image showing the location of the four ROIs on each hemisphere (see Materials and Methods). B, Average power of the four ROIs bilaterally when the cortex was in a high-power state (n = 19). C, Probability of a high-power state occurring in each ROI when the cortex was in a high-power state. M, Motor cortex; PTA, parietal association area; BC, barrel cortex; V, visual cortex.
The relationship between phase distribution and LFO power was investigated using unthresholded phase maps in which all phases within the ROIs used to determine the global power were included regardless of whether the power was above the high-power threshold. For the unthresholded phase maps, we computed the population vector as the mean of the unit vectors at each phase. The phase variability within a map was then characterized by the circular variance, Varc = 1 − R, where R is the magnitude of the population vector. The distribution of circular variance determined during high-power states was best fit by an exponential function, , where a and b are free model parameters and GP is the global power of the LFOs.
In some experiments, flavoprotein optical imaging was acquired simultaneously with extracellular single-unit neural activity. Single-unit extracellular recordings of cerebral cortical neurons used glass-coated, platinum iridium microelectrodes (1–2 MΩ; Alpha Omega) and conventional electrophysiological techniques (Haider et al., 2006; Chen et al., 2009; Cramer et al., 2013). Recordings were conducted throughout the extent of the cerebral cortical layers. The single-cell recordings were digitized at 32 kHz and stored online. Spikes were sorted offline using Spike2 (Cambridge Electronic Design) and periods of spontaneous activity time locked to the image acquisition were exported for analysis in MATLAB (The MathWorks). The spike trains were converted into instantaneous firing rate with 200 ms binning using fractional intervals, a method used to determine the instantaneous firing rate in equal bins based on the inverse of the interspike intervals (Chen et al., 2009; Popa et al., 2012). The mean firing rate for each spike train period was subtracted from the instantaneous firing rate. The spectral content was obtained using a 1024 point FFT transformation of the detrended instantaneous firing rates (Chen et al., 2009). The spectral content of the spontaneous flavoprotein response was determined within a 588 × 588 μm ROI visually aligned over the location of the single-unit recording (see examples in Fig. 5). The correlation coefficient (r) between the spectral content of the optical and single-unit activity signals, simultaneously acquired, was computed in the 0.035–0.11 Hz band using MATLAB. For the examples comparing the power between single-unit firing and the optical activity, the power was normalized due to the different nature of the two signals.
Figure 5.
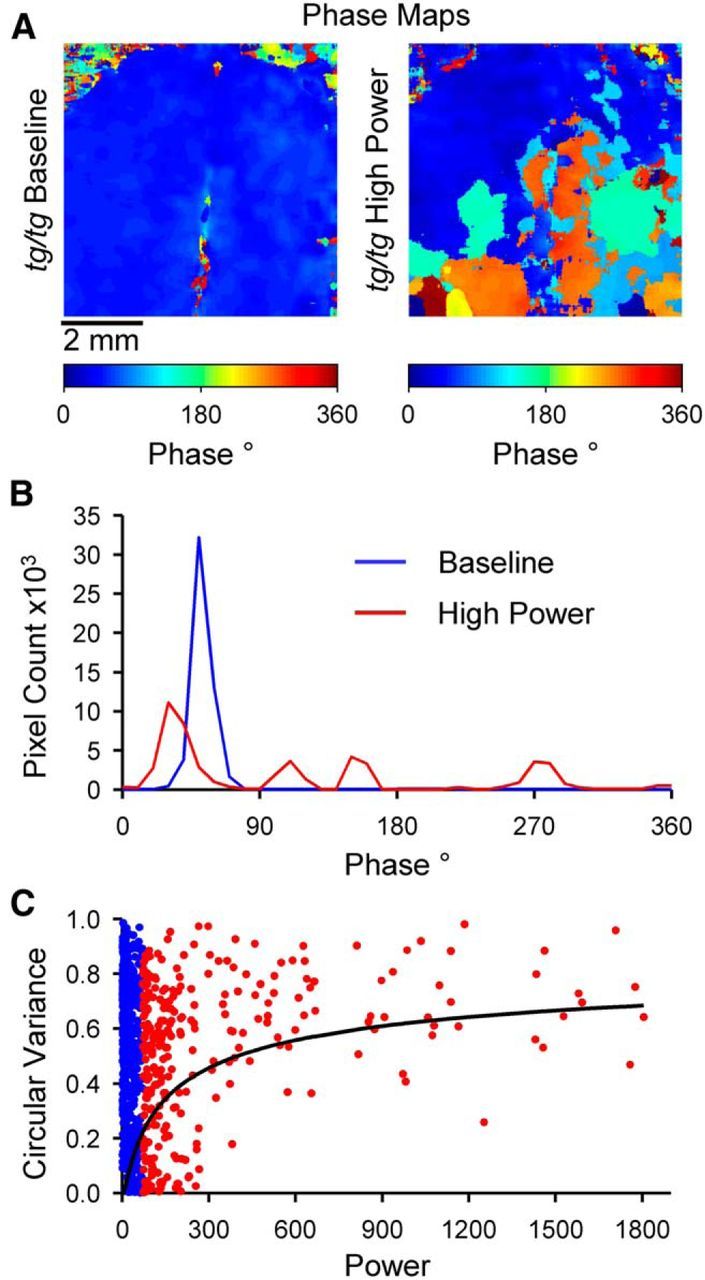
Decrease in phase synchrony during the high-power state. A, Example unthresholded phase maps from a tg/tg mouse in the baseline (left) and high-power (right) states. Note that these maps plot the phase at the dominant frequency at each pixel regardless of the power level. B, Phase distributions calculated from the example maps in A. C, Circular variance for baseline (blue) and high-power (red) states plotted against global power. Solid line represents the single exponential fit to global power in high-power state, given by the following: .
Additional analyses examined whether the high-power state altered the discharge properties of the cerebral cortical neurons in tg/tg mice. For each neuron, the mean and SD were determined from the instantaneous firing rate based on the fractional intervals method with 20 ms binning. Next, the coefficient of variation (CV), defined as the ratio between the SD and the mean firing rate, was determined as a measure of firing variability.
Optical imaging of responses to direct cortical and whisker stimulation.
In experiments examining cerebral cortical responses, direct cortical stimulation was delivered via a paralyene-coated tungsten microelectrode (2–5 MΩ; Frederick Haer) positioned ∼1 mm lateral and ∼1 mm rostral to bregma and lowered to a depth of ∼265 μm below the cortical surface. Stimulation parameters consisted of 5 pulses of 200 μA, 100 μs at 5 Hz. Peripheral whisker stimulation was applied using a pico-injection system (PLI-100; Medical Systems) to deliver a series of air puffs to the left C2 whisker. The whiskers on the left whisker pad were trimmed short except for the C2 whisker and air puffs were delivered perpendicular to the long axis of the C2 whisker to avoid deflecting neighboring whiskers (Bosman et al., 2011). The air-puff stimulus consisted of 5 pulses at 5 Hz (9 psi, 120 ms) applied to the C2 whisker, 1 cm from the whisker base (Devonshire et al., 2010). For both the intracortical and whisker stimulation, the experimental design was to avoid the LFO frequencies. After a 100 s baseline period of images was acquired, direct cortical or air-puff stimulation was delivered continuously at random intervals between 6 and 8 s in duration (program written in Spike2). During the 200 s imaging interval after the baseline period, 25–33 stimuli were delivered. The average (mean ± SD) number of stimuli applied per imaging period was 29 ± 1 for tg/tg mice and 28 ± 1 for WT mice. This interstimulus interval corresponds to a frequency of 0.125–0.167 Hz, outside of the frequency band of interest for the LFOs.
Analysis of the flavoprotein response to direct cortical or whisker stimulation consisted of determining the spectral content within a 700 × 700 μm ROI visually aligned over the cortical response region. For the response evoked by air-puff whisker stimulation, a single ROI was defined over the contralateral somatosensory cortex. For direct cortical stimulation, ROIs were defined over both the ipsilateral and contralateral response regions. The average power between 0.125 and 0.167 Hz (the range of the stimulus frequencies) was then computed for the ROI(s) per image acquisition period, similar to a previous report (Llano et al., 2009). To minimize the effects of the random oscillations, the magnitude of the response was defined as the difference between the average power in the stimulus band and the average power of six neighboring frequencies (three consecutive adjacent frequencies below and above the stimulus band).
Western blotting.
WT and tg/tg cerebral cortices were dissected and then homogenized in brain extraction buffer: 0.25 m Tris-HCl, pH 7.5, with protease inhibitors (1183617001; Roche). Protein concentration was determined with the Bradford method (Bradford regent and technique; Sigma-Aldrich) and 30 μg of neuronal nitric oxide synthase (nNOS) or 60 μg of endothelial nitric oxide synthase (eNOS) of total protein were denatured and run on a 4–15% gradient gel (Bio-Rad) and blotted onto mid-sized nitrocellulose membrane. Membranes were blocked for ∼1 h with 5% blocking buffer (5% w/v milk in 1× PBS) with 0.1% v/v Tween 20. Membranes were incubated with nNOS (ab1376; AbCam) or eNOS (NBP1–19824; Novus Biologicals) antibody overnight at 4°C, washed three times with 1× PBS with 0.1% Tween 20, and incubated with anti-goat (nNOS) or anti-rabbit (eNOS) horseradish peroxidase secondary antibody for 2 h. Samples were also probed with mouse anti-α-tubulin (T5168; Sigma-Aldrich) as a loading control. Images of the membranes were acquired with ImageQuant (General Electric) and protein levels quantified using ImageQuant densitometric analysis software.
Drugs and administration.
Caffeine and acetazolamide (5-acetamido-1,3,4-thiadiazole-2-sulfonamide) were purchased from Sigma-Aldrich. DNQX (6,7-dinitroquinoxaline-2,3-dione), APV (D-(-)-2-amino-5-phosphonopentanoic acid), l-NAME (NG-nitro-l-arginine methyl ester hydrochloride), indomethacin (1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indole), tetrodotoxin (octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethano-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentol), GABAzine (6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide), MPEP (2-methyl-6-(phenylethynyl)-pyridine hydrochloride), LY 367385 ((S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid), ω-agatoxin (ω-agatoxin TK), diltiazem ((2S-cis-)-3-(acetyloxy)-5-[2-(dimethylamino)ethyl]-2,3-dihydro-2-(4-methoxyphenyl)-1,5-benzothiazepin-4(5H)-one hydrochloride), and 4-AP (4-aminopyridine) were purchased from Tocris Bioscience. All drugs except caffeine were dissolved in normal Ringer's solution and applied to the surface of the exposed cerebral cortex by replacing the solution in the optical chamber. Caffeine was dissolved in normal saline and administered intraperitoneally.
Statistical analysis.
The statistical analysis was performed using SAS software (SAS Institute). The effects of various drugs and evoked cortical responses were statistically evaluated with ANOVA (within-subject design with repeated measures, p < 0.05). Statistical evaluation of protein expression levels between WT and tg/tg mice and the three measures of neuronal firing discharges were performed using a Student's t test (p < 0.05). In the text and figures, all values are reported as mean ± SD. When describing the results of an experiment, “n” refers to the number of animals used.
Results
Transient LFOs in the cerebral cortex of tg/tg mice
In the initial experiments, autofluorescence optical imaging was used to examine the baseline activity in the cerebral cortex of the anesthetized tg/tg mouse in vivo. During the course of these experiments, spontaneous and transient oscillations in the flavoprotein fluorescence were observed throughout the exposed cerebral cortex. As shown for an example experiment, the oscillations in the anesthetized tg/tg mouse consist of large-amplitude fluorescence changes (ΔF/F) across most of the exposed cerebral cortex (Fig. 1A,B). The time courses of the ΔF/F (Fig. 1B), based on four ROIs in the frontal and parietal cortices bilaterally (colored circles in Fig. 1A), reveal a periodicity of ∼16 s (∼0.0625 Hz), similar to the LFOs observed in the cerebellar cortex of the tg/tg mouse (Chen et al., 2009). The large-amplitude changes in flavoprotein fluorescence (± 5% ΔF/F) are consistent with extensive shifts in cortical neuronal activity (Reinert et al., 2004; Reinert et al., 2011; Cramer et al., 2013). In contrast, WT mice do not exhibit the large amplitude LFOs observed in tg/tg mice. As shown for a control animal (Fig. 1C,D), the spontaneous activity across the cerebral cortex consists of much smaller-amplitude fluctuations (± 0.7% ΔF/F) at higher frequencies (∼0.2–0.4 Hz).
Because the LFOs in the tg/tg mouse are episodic, the first step was to define quantitatively this abnormal state based on the frequencies and magnitude of the oscillations. A two-step spectral analysis of the autofluorescence signal was performed, similar to the approach used to quantify LFOs in the cerebellar cortex (Chen et al., 2009). In the first step, the power spectrum was determined for 947 imaging periods (each imaging period consisted of 125 s of consecutive images) of spontaneous activity in 34 tg/tg mice and in 308 sessions in 20 WT animals. Because the goal was to define statistically the occurrence of the episodic, large-amplitude LFOs observed in tg/tg mice (Fig. 1A,B), the analysis is based on the activity in both hemispheres. Comparison of the average power versus frequency plots across all periods and animals establishes that strong oscillations occur in a frequency band between 0.035 and 0.11 Hz in the tg/tg mouse, but not in the WT mouse (Fig. 2A). Therefore, we selected this range as the frequency band of interest.
In the second step, the occurrence rate of power in this frequency band was determined across all imaging periods. The power levels in tg/tg mice span three orders of magnitude, with a long tail extending into very-high-power levels (Fig. 2B, white bars). Imaging periods with these higher-power values correspond to the LFOs shown for the example tg/tg mouse in Figure 1, A and B, in which the average power between 0.035 and 0.11 Hz was 541 ± 754. In contrast, the power distribution in WT animals is confined to much lower values (Fig. 2B, gray bars), as shown for the example WT mouse in Figure 1, C and D, in which the average LFO power was 24 ± 17. Therefore, the distributions show the existence of two states, baseline and high power. We defined the high-power state as when the average power between 0.035 and 0.11 Hz exceeded the average power in the WT animal (16.3 ± 17.8) by 3 SDs (> 69.7). This threshold corresponds to a p-value < 0.003. A high-power state was observed in 21 of 34 tg/tg mice and in 311 of 947 (32.8%) imaging periods. Conversely, using this threshold, there was only a single instance of a high-power state in 1 of 20 WT animals and in only 1 of 308 imaging periods (0.3%). Based on this analysis, the power versus frequency plots show the marked differences in power between the high-power and baseline states in tg/tg mice, as well as between the high-power state in tg/tg mice versus average power in WT mice (Fig. 2C).
Using the low-frequency band of interest and the definition of a high-power state, spectral analysis was used to generate maps of the frequency, which were power and phase superimposed onto a background image of the cerebral cortex. In these maps, only pixels with power values greater than the threshold for a high-power state are shown. For a tg/tg mouse in the high-power state, maps of the spontaneous LFOs in the cerebral cortex (Fig. 3, top) show that the oscillation frequencies can be markedly uniform across the cortex; however, power typically varies greatly in different regions (see also Figs. 6,11,13). For the tg/tg mouse in Figure 3, a region of very high power is centered in the right hemisphere, ∼1 mm caudal to bregma and ∼1 mm lateral to the midline. The phase map shows regions with LFOs in phase both within and between the hemispheres. The phase map also illustrates the complex temporal relationships in the oscillations over the cerebral cortex. Conversely, in the example WT mouse (Fig. 3, bottom), there are only small regions within the visual cortex that exceed threshold for the high-power state. Note that because our definition of high-power state is based on the average power across the cerebral cortex, it does not imply that individual pixels or regions do not exceed the threshold in WT animals. The spectral maps illustrate the spatial extent and dynamic nature of the LFOs in the tg/tg mouse during the high-power state. The occurrence of high-power LFOs suggests that the cerebral cortical network is not stable in the tg/tg mouse and can enter an abnormal excitability state.
Figure 3.

Frequency, power, and phase maps of the spontaneous activity in the cerebral cortex of tg/tg and WT mice. Spectral analysis demonstrates the frequency (left), power (middle), and phase (right) maps for an example tg/tg mouse (top) and the WT mouse (bottom) superimposed on background fluorescence images of the cerebral cortex.
Figure 6.

TTX suppresses, but does not block, high-power LFOs in the tg/tg mouse. A, Example frequency, power, and phase maps of spontaneous, high-power cerebral cortical activity in the tg/tg mouse. B, Frequency, power, and phase maps of the same mouse after bath application of 10 μm TTX. C, Summary of the average baseline power in tg/tg mice (gray; for all figures, bars show mean ± SD and asterisk denotes significant difference at p < 0.05) and the resulting power in the presence of TTX (white, n = 5). D, Summary of the average baseline power (gray) in WT mice and upon TTX application (white, n = 4).
Figure 11.
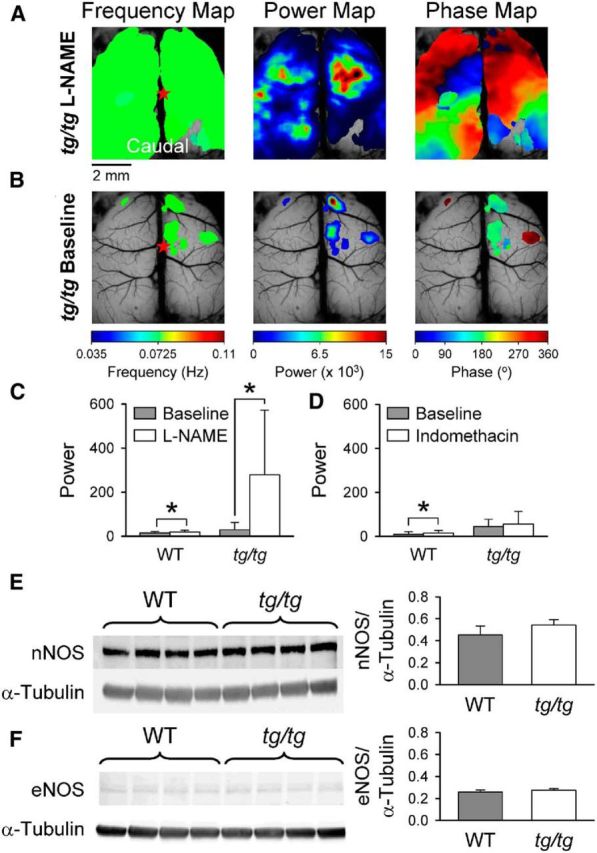
Blocking NO-synthase facilitates LFOs in the cerebral cortex. A, Frequency, power, and phase maps showing large regions of high power in a tg/tg mouse cerebral cortex upon bath application of 1 mm l-NAME, a nonspecific NO synthase antagonist. B, Example maps of the spontaneous activity in the same tg/tg mouse before NO synthase blockade. C, D, Summaries of the average power during the baseline period (gray) in WT and tg/tg mice in the presence (white) of either l-NAME or the cyclooxygenase inhibitor indomethacin (WT, n = 4 for both drug treatments; tg/tg, indomethacin n = 4, l-NAME n = 6). E, Western blot (left) of total cerebral cortical protein probed for neuronal NO-synthase and α-tubulin loading control in WT (n = 4) and tg/tg (n = 4) mice. Summary (right) of densitometric analysis of neuronal NO-synthase protein expression normalized to α-tubulin in WT (gray) and tg/tg (white) mice. F, Similar to E, Western blot (left) of endothelial NO-synthase protein expression in WT (n = 4) and tg/tg (n = 4) mice with α-tubulin control. Densitometric analysis (right) of endothelial NO-synthase expression in WT (gray) and tg/tg (white).
Figure 13.
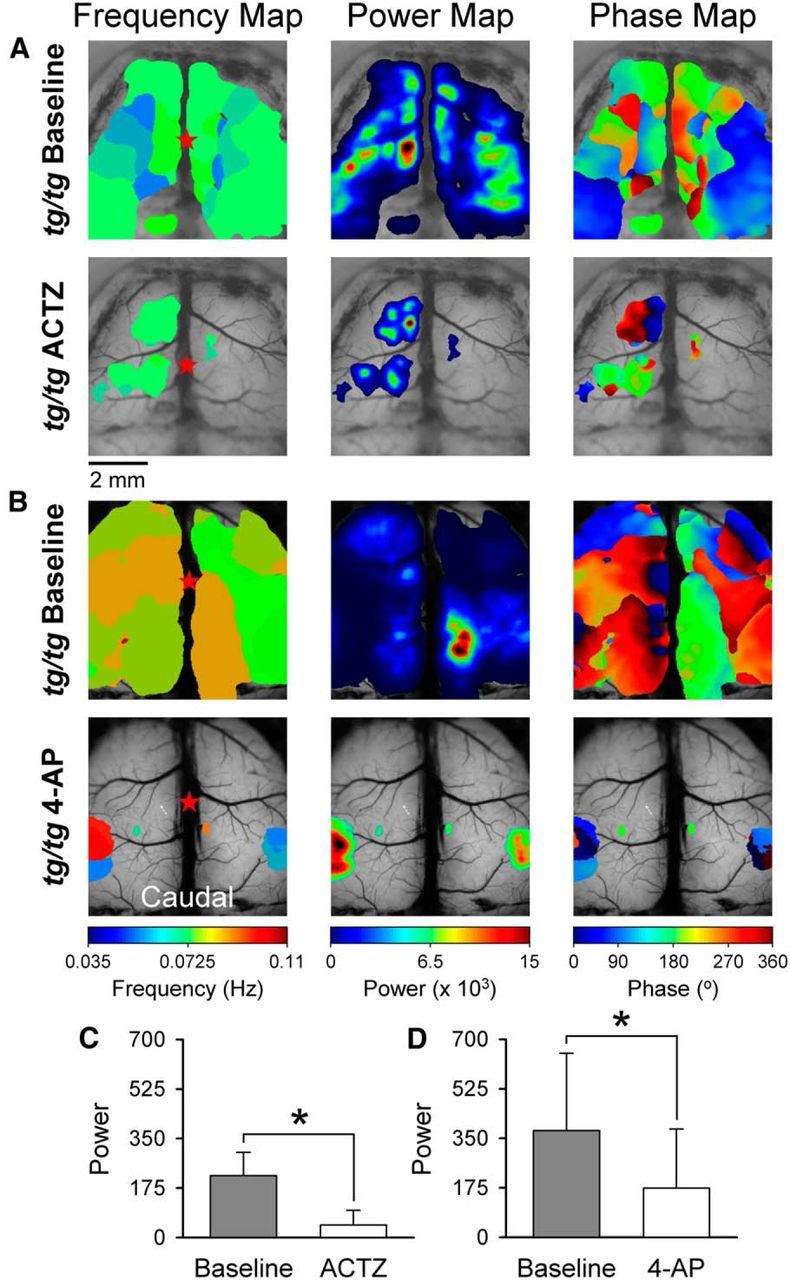
High-power LFOs are reduced by therapeutic agents used to treat EA2. A, B, Examples of the frequency, power, and phase maps demonstrate the high-power LFOs in the tg/tg mouse cerebral cortex in the baseline period (top row) and following (bottom row) application of either ACTZ (A) or 4-AP (B). C, D, Summaries of average baseline (gray bars) power and the average power upon drug application (white bars), either ACTZ (C, n = 4) or 4-AP (D, n = 6).
Regional analysis shows that high-power LFOs occur throughout the entire cerebral cortex in tg/tg mice. In 19 tg/tg mice over 286 imaging periods, the average LFO power in the primary motor, parietal association, barrel, and visual cortices (Fig. 4A) was determined for each hemisphere and then averaged. When in the global high-power state, the average LFO power in each region is well above the high-power threshold (Fig. 4B) and there are no differences in regional power (F(3,54) = 0.2). Furthermore, when in the high-power state, the probability that the LFO power in the regions exceeded the high-power threshold was quite high, ranging from 79 to 92% (Fig. 4C). These results demonstrate that when the high-power state occurs in a tg/tg mouse that most cortical regions are also oscillating at high power.
We also undertook an analysis of the phase distribution for the LFOs in the baseline versus high-power state in the tg/tg mice. Example phase maps from a tg/tg mouse depict the transition from the baseline state to the high-power state (Fig. 5A). Note that, in contrast to the other phase maps shown throughout the Results (Fig. 3), in these maps, the phase at the dominant frequency at each pixel is shown regardless of the power level. These maps show that the phases of the LFOs are less uniform in the high-power than in the baseline state, suggesting a decrease in synchrony with increasing LFO power. This impression is highlighted by the phase distribution plots, in which the phases for the baseline state are concentrated in a single, narrow band, and during the high-power state, in which the phases are spread among several smaller peaks (Fig. 5B). Circular variance was used to quantify the spread of phases in each phase map (see Materials and Methods) and was segregated into two populations: baseline and high-power states (Fig. 5C). The distribution of the circular variance in the baseline state uniformly covers all possible values (between 0 and 1) and shows no obvious trend. In contrast, a bias toward high circular variance was observed at very high global power levels. This distribution fits a single exponential function of the global power (see Materials and Methods, R2 = 0.086, F(1,285) = 22.7, p < 0.0001). Therefore, the results suggest that increasing power decreases the synchrony of the LFOs.
Neural contribution to LFOs in the cerebral cortex
We next evaluated the contribution of neuronal activity to the LFOs by bath application of tetrodotoxin (TTX, 10 μm) to block voltage-gated Na+ channels and to suppress action potentials (Kao, 1966). In tg/tg mice, TTX was applied only after a minimum of six contiguous imaging periods confirmed the presence of the high-power state in the cerebral cortex because we were specifically interested in the neural contribution to the high-power LFOs. Unless stated otherwise, a similar criterion was applied to all subsequent pharmacological experiments to establish the occurrence of the high-power state. As shown in Figure 6, TTX suppresses the high-power LFOs throughout much of the tg/tg cerebral cortex, with only discrete, smaller regions still exhibiting high-power oscillations. Across all experiments, TTX significantly decreased the LFO power by 40% (F(1,8) = 58.8, p < 0.0001; n = 5; Fig. 6C). However, TTX did not completely block the high-power state in tg/tg mice, suggesting that high-power LFOs are not solely dependent on action potential generation or intracortical connections. Application of TTX in WT mice reduces the power by ∼75% (F(1,6) = 174.9; p < 0.0001, n = 4; Fig. 6D) and demonstrates, as expected, that TTX greatly reduces excitability.
In the next experiments, we characterized the relationship between the LFOs and cerebral cortical neural activity in the tg/tg mouse. Extracellular single-unit recordings of the spontaneous discharge of neurons in the cerebral cortex were obtained simultaneously with flavoprotein imaging from seven tg/tg mice. For the single-unit data, we recorded all neurons encountered on tracts beginning at the cortical surface to 860 μm, a depth corresponding to cortical layer VI in the mouse (DeFelipe et al., 2002). In five of seven tg/tg mice, spectral analysis was performed on the simultaneously obtained optical and neuronal firing data for each imaging period (131 neurons in 278 imaging periods). The analysis was restricted to an ∼290 μm2 ROI aligned over the recording electrode (see example ROIs in Fig. 7). As shown for several example cell recordings, the neuronal firing contains frequencies within the 0.035–0.11 Hz band of interest and the normalized power spectrum closely mirrors the spectral content in the flavoprotein signal in the same region (Fig. 7C,D). The firing of other cortical neurons had less power at these low frequencies or did not match the power in the optical signal (Fig. 7B,E). To quantify the neural–optical relationship, the correlation was computed between the power spectrum of the optical and neural data within the 0.035–0.11 Hz band of interest. Although there is a large range of correlation coefficients (r-values), for the vast majority of neurons, the r-values are positive. Of the 278 recording periods, 148 (53%) exhibited r > 0.7 and the overall mean r-value was 0.54 ± 0.47 (critical value for significance at p ≤ 0.05 is r15 ≥ ± 0.48). Furthermore, neurons in all cortical layers exhibit LFO firing patterns with no obvious relationship between cortical recording depth and firing pattern. Therefore, the firing of the majority of cortical neurons in tg/tg mice oscillate at the same low frequencies observed in the optical recordings. The results from application of TTX and the single-unit recordings show that neuronal activity contributes to the LFOs.
Figure 7.

Power comparison between simultaneously acquired flavoprotein fluorescence and single-unit extracellular recordings in the tg/tg mouse. A, Background image of the cerebral cortex with colored ROIs used to compute the power in the optical signal (see Materials and Methods) shown in the example traces in B–E. B–E, Four examples of the normalized power for the optical (black) and the single-unit firing (red) and the computed r-value. F, Histogram of occurrence rate versus r-values for the comparison of the optical and single-unit power for each of the 278 imaging periods (125 s duration) acquired with single-unit recordings of 131 cerebral cortical neurons in five tg/tg mice.
The firing properties of 137 cortical neurons from all of the seven tg/tg mice were compared in relation to baseline and high-power states. The mean firing during the high-power state, 17.67 ± 12.37 spikes/s, is not significantly different from the mean firing during baseline, 14.17 ± 9.66 spikes/s (t135 = 1.67, p = 0.096). However, the SD increases from 29.82 ± 17.15 spikes/s during baseline to 41.62 ± 23.15 spikes/s during the high-power state (t135 = 3.10, p = 0.002). CV, a global measure of firing regularity, also increases from 2.25 ± 0.49 to 3.07 ± 1.73 (t135 = 4.43, p < 0.0001) during high-power states. Therefore, the high-power state is associated with increased firing variability.
Contribution of P/Q-type Ca2+ channels and synaptic transmission to the LFOs
Next, we evaluated whether a decrease in P/Q-type Ca2+ channel function underlies the high-power LFOs observed in the tg/tg mouse. Therefore, we tested whether acute reduction in P/Q-type Ca2+ current in the WT mouse with the specific P/Q-type Ca2+ channel blocker ω-agatoxin (1.5 μm) (Teramoto et al., 1993) would recapitulate the LFOs observed in the tg/tg mouse. Based on previous reports of the in vivo efficacy of 100 nm ω-agatoxin (Hoebeek et al., 2005), 1.5 μm likely blocks the vast majority of cortical P/Q-type channels. In WT animals, ω-agatoxin produces a significant increase in the power of LFOs from 17.2 ± 13.3 to 33.1 ± 32.3 (F(1,6) = 18.4, p < 0.0001, n = 4; Fig. 8C). As shown in the example spectral maps (Fig. 8A), there are regions within the cerebral cortex with increased power in the frequency band of interest; however, the global power levels did not enter the high-power state. In addition, we tested whether a further reduction in P/Q-type Ca2+ channel function in the tg/tg mouse results in the transition to a high-power state. Because we were specifically interested in the transition from baseline to a high-power state in this experiment, the ω-agatoxin was applied only after a minimum of six contiguous imaging periods had confirmed the presence of a baseline state. Both the example experiment (Fig. 8B) and the population results (Fig. 8D) show that ω-agatoxin does not result in high-power LFOs in the tg/tg mouse. In the presence of ω-agatoxin, the power of the LFOs (60.7 ± 49.6) is nearly identical to baseline power (60.2 ± 46.1) and not significantly different (F(1,6) = 0.01, p = 0.92, n = 4; Fig. 8D). Together, these two experiments suggest that a chronic reduction in the P/Q-type Ca2+ current favors the development of network instabilities in the cerebral cortex, as evidenced by the occurrence of the high-power state in the tg/tg mouse. However, because ω-agatoxin does not produce a high-power state in WT or tg/tg mice, the mechanisms underlying high-power LFOs are not due to an acute reduction in P/Q-type Ca2+ channel function and likely involve compensatory changes.
Figure 8.
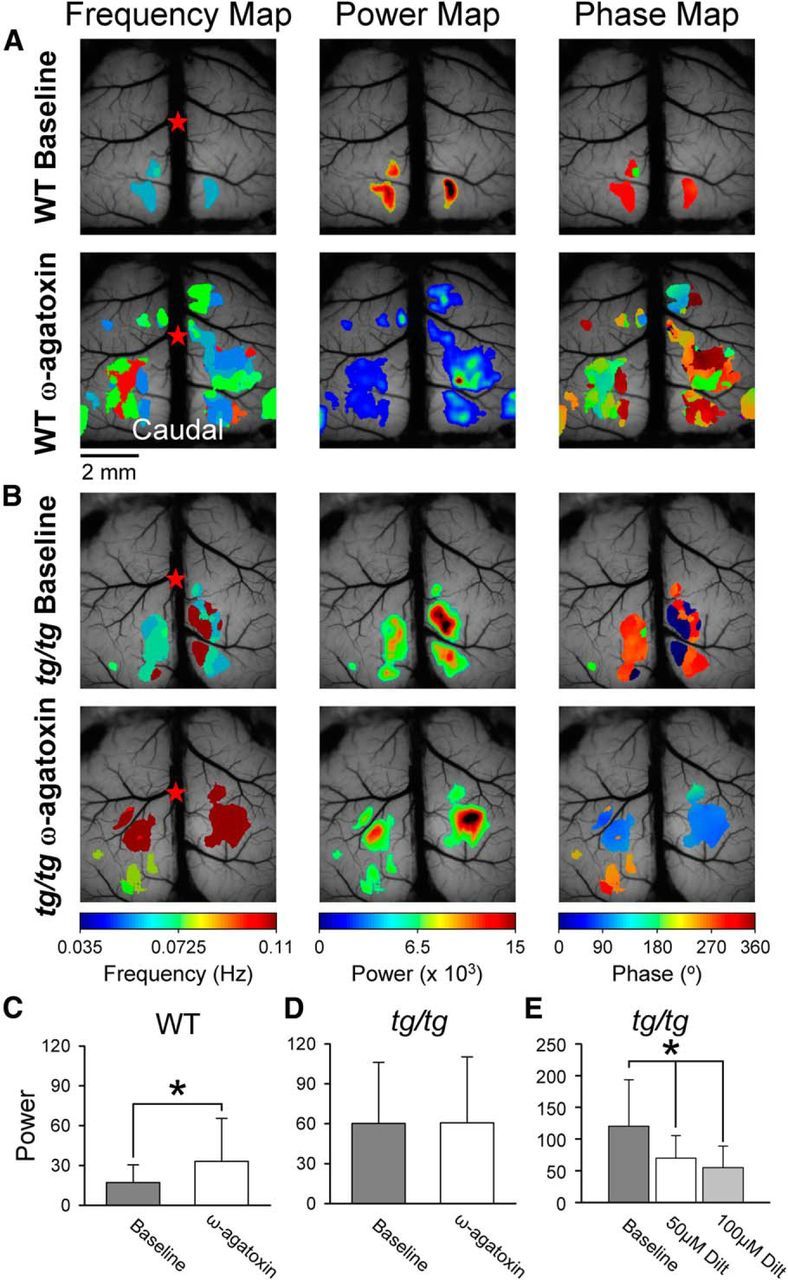
ω-Agatoxin blockade of P/Q-type Ca2+ channels increases LFO power in WT, but not in tg/tg mice. A, Example maps from a baseline imaging period (top row) in a WT mouse show small regions of high power that expand upon application of 1.5 μm ω-agatoxin. B, Example maps for the same experiment in a tg/tg mouse. ω-Agatoxin application does not alter the power of the spontaneous activity in the tg/tg mouse cerebral cortex. C, D, Average power in the baseline period (gray) and upon application of ω-agatoxin (white) in the WT mouse (C, n = 4) and the tg/tg mouse (D, n = 4). E, Average power for tg/tg mice (n = 3) in the high-power state and upon application of 50 μm (white) and 100 μm (light gray) diltiazem.
To further examine for compensatory changes that contribute to the high-power LFOs, the next experiment evaluated the involvement of l-type voltage-gated Ca2+ channels. In tg/tg mice, l-type Ca2+ channel expression is increased and this increased expression is involved in the motor and cerebellar abnormalities (Campbell and Hess, 1999; Chen et al., 2009). Blocking l-type Ca2+ channels with diltiazem (50 or 100 μm) in the high-power state significantly decreases LFO power (F(1,2) = 21.5, p < 0.0001 followed by Bonferroni post hoc testing at p < 0.05; Fig. 8E). The higher concentration of diltiazem terminates the high-power state in each animal tested (n = 3). Therefore, increased expression of l-type Ca2+ channels plays a role in producing high-power LFOs in both the cerebellum and cerebral cortex.
The next series of experiments assessed whether the instability in the tg/tg cerebral cortex is unmasked by altering the balance of excitation or inhibition. As noted in the Introduction, abnormalities in both glutamate and GABA neurotransmission are present in the tg/tg mouse cerebral cortex (Tehrani and Barnes, 1995; Tehrani et al., 1997; Ayata et al., 2000). Given that the majority of fast excitatory inputs, either via thalamocortical or corticocortical projections, are mediated by postsynaptic ionotropic glutamate receptors (AMPA and NMDA) (Sherman and Guillery, 2011), we bath applied 100 μm DNQX and 200 μm APV to block fast glutamatergic neurotransmission. As for the experiments blocking P/Q-type Ca2+ channels, we applied the antagonists in the baseline state. In WT mice, DNQX and APV result in a significant reduction in the low-frequency power (see example maps in Fig. 9B), decreasing baseline power from 22.0 ± 23.0 to 13.0 ± 20.3 (F(1,6) = 10.2, p = 0.0023, n = 4; Fig. 10A). Surprisingly, in the tg/tg mouse, DNQX/APV produces a significant increase in power from 11.2 ± 13.8 to 61.2 ± 117.0 (F(1,18) = 177.4, p < 0.0001, n = 10; Figs. 9A,10B) and resulted in a high-power state in 3 of 10 tg/tg mice. These findings suggest that a reduction in postsynaptic, ionotropic glutamatergic activation contributes to the LFOs and increases the likelihood of a high-power state.
Figure 9.
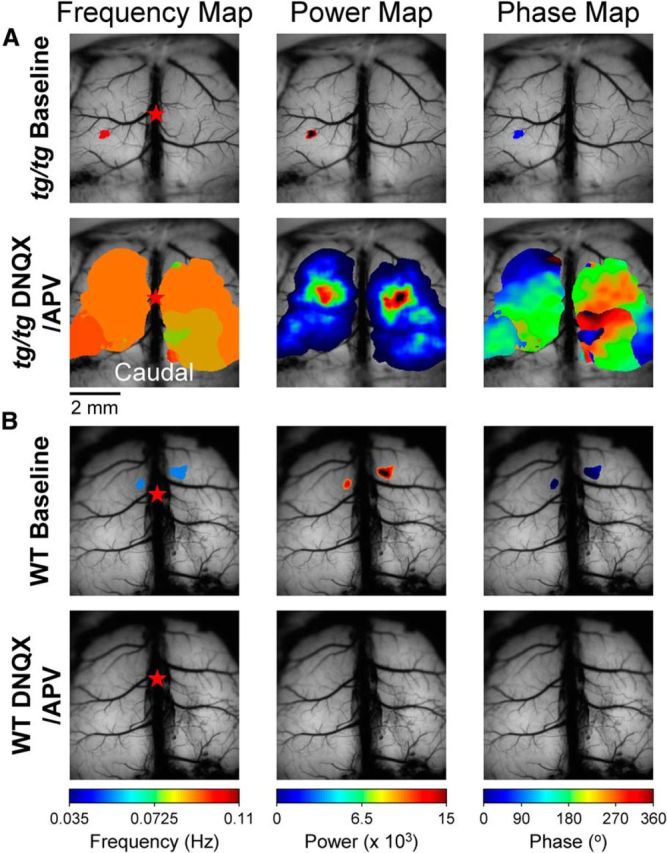
DNQX/APV application increases the high-power domains in the cortex of tg/tg mice, but not WT mice. A, Spectral analysis demonstrates a large increase in high-power domains upon application of DNQX and APV (100 and 200 μm, respectively) compared with baseline activity (top row) in the tg/tg mouse. B, Baseline power (top row) is suppressed by DNQX/APV application (bottom row) in the WT mouse.
Figure 10.

Blocking ionotropic and mGluRs facilitates high-power LFOs in the tg/tg mouse. A, Bath application of DNQX/APV (white) to the cerebral cortex causes a significant decrease in the power compared with the baseline (gray) state in WT mice (n = 4). B, Power is significantly increased from baseline (gray) in the presence of DNQX/APV (white) in the tg/tg mouse (n = 10). C, Bath application of the mGluR type 5 antagonist MPEP (30 μm) and the mGluR type 1 antagonist LY367385 (50 μm) results in a significant decrease in power (white) compared with baseline (gray) in WT mice (n = 4). D, In contrast, blockade of mGluRs in tg/tg mice results in a significant elevation of the power (white) compared with baseline (gray, n = 4).
In the cerebral cortex, excitatory glutamatergic synaptic transmission also involves metabotropic glutamate receptors (mGluRs), mainly types 1 and 5 (Alexander and Godwin, 2006; Ferraguti and Shigemoto, 2006). Furthermore, given the slower time of action of these receptors on postsynaptic neurons, one could hypothesize a role in the LFOs. Therefore, we also tested the effect of blocking mGluRs by applying LY36738 (50 μm), a mGluR type 1 antagonist, and MPEP (30 μm), a mGluR type 5 antagonist, into the optical chamber. This mixture of LY367385 and MPEP reliably blocks mGluR activity in the cerebral cortex of mice (De Pasquale and Sherman, 2012) and significantly increases power in the LFOs in tg/tg mice (39.4 ± 22.6 to 115.2 ± 98.7, F(1,6) = 52.5, p < 0.0001, n = 4; Fig. 10D). Furthermore, LY367385 and MPEP resulted in the high-power state in three of four tg/tg mice. In contrast, inhibition of mGluRs in WT mice significantly reduces the power of spontaneous cerebral cortical activity from 21.0 ± 13.5 to 15.3 ± 8.5 (F(1,6) = 12.7, p = 0.0008, n = 4; Fig. 10C). Similar to blocking ionotropic glutamate receptors, although more effective, reducing mGluR activation increases the likelihood of entering a high-power state.
Given the importance of cerebral cortical inhibitory interneurons in neuronal oscillations (Wang, 2010; Buzsáki and Wang, 2012) and in controlling excitability and seizure production (Dichter and Ayala, 1987; Chagnac-Amitai and Connors, 1989), and because GABAergic transmission is impaired in tg/tg mice (Tehrani and Barnes, 1995; Sasaki et al., 2006), one might expect that that inhibitory networks play a role in the LFOs. Therefore, we tested whether suppression of GABAAergic transmission in the tg/tg cortex facilitates a transition to the high-power state. Application of the GABAA receptor antagonist GABAzine (1 μm) does not change the baseline LFO power (9.6 ± 7.3 to 7.2 ± 5.4, F(1,8) = 5.57; p = 0.0211; n = 5, data not shown). The results based on blocking glutamatergic and GABAergic receptors show that high-power LFOs are not due to an increase in excitability. Instead, the LFOs are facilitated by a reduction in excitatory synaptic transmission.
Contribution of nitric oxide signaling to cerebral cortical LFO activity
Given the high incidence of migraine in patients with CACNA1A mutations (Jen et al., 2004) and reports of altered nitric oxide (NO) signaling in the cerebellum of the tg/tg mouse (Rhyu et al., 2003; Frank-Cannon et al., 2007), we tested whether blood flow changes contribute to the LFOs. Two prominent pathways exist in the cerebral cortex to regulate neurovascular coupling: the nNOS pathway, which produces NO in response to neural activation, and the cyclooxygenase (COX) pathway, which produces multiple signaling molecules, including prostaglandins, in response to astrocyte activation (Attwell et al., 2010).
To parse their relative contributions to the LFOs, we separately blocked the nNOS and prostaglandin pathways, applying the blockers in the baseline state. l-NAME (1 mm) was applied to inhibit nNOS-mediated production of NO (Moore and Handy, 1997). Unexpectedly, blocking nNOS in tg/tg mice results in a dramatic increase in the LFOs (Fig. 11A). The spectral maps from an example experiment show high-power LFOs across most of the cerebral cortex, as well as large regions of very high power (Fig. 11A) similar to that observed spontaneously (Fig. 1). On average, l-NAME increased LFO power 10-fold in tg/tg mice (F(1,10) = 41.1, p < 0.0001; n = 6; Fig. 11C) and produced a high-power state in 6 of 6 tg/tg mice tested. In contrast, application of l-NAME in WT mice resulted in only a modest increase in baseline power (F(1,6) = 4.15, p = 0.0465; n = 4; Fig. 11C) and never resulted in a high-power state.
The observation that l-NAME evokes LFOs in the cerebral cortex of the tg/tg mouse, coupled with the demonstration that nNOS expression is elevated in the cerebellum of the tg/tg mouse (Rhyu et al., 2003; Frank-Cannon et al., 2007), led us to examine the protein expression levels of nNOS and eNOS in the cerebral cortices of tg/tg and WT mice. We did not examine cerebral cortical expression of inducible NOS because previous studies have demonstrated very low expression levels in the absence of acute pathological insult (Calabrese et al., 2007). Densitometric analysis of nNOS protein levels normalized to the α-tubulin control shows that protein expression is unchanged in the cerebral cortex of the tg/tg mouse versus WT mice (t6 = −1.85, p = 0.11, n = 4 each of WT and tg/tg; Fig. 11E). Similar to the nNOS results, Western blot analysis of eNOS levels in the cerebral cortex shows no difference between tg/tg and WT mice (t6 = −1.35, p = 0.23, n = 4 each; Fig. 11F). Although this analysis would not detect a transient increase in nNOS expression or NO production, there is no evidence for a basal change in nNOS levels that could account for the effect of l-NAME on the LFOs.
In addition, bath application of indomethacin (100 μm) to inhibit COX-mediated production of prostaglandins (Vane, 1971; Faraci, 1992) does not result in the transition from the baseline to the high-power state in the tg/tg cerebral cortex. Indomethacin results in a modest, yet significant, increase in LFO power (F(1,6) = 4.4, p = 0.0403; n = 4; Fig. 11D). Similarly, indomethacin in WT mice does not result in a high-power state, producing a very modest, yet significant, increase in LFO power from 8.9 ± 12.2 to 15.0 ± 11.9 (F(1,6) = 7.3, p = 0.0091; n = 4; Fig. 11D). It should be noted that 10 μm indomethacin perfused over the cerebral cortex produces a marked reduction in the increased blood flow evoked by somatosensory stimuli (Kitaura et al., 2007), supporting the effectiveness in the use of 100 μm indomethacin in this study. Together, these results suggest that the NO signaling plays a major role in the transition from baseline to high-power state in the tg/tg mouse; in contrast, the prostaglandin pathway does not. Because neither nNOS nor eNOS expression differs between the tg/tg and WT mice, the effect of l-NAME in tg/tg mice in generating abnormal LFOs is not due to the disruption of a high basal level of NO production.
Abnormal cerebral cortical responses to sensory input and direct cortical stimulation in tg/tg mice
Given the highly abnormal activity in the cerebral cortex of the tg/tg mouse, the next experiments tested whether the responses to sensory stimuli or direct cortical stimulation in tg/tg mice differ from WT mice. The C2 whisker was stimulated using a 5 Hz train at random intervals between 6 and 8 s to avoid the frequency band of the LFOs (see Materials and Methods). The response amplitude was defined as the power evoked in the stimulus frequency range (0.125–0.167 Hz) (Llano et al., 2009). In both WT and tg/tg mice, air puffs delivered to the C2 whisker evokes a patch of activity in the contralateral barrel cortex (Fig. 12A). The response in tg/tg mice is reduced relative to WT (F(1,6) = 7.0, p = 0.0089, n = 4 each for WT and tg/tg; Fig. 12B) and demonstrates a deficit in the processing of sensory inputs into the somatosensory cortex.
The reduction in the response to C2 whisker puff could have occurred outside of the cerebral cortex. Therefore, we investigated direct intracortical stimulation in the motor cortex using the same stimulation paradigm used for the whiskers. The ipsilateral and contralateral responses to intracortical stimulation are lower in the tg/tg compared with the WT mice, as shown in the example power maps (Fig. 12C). The tg/tg population data confirm the significant reduction in the responses, both ipsilaterally (F(1,6) = 24.0, p < 0.0001, n = 4) and contralaterally (F(1,6) = 8.7, p = 0.0037, n = 4), compared with WT mice (Fig. 12D). Therefore, the responses to both sensory input and direct cortical simulation are reduced in the tg/tg mouse cerebral cortex and the latter result demonstrates a deficit in information processing that is intrinsic to the cerebral cortex.
LFOs are decreased by therapeutic agents used to treat EA2
We hypothesized that, if the LFOs in the cerebral cortex are important in the neurological abnormalities in patients with CACNA1A mutations, then the agents effective in treating EA2, acetazolamide (ACTZ) and 4-aminopyridine (4-AP), should reduce the LFO activity in tg/tg mice. As shown in the example experiment, bath application of the carbonic anhydrase inhibitor ACTZ (4 mm) nearly eliminates the LFO activity in a high-power state to within the baseline range (Fig. 13A,C). On average, ACTZ significantly reduced the LFO power, from 218.7 ± 82.0 to 44.0 ± 51.9 (F(1,6) = 173.9, p < 0.0001, n = 4; Fig. 13C), and converted the high-power state to a baseline state in all tg/tg mice tested. Similarly, as shown in an example experiment (Fig. 13B) and the population data, bath application of 500 μm 4-AP, a K+ channel antagonist, during the high-power state also greatly reduces the power of the LFOs from 377.2 ± 273.0 to 173.7 ± 208.6 (F(1,10) = 57.0, p < 0.0001, n = 6; Fig. 13D), although not quite as effectively as ACTZ. Therefore, the high-power LFOs that characterize the tg/tg mice are markedly decreased by the two common EA2 therapies.
Discussion
Cerebral cortical oscillations represent an abnormal excitability state
This study is the first report of episodic and highly abnormal LFOs in the cerebral cortex of tg/tg mice. The oscillations are at the same frequencies observed previously in the cerebellar cortex (Chen et al., 2009), highlighting that the tg/tg mutation results in similar instabilities in widely different circuits within the CNS. Several observations demonstrate that the LFOs represent pathological CNS activity. The high-power state is rarely observed in WT mice and, when present in tg/tg mice, high-power LFOs are found throughout most of the cerebral cortex. The high-power state is associated with increased firing variability and decreased LFO synchrony. Furthermore, the responses to several pharmacological agents are strikingly aberrant in tg/tg mice. For example, blocking ionotropic or mGluRs reduces LFO power in WT, but increases LFO power in tg/tg mice. Inhibition of NOS results in high-power LFOs in the tg/tg cerebral cortex, but not in WT mice. In WT controls, TTX virtually eliminates LFO power because blocking action potentials greatly suppresses neuronal activity in normal animals, including sensory input (Sachidhanandam et al., 2013) and corticocortical connections (Khalilov et al., 2003; Chen et al., 2013). Although TTX greatly reduces the LFOs in tg/tg mice, the high-power state was not eliminated, suggesting that local cellular, action-potential-independent mechanisms can support the oscillations, as observed in the cerebellar cortex (Chen et al., 2009). Overall, the LFOs reflect a highly abnormal excitability state in the cerebral cortex of tg/tg mice.
Role of synaptic transmission in the LFOs
The high-power LFOs in the tg/tg mouse are not solely due to an acute decrease in P/Q-type Ca2+ channel function. In both WT and tg/tg mice, acutely blocking P/Q-type Ca2+ channels with ω-agatoxin does not result in high-power LFOs. This likely reflects changes in the makeup of voltage-gated Ca2+ channels at axon terminals such that additional antagonism of P/Q-type Ca2+ channels does lead to high-power LFOs (Campbell and Hess, 1999; Qian and Noebels, 2000; Zhou et al., 2003; Pardo et al., 2006; Kaja et al., 2007). One change in voltage-gated Ca2+ channels that plays an important role in tg/tg pathophysiology is the upregulation of l-type Ca2+ channels (Campbell and Hess, 1999). Here, we show that blocking l-type Ca2+ channels suppresses the LFOs in the cerebral cortex. l-type Ca2+ channel antagonists reduce the episodic dystonia (Campbell and Hess, 1999) and the LFOs in the cerebellum (Chen et al., 2009). Conversely, l-type Ca2+ channel agonists increase the LFOs in the cerebellum and the motor attacks (Campbell and Hess, 1999; Chen et al., 2009). These findings, in combination with the observation that the episodic dystonia does not appear before ∼21 d of age in the tg/tg mouse (Green and Sidman, 1962; Meier and MacPike, 1971; Seyfried and Glaser, 1985), suggest that developmental/compensatory mechanisms occur that bias the cerebral cortex to generate high-power LFOs.
Although there is compensation for the reduction in P/Q-type Ca2+ channel function in tg/tg mice, synaptic transmission is still abnormal. Reductions in glutamatergic synaptic transmission occurs in the cerebellum (Matsushita et al., 2002; Chen et al., 2009), thalamus (Caddick et al., 1999), and cerebral cortex (Ayata et al., 2000; Sasaki et al., 2006). Consistent with previous in vitro demonstrations, the present in vivo results show reduced cerebral cortical responses to both somatosensory inputs and to intracortical stimulation. We hypothesize that this reduction in glutamatergic synaptic transmission in tg/tg mice produces instability in the cerebral cortical circuitry and that further compromise of glutamatergic synaptic transmission increases the instability. In support of this view, LFO power and the probability of entering the high-power state increases with blocking ionotropic or mGluRs. These observations suggest that excitatory synaptic transmission constrains the cerebral cortical network from entering the high-power state in tg/tg mice.
As stated in the Introduction, a developmental change in tg/tg mice is increased norepinephrine (NE) innervation (Levitt and Noebels, 1981). Previous reports have shown little effect of increased NE innervation on the episodic dystonia (Noebels, 1984; Campbell et al., 1999) and the effects of manipulating NE signaling on the motor phenotype are complex (Fureman and Hess, 2005). Interestingly, NO facilitates NE reuptake (Simaan and Sabra, 2011). Blocking NO production, which produces the high-power state in tg/tg mice, is likely to increase NE activity in the cerebral cortex. Further studies are needed to determine the effects of NE activity on the LFOs in tg/tg mice.
Role of NO signaling in the LFOs
NO is an additional factor involved in the cerebral cortical LFOs, because l-NAME-mediated inhibition of NOS reliably produces the high-power state in tg/tg, but not in WT mice. The contribution of NO signaling likely goes beyond modulation of cerebral blood flow because blocking COX-mediated cerebral blood flow regulation does not produce high-power LFOs. NO signaling influences multiple pathways, resulting in many downstream effects, including modulating neurotransmission, transcription, and vascular tone (Calabrese et al., 2007). Despite similar levels of nNOS and eNOS expression in the tg/tg and WT mice, there is a fundamental difference in the cerebral cortical response to NOS blockade between the WT and tg/tg mice. Because a reduction in basal NO production reliably increases LFO activity in tg/tg mice, downstream signaling likely serves to dampen the aberrant cerebral cortical activity that occurs during the high-power state. Although there is not a consensus among studies, it is notable that NO modulates the activity of voltage-gated Ca2+ channels, including facilitating channel activity (Chen and Schofield, 1995; Ohkuma et al., 1998; but see Yoshimura et al., 2001). In the tg/tg mouse, NO may facilitate voltage-gated Ca2+ channel conductance and help compensate for the reduced activity of the mutant P/Q-type Ca2+ channels and impaired synaptic transmission.
Role for LFOs in episodic cortical dysfunction
The transient nature of the LFOs in the tg/tg cerebellar and cerebral cortices emphasizes the multifaceted, episodic dysfunction characteristic of the P/Q-type Ca2+ channelopathies (Rajakulendran et al., 2012). In human CACNA1A diseases, cerebral cortical symptoms, including migraine and epilepsy, are common, as are sensory motor and cognitive impairments (Van Bogaert and Szliwowski, 1996; Baloh et al., 1997; Jouvenceau et al., 2001; Jen et al., 2004; Rajakulendran et al., 2012; Nachbauer et al., 2014). These diverse findings are thought to reflect abnormalities in either neurovascular coupling (migraine) or abnormal synaptic transmission (epilepsy). The LFOs in tg/tg mice provide a possible mechanism for transient cerebral cortical dysfunction and the reduction of LFOs by ACTZ and 4-AP argues for a connection between the oscillations in tg/tg mice and the human P/Q-type Ca2+ channelopathies. In EA2 patients, ACTZ reduces the frequency and severity of ataxic bouts (Griggs et al., 1978) and ameliorates noncerebellar features, including migraine attacks and abnormal EEG activity (Zasorin et al., 1983; Neufeld et al., 1996). Similar to ACTZ, 4-AP effectively decreases attack frequency and severity, interictal ataxia, and migraine occurrence in EA2 patients (Strupp et al., 2004; Löhle et al., 2008; Jung et al., 2010).
The relationship between the LFOs and CSD is also of interest. The threshold for evoking CSD is increased (Ayata et al., 2000) and decreased (Tottene et al., 2009; van den Maagdenberg et al., 2010; Gao et al., 2012) in tg/tg and FHM1 mice, respectively. Although the high-power LFOs may appear to contradict the increased threshold for CSD, we speculate that the two are linked. The present results show that the LFOs are triggered by a decrease in excitatory neurotransmission and the increased threshold for CSD in the tg/tg mouse is likely due to the decrease in glutamate release and reduced excitatory synaptic transmission (Ayata et al., 2000; Qian and Noebels, 2000). Therefore, a common deficit may underlie both the high-power LFOs and the decrease in CSD.
Although the mutations in EA2 and FHM1 have opposite effects on P/Q-type Ca2+ channel function, both result in ataxia and epilepsy in patients and mouse models (Tottene et al., 2005; Walter et al., 2006; Gao et al., 2012). In both tg/tg and the S218L mouse model of FHM1, the ataxia has been attributed to irregular Purkinje cell firing (Hoebeek et al., 2005; Walter et al., 2006; Gao et al., 2012). Intriguingly, Ca2+-dependent K+ channel activators counteract abnormal neuronal firing and improve motor performance in both disorders, suggesting a possible general therapeutic approach. It will be important to determine whether the cerebral cortical and cerebellar LFOs are also suppressed by Ca2+-dependent K+ channel activators.
In conclusion, abnormal synaptic transmission is central to the episodic LFOs in the cerebral cortex of the tg/tg mouse. This conclusion is based on the observations that decreased glutamatergic synaptic transmission and NO synthesis facilitate high-power LFOs. The conclusion is reinforced by the findings that LFOs are reduced by 4-AP, an agent that enhances intracellular Ca2+ influx and neurotransmitter release (Smith et al., 2000), as well as by ACTZ, a drug with multiple effects, including the enhancement of NO production (Aamand et al., 2009). The highly abnormal excitability state represented by the high-power LFOs in the cerebral cortex is a novel finding and offers a potential mechanism for the noncerebellar dysfunction in P/Q-type Ca2+ channelopathies.
Footnotes
This work was supported in part by the National Institutes of Health (Grants R01 NS18338, F31 NS080338, P30 NS062158, T32 GM008244, and T32 GM008471). We thank Michael McPhee for generating the graphics; Kris Bettin and Lauren Skalicky for preparation of the manuscript; Ellen Hess at Emory University for generously providing the breeding stock for the tottering mice; and Jillian L. Frisch, Lisa Duvick, Brennon O'Callaghan, and Harry T. Orr for assistance in the Western blot experiments.
The authors declare no competing financial interests.
References
- Aamand R, Dalsgaard T, Jensen FB, Simonsen U, Roepstorff A, Fago A. Generation of nitric oxide from nitrite by carbonic anhydrase: a possible link between metabolic activity and vasodilation. Am J Physiol Heart Circ Physiol. 2009;297:H2068–H2074. doi: 10.1152/ajpheart.00525.2009. [DOI] [PubMed] [Google Scholar]
- Alexander GM, Godwin DW. Metabotropic glutamate receptors as a strategic target for the treatment of epilepsy. Epilepsy Res. 2006;71:1–22. doi: 10.1016/j.eplepsyres.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Shimizu-Sasamata M, Lo EH, Noebels JL, Moskowitz MA. Impaired neurotransmitter release and elevated threshold for cortical spreading depression in mice with mutations in the alpha1A subunit of P/Q type calcium channels. Neuroscience. 2000;95:639–645. doi: 10.1016/s0306-4522(99)00446-7. [DOI] [PubMed] [Google Scholar]
- Baloh RW. Episodic ataxias 1 and 2. Handb Clin Neurol. 2012;103:595–602. doi: 10.1016/B978-0-444-51892-7.00042-5. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Yue Q, Furman JM, Nelson SF. Familial episodic ataxia: clinical heterogeneity in four families linked to chromosome 19p. Ann Neurol. 1997;41:8–16. doi: 10.1002/ana.410410105. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Houweling AR, Owens CB, Tanke N, Shevchouk OT, Rahmati N, Teunissen WH, Ju C, Gong W, Koekkoek SK, De Zeeuw CI. Anatomical pathways involved in generating and sensing rhythmic whisker movements. Front Integr Neurosci. 2011;5:53. doi: 10.3389/fnint.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Wang XJ. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–225. doi: 10.1146/annurev-neuro-062111-150444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caddick SJ, Wang C, Fletcher CF, Jenkins NA, Copeland NG, Hosford DA. Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4(lh)) and tottering (Cacna1a(tg)) mouse thalami. J Neurophysiol. 1999;81:2066–2074. doi: 10.1152/jn.1999.81.5.2066. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- Campbell DB, Hess EJ. L-type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol. 1999;55:23–31. doi: 10.1124/mol.55.1.23. [DOI] [PubMed] [Google Scholar]
- Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–278. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium. 1998;24:307–323. doi: 10.1016/S0143-4160(98)90055-0. [DOI] [PubMed] [Google Scholar]
- Chagnac-Amitai Y, Connors BW. Horizontal spread of synchronized activity in neocortex and its control by GABA-mediated inhibition. J Neurophysiol. 1989;61:747–758. doi: 10.1152/jn.1989.61.4.747. [DOI] [PubMed] [Google Scholar]
- Chance B, Ernster L, Garland PB, Lee CP, Light PA, Ohnishi T, Ragan CI, Wong D. Flavoproteins of the mitochondrial respiratory chain. Proc Natl Acad Sci U S A. 1967;57:1498–1505. doi: 10.1073/pnas.57.5.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Schofield GG. Nitric oxide donors enhanced Ca2+ currents and blocked noradrenaline-induced Ca2+ current inhibition in rat sympathetic neurons. J Physiol. 1995;482:521–531. doi: 10.1113/jphysiol.1995.sp020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Popa LS, Wang X, Gao W, Barnes J, Hendrix CM, Hess EJ, Ebner TJ. Low frequency oscillations in the cerebellar cortex of the tottering mouse. J Neurophysiol. 2009;101:234–245. doi: 10.1152/jn.90829.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Cramer SW, Ranum LPW, Swanson MS, Ebner TJ. Autofluorescence optical imaging of the responses to intracortical stimulation in normal and mouse models of myotonic dystrophy. Soc Neurosci Abstr. 2013;39:530.09. [Google Scholar]
- Cramer SW, Gao W, Chen G, Ebner TJ. Reevaluation of the beam and radial hypotheses of parallel fiber action in the cerebellar cortex. J Neurosci. 2013;33:11412–11424. doi: 10.1523/JNEUROSCI.0711-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pasquale R, Sherman SM. Modulatory effects of metabotropic glutamate receptors on local cortical circuits. J Neurosci. 2012;32:7364–7372. doi: 10.1523/JNEUROSCI.0090-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, Alonso-Nanclares L, Arellano JI. Microstructure of the neocortex: comparative aspects. J Neurocytol. 2002;31:299–316. doi: 10.1023/A:1024130211265. [DOI] [PubMed] [Google Scholar]
- Denier C, Ducros A, Vahedi K, Joutel A, Thierry P, Ritz A, Castelnovo G, Deonna T, Gérard P, Devoize JL, Gayou A, Perrouty B, Soisson T, Autret A, Warter JM, Vighetto A, Van Bogaert P, Alamowitch S, Roullet E, Tournier-Lasserve E. High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology. 1999;52:1816–1821. doi: 10.1212/WNL.52.9.1816. [DOI] [PubMed] [Google Scholar]
- Devonshire IM, Dommett EJ, Grandy TH, Halliday AC, Greenfield SA. Environmental enrichment differentially modifies specific components of sensory-evoked activity in rat barrel cortex as revealed by simultaneous electrophysiological recordings and optical imaging in vivo. Neuroscience. 2010;170:662–669. doi: 10.1016/j.neuroscience.2010.07.029. [DOI] [PubMed] [Google Scholar]
- Dichter MA, Ayala GF. Cellular mechanisms of epilepsy: a status report. Science. 1987;237:157–164. doi: 10.1126/science.3037700. [DOI] [PubMed] [Google Scholar]
- Faraci FM. Regulation of the cerebral circulation by endothelium. Pharmacol Ther. 1992;56:1–22. doi: 10.1016/0163-7258(92)90035-X. [DOI] [PubMed] [Google Scholar]
- Ferraguti F, Shigemoto R. Metabotropic glutamate receptors. Cell Tissue Res. 2006;326:483–504. doi: 10.1007/s00441-006-0266-5. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/S0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Frank-Cannon TC, Zeve DR, Abbott LC. Developmental expression of neuronal nitric oxide synthase in P/Q-type voltage-gated calcium ion channel mutant mice, leaner and tottering. Brain Res. 2007;1140:96–104. doi: 10.1016/j.brainres.2005.10.082. [DOI] [PubMed] [Google Scholar]
- Fureman BE, Hess EJ. Noradrenergic blockade prevents attacks in a model of episodic dysfunction caused by a channelopathy. Neurobiol Dis. 2005;20:227–232. doi: 10.1016/j.nbd.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Gao W, Chen G, Reinert KC, Ebner TJ. Cerebellar cortical molecular layer inhibition is organized in parasagittal zones. J Neurosci. 2006;26:8377–8387. doi: 10.1523/JNEUROSCI.2434-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Todorov B, Barrett CF, van Dorp S, Ferrari MD, van den Maagdenberg AM, De Zeeuw CI, Hoebeek FE. Cerebellar ataxia by enhanced Ca(V)2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a(S218L) mutant mice. J Neurosci. 2012;32:15533–15546. doi: 10.1523/JNEUROSCI.2454-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MC, Sidman RL. Tottering–a neuromusclar mutation in the mouse and its linkage with oligosyndacylism. J Hered. 1962;53:233–237. doi: 10.1093/oxfordjournals.jhered.a107180. [DOI] [PubMed] [Google Scholar]
- Griggs RC, Moxley RT, 3rd, Lafrance RA, McQuillen J. Hereditary paroxysmal ataxia: response to acetazolamide. Neurology. 1978;28:1259–1264. doi: 10.1212/WNL.28.12.1259. [DOI] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helekar SA, Noebels JL. Synchronous hippocampal bursting reveals network excitability defects in an epilepsy gene mutation. Proc Natl Acad Sci U S A. 1991;88:4736–4740. doi: 10.1073/pnas.88.11.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmich RC, Siebner HR, Giffin N, Bestmann S, Rothwell JC, Bloem BR. The dynamic regulation of cortical excitability is altered in episodic ataxia type 2. Brain. 2010;133:3519–3529. doi: 10.1093/brain/awq315. [DOI] [PubMed] [Google Scholar]
- Hewitt AL, Popa LS, Pasalar S, Hendrix CM, Ebner TJ. Representation of limb kinematics in Purkinje cell simple spike discharge is conserved across multiple tasks. J Neurophysiol. 2011;106:2232–2247. doi: 10.1152/jn.00886.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebeek FE, Stahl JS, van Alphen AM, Schonewille M, Luo C, Rutteman M, van den Maagdenberg AM, Molenaar PC, Goossens HH, Frens MA, De Zeeuw CI. Increased noise level of purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 2005;45:953–965. doi: 10.1016/j.neuron.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Husson TR, Mallik AK, Zhang JX, Issa NP. Functional imaging of primary visual cortex using flavoprotein autofluorescence. J Neurosci. 2007;27:8665–8675. doi: 10.1523/JNEUROSCI.2156-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW, CINCH investigators Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130:2484–2493. doi: 10.1093/brain/awm126. [DOI] [PubMed] [Google Scholar]
- Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology. 2004;62:17–22. doi: 10.1212/01.WNL.0000101675.61074.50. [DOI] [PubMed] [Google Scholar]
- Jouvenceau A, Eunson LH, Spauschus A, Ramesh V, Zuberi SM, Kullmann DM, Hanna MG. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. 2001;358:801–807. doi: 10.1016/S0140-6736(01)05971-2. [DOI] [PubMed] [Google Scholar]
- Jung J, Testard H, Tournier-Lasserve E, Riant F, Vallet AE, Berroir S, Broussolle E. Phenotypic variability of episodic ataxia type 2 mutations: a family study. Eur Neurol. 2010;64:114–116. doi: 10.1159/000315145. [DOI] [PubMed] [Google Scholar]
- Kaja S, van de Ven RC, Broos LA, Frants RR, Ferrari MD, van den Maagdenberg AM, Plomp JJ. Characterization of acetylcholine release and the compensatory contribution of non-Ca(v)2.1 channels at motor nerve terminals of leaner Ca(v)2.1-mutant mice. Neuroscience. 2007;144:1278–1287. doi: 10.1016/j.neuroscience.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Kao CY. Tetrodotoxin, saxitoxin and their significance in the study of excitation phenomena. Pharmacol Rev. 1966;18:997–1049. [PubMed] [Google Scholar]
- Khalilov I, Holmes GL, Ben-Ari Y. In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nat Neurosci. 2003;6:1079–1085. doi: 10.1038/nn1125. [DOI] [PubMed] [Google Scholar]
- Kitaura H, Uozumi N, Tohmi M, Yamazaki M, Sakimura K, Kudoh M, Shimizu T, Shibuki K. Roles of nitric oxide as a vasodilator in neurovascular coupling of mouse somatosensory cortex. Neurosci Res. 2007;59:160–171. doi: 10.1016/j.neures.2007.06.1469. [DOI] [PubMed] [Google Scholar]
- Kramer PL, Yue Q, Gancher ST, Nutt JG, Baloh R, Smith E, Browne D, Bussey K, Lovrien E, Nelson S. A locus for the nystagmus-associated form of episodic ataxia maps to an 11-cM region on chromosome 19p. Am J Hum Genet. 1995;57:182–185. doi: 10.1002/ajmg.1320570215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM. Neurological channelopathies. Annu Rev Neurosci. 2010;33:151–172. doi: 10.1146/annurev-neuro-060909-153122. [DOI] [PubMed] [Google Scholar]
- Levitt P, Noebels JL. Mutant mouse tottering: selective increase of locus ceruleus axons in a defined single-locus mutation. Proc Natl Acad Sci U S A. 1981;78:4630–4634. doi: 10.1073/pnas.78.7.4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DH, Mohajerani MH, Ledue J, Boyd J, Chen S, Murphy TH. In vivo large-scale cortical mapping using channelrhodopsin-2 stimulation in transgenic mice reveals asymmetric and reciprocal relationships between cortical areas. Front Neural Circuits. 2012;6:11. doi: 10.3389/fncir.2012.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano DA, Theyel BB, Mallik AK, Sherman SM, Issa NP. Rapid and sensitive mapping of long-range connections in vitro using flavoprotein autofluorescence imaging combined with laser photostimulation. J Neurophysiol. 2009;101:3325–3340. doi: 10.1152/jn.91291.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhle M, Schrempf W, Wolz M, Reichmann H, Storch A. Potassium channel blocker 4-aminopyridine is effective in interictal cerebellar symptoms in episodic ataxia type 2–a video case report. Mov Disord. 2008;23:1314–1316. doi: 10.1002/mds.22071. [DOI] [PubMed] [Google Scholar]
- Matsushita K, Wakamori M, Rhyu IJ, Arii T, Oda S, Mori Y, Imoto K. Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J Neurosci. 2002;22:4388–4398. doi: 10.1523/JNEUROSCI.22-11-04388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier H, MacPike AD. Three syndromes produced by two mutant genes in the mouse: clinical, pathological, and ultrastructural bases of tottering, leaner, and heterozygous mice. J Hered. 1971;62:297–302. doi: 10.1093/oxfordjournals.jhered.a108176. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-Z. [DOI] [PubMed] [Google Scholar]
- Moore PK, Handy RL. Selective inhibitors of neuronal nitric oxide synthase–is no NOS really good NOS for the nervous system? Trends Pharmacol Sci. 1997;18:204–211. doi: 10.1016/s0165-6147(97)01064-x. [DOI] [PubMed] [Google Scholar]
- Nachbauer W, Nocker M, Karner E, Stankovic I, Unterberger I, Eigentler A, Schneider R, Poewe W, Delazer M, Boesch S. Episodic ataxia type 2: phenotype characteristics of a novel CACNA1A mutation and review of the literature. J Neurol. 2014;261:983–991. doi: 10.1007/s00415-014-7310-2. [DOI] [PubMed] [Google Scholar]
- Neufeld MY, Nisipeanu P, Chistik V, Korczyn AD. The electroencephalogram in acetazolamide-responsive periodic ataxia. Mov Disord. 1996;11:283–288. doi: 10.1002/mds.870110312. [DOI] [PubMed] [Google Scholar]
- Neychev VK, Fan X, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain. 2008;131:2499–2509. doi: 10.1093/brain/awn168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL. A single gene error of noradrenergic axon growth synchronizes central neurones. Nature. 1984;310:409–411. doi: 10.1038/310409a0. [DOI] [PubMed] [Google Scholar]
- Noebels JL, Sidman RL. Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse tottering. Science. 1979;204:1334–1336. doi: 10.1126/science.572084. [DOI] [PubMed] [Google Scholar]
- Ohkuma S, Katsura M, Hibino Y, Xu J, Shirotani K, Kuriyama K. Multiple actions of nitric oxide on voltage-dependent Ca2+ channels in mouse cerebral cortical neurons. Brain Res Mol Brain Res. 1998;54:133–140. doi: 10.1016/S0169-328X(97)00331-8. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/S0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Pardo NE, Hajela RK, Atchison WD. Acetylcholine release at neuromuscular junctions of adult tottering mice is controlled by N-(cav2.2) and R-type (cav2.3) but not L-type (cav1.2) Ca2+ channels. J Pharmacol Exp Ther. 2006;319:1009–1020. doi: 10.1124/jpet.106.108670. [DOI] [PubMed] [Google Scholar]
- Pietrobon D. CaV2.1 channelopathies. Pflugers Arch. 2010;460:375–393. doi: 10.1007/s00424-010-0802-8. [DOI] [PubMed] [Google Scholar]
- Popa LS, Hewitt AL, Ebner TJ. Predictive and feedback performance errors are signaled in the simple spike discharge of individual Purkinje cells. J Neurosci. 2012;32:15345–15358. doi: 10.1523/JNEUROSCI.2151-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Presynaptic Ca(2+) influx at a mouse central synapse with Ca(2+) channel subunit mutations. J Neurosci. 2000;20:163–170. doi: 10.1523/JNEUROSCI.20-01-00163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raike RS, Weisz C, Hoebeek FE, Terzi MC, De Zeeuw CI, van den Maagdenberg AM, Jinnah HA, Hess EJ. Stress, caffeine and ethanol trigger transient neurological dysfunction through shared mechanisms in a mouse calcium channelopathy. Neurobiol Dis. 2013;50:151–159. doi: 10.1016/j.nbd.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran S, Graves TD, Labrum RW, Kotzadimitriou D, Eunson L, Davis MB, Davies R, Wood NW, Kullmann DM, Hanna MG, Schorge S. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. 2010;588:1905–1913. doi: 10.1113/jphysiol.2009.186437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran S, Kaski D, Hanna MG. Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat Rev Neurol. 2012;8:86–96. doi: 10.1038/nrneurol.2011.228. [DOI] [PubMed] [Google Scholar]
- Reinert KC, Dunbar RL, Gao W, Chen G, Ebner TJ. Flavoprotein autofluorescence imaging of neuronal activation in the cerebellar cortex in vivo. J Neurophysiol. 2004;92:199–211. doi: 10.1152/jn.01275.2003. [DOI] [PubMed] [Google Scholar]
- Reinert KC, Gao W, Chen G, Wang X, Peng YP, Ebner TJ. Cellular and metabolic origins of flavoprotein autofluorescence in the cerebellar cortex in vivo. Cerebellum. 2011;10:585–599. doi: 10.1007/s12311-011-0278-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyu IJ, Nahm SS, Hwang SJ, Kim H, Suh YS, Oda SI, Frank TC, Abbott LC. Altered neuronal nitric oxide synthase expression in the cerebellum of calcium channel mutant mice. Brain Res. 2003;977:129–140. doi: 10.1016/S0006-8993(03)02403-X. [DOI] [PubMed] [Google Scholar]
- Ryan DP, Ptácek LJ. Episodic neurological channelopathies. Neuron. 2010;68:282–292. doi: 10.1016/j.neuron.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Sachidhanandam S, Sreenivasan V, Kyriakatos A, Kremer Y, Petersen CC. Membrane potential correlates of sensory perception in mouse barrel cortex. Nat Neurosci. 2013;16:1671–1677. doi: 10.1038/nn.3532. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Huda K, Inoue T, Miyata M, Imoto K. Impaired feedforward inhibition of the thalamocortical projection in epileptic Ca2+ channel mutant mice, tottering. J Neurosci. 2006;26:3056–3065. doi: 10.1523/JNEUROSCI.5422-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried TN, Glaser GH. A review of mouse mutants as genetic models of epilepsy. Epilepsia. 1985;26:143–150. doi: 10.1111/j.1528-1157.1985.tb05398.x. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. Distinct functions for direct and transthalamic corticocortical connections. J Neurophysiol. 2011;106:1068–1077. doi: 10.1152/jn.00429.2011. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Hishida R, Murakami H, Kudoh M, Kawaguchi T, Watanabe M, Watanabe S, Kouuchi T, Tanaka R. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol. 2003;549:919–927. doi: 10.1113/jphysiol.2003.040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simaan J, Sabra R. In-vivo evidence of a role for nitric oxide in regulating the activity of the norepinephrine transporter. Eur J Pharmacol. 2011;671:102–106. doi: 10.1016/j.ejphar.2011.09.165. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Felts PA, John GR. Effects of 4-aminopyridine on demyelinated axons, synapses and muscle tension. Brain. 2000;123:171–184. doi: 10.1093/brain/123.1.171. [DOI] [PubMed] [Google Scholar]
- Spacey SD, Hildebrand ME, Materek LA, Bird TD, Snutch TP. Functional implications of a novel EA2 mutation in the P/Q-type calcium channel. Ann Neurol. 2004;56:213–220. doi: 10.1002/ana.20169. [DOI] [PubMed] [Google Scholar]
- Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, Brandt T. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. 2004;62:1623–1625. doi: 10.1212/01.WNL.0000125691.74109.53. [DOI] [PubMed] [Google Scholar]
- Tehrani MH, Barnes EM., Jr Reduced function of gamma-aminobutyric acidA receptors in tottering mouse brain: role of cAMP-dependent protein kinase. Epilepsy Res. 1995;22:13–21. doi: 10.1016/0920-1211(95)00033-7. [DOI] [PubMed] [Google Scholar]
- Tehrani MH, Baumgartner BJ, Liu SC, Barnes EM., Jr Aberrant expression of GABAA receptor subunits in the tottering mouse: an animal model for absence seizures. Epilepsy Res. 1997;28:213–223. doi: 10.1016/S0920-1211(97)00058-2. [DOI] [PubMed] [Google Scholar]
- Teramoto T, Kuwada M, Niidome T, Sawada K, Nishizawa Y, Katayama K. A novel peptide from funnel web spider venom, omega-Aga-TK, selectively blocks, P-type calcium channels. Biochem Biophys Res Commun. 1993;196:134–140. doi: 10.1006/bbrc.1993.2225. [DOI] [PubMed] [Google Scholar]
- Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, Pietrobon D. Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci U S A. 2002;99:13284–13289. doi: 10.1073/pnas.192242399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottene A, Pivotto F, Fellin T, Cesetti T, van den Maagdenberg AM, Pietrobon D. Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem. 2005;280:17678–17686. doi: 10.1074/jbc.M501110200. [DOI] [PubMed] [Google Scholar]
- Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M, van den Maagdenberg AM, Ferrari MD, Pietrobon D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron. 2009;61:762–773. doi: 10.1016/j.neuron.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Van Bogaert P, Szliwowski HB. EEG findings in acetazolamide-responsive hereditary paroxysmal ataxia. Neurophysiol Clin. 1996;26:335–340. doi: 10.1016/S0987-7053(97)85101-5. [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, Barrett CF, Gherardini L, van de Ven RC, Todorov B, Broos LA, Tottene A, Gao Z, Fodor M, De Zeeuw CI, Frants RR, Plesnila N, Plomp JJ, Pietrobon D, Ferrari MD. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol. 2010;67:85–98. doi: 10.1002/ana.21815. [DOI] [PubMed] [Google Scholar]
- Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alviña K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Wang XJ. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev. 2010;90:1195–1268. doi: 10.1152/physrev.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wappl E, Koschak A, Poteser M, Sinnegger MJ, Walter D, Eberhart A, Groschner K, Glossmann H, Kraus RL, Grabner M, Striessnig J. Functional consequences of P/Q-type Ca2+ channel Cav2.1 missense mutations associated with episodic ataxia type 2 and progressive ataxia. J Biol Chem. 2002;277:6960–6966. doi: 10.1074/jbc.M110948200. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura N, Seki S, de Groat WC. Nitric oxide modulates Ca(2+) channels in dorsal root ganglion neurons innervating rat urinary bladder. J Neurophysiol. 2001;86:304–311. doi: 10.1152/jn.2001.86.1.304. [DOI] [PubMed] [Google Scholar]
- Zasorin NL, Baloh RW, Myers LB. Acetazolamide-responsive episodic ataxia syndrome. Neurology. 1983;33:1212–1214. doi: 10.1212/WNL.33.9.1212. [DOI] [PubMed] [Google Scholar]
- Zhou YD, Turner TJ, Dunlap K. Enhanced G-protein-dependent modulation of excitatory synaptic transmission in the cerebellum of the Ca2+ channel-mutant mouse, tottering. J Physiol. 2003;547:497–507. doi: 10.1113/jphysiol.2002.033415. [DOI] [PMC free article] [PubMed] [Google Scholar]