

Abstract
Accumulating evidence from genetic and biochemical studies implicates dysfunction of the autophagic-lysosomal pathway as a key feature in the pathogenesis of Parkinson's disease (PD). Most studies have focused on accumulation of neurotoxic α-synuclein secondary to defects in autophagy as the cause of neurodegeneration, but abnormalities of the autophagic-lysosomal system likely mediate toxicity through multiple mechanisms. To further explore how endolysosomal dysfunction causes PD-related neurodegeneration, we generated a murine model of Kufor–Rakeb syndrome (KRS), characterized by early-onset Parkinsonism with additional neurological features. KRS is caused by recessive loss-of-function mutations in the ATP13A2 gene encoding the endolysosomal ATPase ATP13A2. We show that loss of ATP13A2 causes a specific protein trafficking defect, and that Atp13a2 null mice develop age-related motor dysfunction that is preceded by neuropathological changes, including gliosis, accumulation of ubiquitinated protein aggregates, lipofuscinosis, and endolysosomal abnormalities. Contrary to predictions from in vitro data, in vivo mouse genetic studies demonstrate that these phenotypes are α-synuclein independent. Our findings indicate that endolysosomal dysfunction and abnormalities of α-synuclein homeostasis are not synonymous, even in the context of an endolysosomal genetic defect linked to Parkinsonism, and highlight the presence of α-synuclein-independent neurotoxicity consequent to endolysosomal dysfunction.
Keywords: ATP13A2, autophagy, endolysosomal system, genetics, Parkinson's disease
Introduction
Parkinson's disease (PD) is a common, progressively debilitating neurodegenerative illness, the therapy of which is limited to symptom suppression. The inability to slow or halt parkinsonian neurodegeneration results, in part, from a limited understanding of the underlying mechanisms (Dauer and Przedborski, 2003). The discovery that familial forms of PD are caused by mutations in genes encoding proteins that function in or are processed by the autophagic-lysosomal pathway (ALP) highlights a key role for these processes in disease pathogenesis. These discoveries include mutations in α-synuclein—degraded in part by the ALP (Webb et al., 2003; Cuervo et al., 2004)—and the endolysosomal proteins glucocerebrosidase (Sidransky et al., 2009), VPS35 (Vilariño-Güell et al., 2011; Zimprich et al., 2011), RAB7L1 (Satake et al., 2009; MacLeod et al., 2013), synaptojanin 1 (Krebs et al., 2013; Quadri et al., 2013), and ATP13A2 (Ramirez et al., 2006). These findings suggest ALP dysfunction causes PD through abnormal increases in α-synuclein levels. However, loss of ALP proteins causes a variety of trafficking abnormalities and accumulation of substrates other than α-synuclein, which may contribute to neurodegeneration.
Recessive mutations in ATP13A2 encoding ATP13A2 cause Kufor–Rakeb syndrome (KRS; Ramirez et al., 2006), an autosomal-recessive form of levodopa-responsive Parkinsonism with additional neurological features, accompanied by diffuse cerebral and cerebellar atrophy (Williams et al., 2005; Ramirez et al., 2006; Behrens et al., 2010; Brüggemann et al., 2010). No human neuropathological data exist, but skin and muscle of Kufor–Rakeb subjects contain electron-dense lamellated structures (Malandrini et al., 2013). A recessive mutation in canine ATP13A2 causes adult-onset neuronal ceroid lipofuscinosis (NCL) in Tibetan terriers (Farias et al., 2011; Wohlke et al., 2011). Similarly, Atp13a2 null mice exhibit increased autofluorescence in multiple brain regions (Schultheis et al., 2013), indicative of lipofuscin accumulation.
ATP13A2 is a poorly understood Type 5 P-type ATPase that appears to localize to lysosomes and/or late endosomes (Kong et al., 2014) and has been implicated in zinc transport (Kong et al., 2014; Tsunemi and Krainc, 2014). A loss-of-function mechanism of disease pathogenesis is suggested by the recessive inheritance pattern and in vitro studies (Park et al., 2011; Podhajska et al., 2012). ATP13A2 knockdown in cell culture causes increased lysosome size and number, reduced protein degradation through the ALP, modestly elevated levels of α-synuclein, and neurotoxicity (Dehay et al., 2012; Usenovic et al., 2012).
To dissect the relationship between endolysosomal dysfunction, α-synuclein accumulation, and neurodegeneration, we generated and characterized Atp13a2 null mice. These mutants exhibit motor deficits, widespread gliosis, endolysosomal abnormalities, and deposition of lipofuscin and ubiquitinated protein aggregates. Strikingly, we did not observe any α-synuclein-related abnormalities in mice up to 18 months of age, and modulating α-synuclein levels by intercrossing Atp13a2 null mice with α-synuclein null or overexpressing transgenic lines did not change the onset or extent of pathological change. These are the first studies to test directly, either in vivo or in vitro, a role for α-synuclein in the pathogenesis of Atp13a2-related neurotoxicity. Our findings highlight that α-synuclein homeostasis may remain normal in the presence of marked disruption of the endolysosomal system, and emphasize that ALP dysfunction, even when provoked by Parkinson-related triggers, can cause neurotoxicity through α-synuclein-independent mechanisms.
Materials and Methods
Antibodies and materials
The following antibodies were used: cathepsin B (1:500; Santa Cruz Biotechnology, catalog #sc-6493; Research resource identifier (RRID):AB_2086939), cathepsin D (1:300; Santa Cruz Biotechnology, catalog #sc-6486; RRID:AB_637896), cathepsin L (1:200; Santa Cruz Biotechnology, catalog #sc-10778; RRID:AB_2087827), EEA1 (1:5000; BD Biosciences, catalog #610456; RRID:AB_397829), glial fibrillary acidic protein (GFAP; immunohistochemistry (IHC), 1:2000; Western blot (WB), 1:50,000; Dako Cytomation, catalog #Z0334; RRID:AB_10013382), GAPDH (1:3000; Abcam, catalog #ab8245; RRID:AB_2107448), GM130 (1:500; BD Biosciences, catalog #610822; RRID: AB_398141), hsc-70 (1:5000; Novus Biologicals, catalog #NB120-2788; RRID:AB_2120309), ionized calcium-binding adaptor molecule 1 (Iba1; 1:500; Wako Chemicals, catalog #019-19741; RRID:AB_839504), LAMP1 (1:1000; Developmental Studies Hybridoma Bank, 1D4B-C; RRID:AB_2134500), LAMP2 (1:1000; Developmental Studies Hybridoma Bank, ABL-93-C; RRID:AB_2134767), LAMP2A (1:1000; Invitrogen, catalog #51-2200; RRID:AB_87643), LC3 (1:5000; Novus Biologicals, catalog #NB100-2220; RRID:AB_10003146), NeuN (1:1000; Millipore, catalog #MAB377; RRID:AB_2298772), p62/SQSTM1 (WB, 1:10,000; AbNova, catalog #H00008878-01, RRID:AB_548364; immunofluorescence (IF), 1:1000; American Research, catalog #03-GP62-C, RRID:AB_1542690), Rab9 (1:3000; Santa Cruz Biotechnology, catalog #sc-28573; RRID:AB_2175330), α-synuclein (1:10,000, Santa Cruz Biotechnology, catalog #sc-7011-R, RRID:AB_2192953 or 1:500, BD Biosciences, catalog #610786, RRID:AB_398107), human α-synuclein (LB509; 1:10,000; Santa Cruz Biotechnology, catalog #sc-58480; RRID:AB_785898), tyrosine hydroxylase (TH; IHC, 1:2000; Millipore Bioscience Research Reagents, catalog #AB152, RRID:AB_390204; IF, 1:2000; Millipore Bioscience Research Reagents, catalog #AB1542, RRID:AB_90755), ubiquitin (1:500; Dako Cytomation, catalog #Z0458), and Vps4 (1:2000; Sigma-Aldrich, catalog #SAB4200025; RRID:AB_10603251).
Mouse lines
Atp13a2 null mice were generated by inserting LoxP sites around Exons 2 and 3 (Fig. 1A) to create an Atp13a2-flx line on a mixed C57BL6/129 background. Floxed Atp13a2 mice were intercrossed with a germline-expressing “Cre-deletor” mouse to create Atp13a2-KO/+ heterozygous mice. Intercrossing KO/+ heterozygotes produced Atp13a2 null mice and wild-type littermates. The α-synuclein null and transgenic mouse lines have been characterized previously (Dauer et al., 2002; Giasson et al., 2002; RRID:IMSR_JAX:004479). Double mutants were generated by crossing Atp13a2 null mice to α-synuclein null mice to generate double heterozygotes. Double heterozygotes were then intercrossed to generate double null mice and the appropriate control mice. Atp13a2 null/SNCA transgenics were generated in a similar manner.
Figure 1.

Atp13a2 null mice display age-related motor abnormalities. A, Targeting strategy to generate Atp13a2 null mice. Diagram shows the Atp13a2 locus and insertion of LoxP sites around exons 2 and 3. Crossing to a mouse that expresses Cre recombinase in the germline resulted in deletion of exons 2 and 3 and the insertion of a premature stop codon in exon 4. B, Left, Levels of Atp13a2 cDNA generated from RNA isolated from 6-month-old wild-type, heterozygous, or Atp13a2 null mouse brain. Right, cDNA sequencing of Atp13a2 locus shows successful recombination resulting in a premature stop codon. C, Eighteen-month-old wild-type (left) or Atp13a2 null mouse (right) during tail suspension. Atp13a2 null mice adopted an abnormal clasping position, which was quantified as the percentage of mice displaying clasping at 12 and 18 months (χ2 = 7.27, N = 18 wild-type, 15 Atp13a2 null, p < 0.05). D, Spontaneous beam breaks during 1 h in the open field apparatus. Wild-type and Atp13a2 null mice were tested every 3 months from 9 to 18 months of age, with beam breaks measured in 5 min bins for 1 h. There was an effect of genotype, but not of time (repeated-measures ANOVA, Fgenotype(1,21) = 7.09, p = 0.015; Ftime(3,63) = 1.31, p = 0.28; Finteraction(3,63) = 0.16, p = 0.92). †t = 2.42, p = 0.024; a Bonferroni's post hoc test showed a decrease in movements at 15 months. E, Wild-type and Atp13a2 null mice were tested for time to traverse a 5 mm balance beam (left) and latency to fall from an accelerating rotarod (right) every 3 months from 9 to 18 months old. Error bars represent SEM. *p < 0.05; ***p < 0.001.
Motor behavior
Mice were housed four per cage and maintained on a 12 h light/dark schedule (lights on at 6:00 A.M.). Food and water were provided ad libitum. Behavioral testing was conducted in accordance with the National Institutes of Health laboratory animal care guidelines and with the approval of the University Committee of Use and Care of Animals at the University of Michigan. Male littermates (n = 16 wild type, 13 Atp13a2 null) were tested for motor behavior every 3 months from 9 to 18 months of age in the open field, rotarod, balance beam, and tail suspension tests.
Open field.
Mice at the indicated ages were tested for spontaneous activity during a 60 min period using the Open Field 16 × 16 Photobeam Activiation System and Flex-Field software (San Diego Instruments). Data were analyzed as number of beam breaks and number of rears over time.
Rotarod.
Mice were placed in one of five slots on an accelerating rotarod apparatus (Ugo Basile), and latency to fall was recorded using an accelerating rotarod protocol from 3 to 30 rpm over 5 min. Mice were trained on 2 consecutive days with each trial ending with the mouse's fall or when 300 s elapsed. On the third day, mice were tested with an accelerating rotarod from 3 to 30 rpm over 5 min, for three trials, with the end point being latency to fall in each trial up to 300 s.
Balance beam.
Mice were trained to cross a square, 5-mm-wide, 41-cm-long Plexiglas beam. The beam was placed horizontally 50 cm above a table with a bright light illuminating the start platform and a darkened enclosed 8000 cm3 escape box (20 × 20 × 20 cm; PlasticTech) at the end of the beam. Mice were trained for 3 consecutive days with four trials per day. The fourth day, mice were tested with three consecutive trials. The time to traverse was recorded for each trial with a 30 s maximum cutoff and falls scored as 30 s. The number of footslips was also recorded.
Tail suspension test.
At 12 and 18 months of age, mice were suspended by the tail and videotaped for 30 s. An observer blinded to genotype scored the videos for the presence of clasping behavior of at least 2 s duration.
cDNA synthesis and sequencing
Total RNA was extracted from whole brains from 6-month-old wild-type Atp13a2 heterozygous or Atp13a2 null mice using TRIzol Reagent (Invitrogen) according to manufacturer's instructions. Complementary DNA (cDNA) was synthesized from 5 μg of purified total RNA using the SuperScript III One-Step RT-PCR System (Invitrogen). cDNA was amplified with primers specific for Atp13a2 RNA (forward, 5′-GCGAGGAGCCGAAATGAG-3′; reverse, 5′-CGTAAATGGTCTTCCTCGTAGC-3′). The resulting PCR product was sequenced at the University of Michigan Sequencing Core.
Immunocytochemistry
Wild-type and Atp13a2 null male mice at the indicated ages were anesthetized with a lethal dose of xylazine and ketamine HCl and transcardially perfused with a 0.9% saline solution followed by 4% paraformaldehyde in phosphate buffer. Whole brains were postfixed overnight, equilibrated in 30% sucrose, and embedded in OCT (Tissue-Tek; Sakura Finetek). Forty micrometer frozen sagittal sections were collected on a Leica CM1850 cryostat. For immunofluorescence, sections were blocked in 5% normal donkey serum (NDS) in PBS-Tx (PBS plus 0.01% Triton X-100). Following overnight incubation at 4°C in primary antibodies in 1.5% NDS/PBS-Tx, sections were incubated in an AlexaFluor 488- or 555-conjugated secondary antibodies (Invitrogen), incubated with 0.3% Sudan Black in 70% ethanol to quench autofluorescence, and mounted using ProLong Gold. For DAB immunohistochemistry, the following modifications were made to the above protocol. Before blocking, sections were first subjected to 30 min in 0.3% H2O2 in PBS to quench endogenous peroxidase activity. After a 1 h incubation in a biotinylated secondary antibody (Jackson ImmunoResearch), sections were incubated in ABC for 1 h (Vector Laboratories) followed by DAB reaction (Sigma-Aldrich). Images were acquired with a Zeiss Axioskop microscope. Confocal images were acquired on an Olympus FluoView 500 in the University of Michigan Microscope and Image Analysis Laboratory. For each experiment, a representative image was selected after examination of 4 to 10 animals.
Stereology
Dopamine cell number in the substantia nigra pars compacta (SNpC) of 18-month-old Atp13a2 null or wild-type mice was assessed by unbiased stereology using TH immunocytochemistry and Nissl stain as described previously (Tieu et al. 2003).
Tissue homogenization and Western blot
To prepare brain lysates, microdissected frozen brains were homogenized with a Dounce homogenizer in an appropriate volume of ice-cold RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% NP-40) containing protease inhibitors (Roche Molecular Biochemicals). The samples were then sonicated to solubilize all proteins. Protein concentrations were determined using the BCA assay (Pierce Biotechnology). Proteins were separated by SDS-PAGE using Tris-HCl 4–20% gradient gels (Bio-Rad) and transferred onto PVDF membranes (Bio-Rad). Membranes were blocked in 5% nonfat milk in TBS-T (TBS plus 0.1% (v/v) Tween 20) for 1 h, followed by overnight incubation at 4°C in primary antibodies in 5% nonfat milk in TBS-T. After a 1 h incubation in an HRP-conjugated secondary antibody, immunoreactive bands were visualized on film by enhanced chemiluminescence (SuperSignal West Pico, Pierce Biotechnology).
Cortical homogenates were sequentially extracted in increasing detergents using a protocol described previously (Luk et al., 2012). Briefly, cortices from 18-month-old mice were microdissected and sequentially extracted in buffers containing high salt (HS; 50 mm Tris, pH 7.5, 750 mm NaCl, and 5 mm EDTA), HS buffer containing 1% Triton X-100, and 1% SDS buffer (50 mm Tris, pH 7.5, and 1% SDS). Protease and phosphatase inhibitors (Roche) were added to buffers before use. Following extraction, samples were sonicated and subjected to ultracentrifugation at 100,000 × g for 30 min in a 70.1 Ti rotor (Beckman Coulter). At each step, the supernatant was removed and the pellet extracted in 3 ml of buffer per gram of tissue. Proteins were then subjected to SDS-PAGE analysis as described above.
Transmission electron microscopy
One- or 12-month-old Atp13a2 null or wild-type mice were prepared for ultrastructural analysis as described previously (Goodchild et al., 2005). Briefly, mice were transcardially perfused with 4% paraformaldehyde/2.5% glutaraldehyde in 100 mm sodium phosphate buffer, pH 7.4, followed by overnight postfixation in the same solution. Samples were postfixed with 1% osmium tetroxide in 0.1 m cacodylate buffer, pH 7.4, for 1 h, followed by treatment with 3% uranyl acetate, dehydration with alcohol, propylene oxide treatment, and embedding in Embed 812 (Electron Microscopy Sciences). The resin was polymerized in a 60°C oven for 2 d, and sections were cut with a DuPont diamond knife on a Reichart Ultracut-E microtome (Leica), collected on copper grids, and double stained with saturated aqueous uranyl acetate and lead citrate. Images were taken using a Philips CM-100 transmission electron microscope (Philips Research) and AMTV540 image capture software (Advanced Microscopy Techniques) in the University of Michigan Microscope and Image Analysis Laboratory. Golgi structure was analyzed by a blinded observer calculating the Golgi length and the number of cisternae/stack for each Golgi identified. The blinded observer also identified swollen Golgi and Golgi misaligned relative to the nucleus of the cell.
Lysosomal pH
Lysosomal pH on mouse embryonic fibroblasts (MEFs) isolated from wild-type or Atp13a2 null E12 embryos was determined using ratiometric imaging of endocytosed Oregon Green dextran as described previously (Elrick et al., 2012). Oregon Green dextran has both a pH-independent (440 nm) and pH-dependent (492 nm) excitation maximum, with a pKa of 4.7, allowing the measurement of pH in living cells. Briefly, mouse embryonic fibroblasts grown on glass coverslips were pulsed overnight with 150 μg/ml Oregon Green dextran in culture medium, which was then chased into lysosomes by washing three times and incubating in regular medium for 3–5 h. Cells were then rinsed and imaged in Ringer's buffer [containing (in mm) 155 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 2 NaH2PO4, 10 HEPES, and 10 glucose] using a Nikon TE300 inverted microscope with two filter pairs (excitation/emission wavelengths, C1, 436/535 nm; C2, 492/535 nm). Calibration was performed as described previously (Elrick et al., 2012). Using GraphPad Prism 5.0, the standards were fit to a sigmoidal curve by the least squares method, and unknown pH values were interpolated from this curve.
Lipid analysis using high-performance liquid chromatography-mass spectrometry
Lipid extracts were prepared from 18-month-old cortical lysates using a modified Bligh/Dyer extraction procedure as described previously (Chan et al., 2012). Samples were analyzed using an Agilent Technologies 6490 Ion Funnel LC/MS Triple Quadrupole system with front-end 1260 Infinity HPLC. Phospholipids and sphingolipids were separated by normal-phase HPLC, whereas neutral lipids were separated using reverse-phase HPLC. For normal-phase analysis, lipids were separated on an Agilent Rx-Sil column [inner diameter (i.d.), 2.1 × 100 mm] using a gradient consisting of (1) chloroform, methanol, and ammonium hydroxide (89.9:10:0.1) and (2) chloroform, methanol, water, and ammonium hydroxide (55:39:5.9:0.1) starting at 5% and ramping to 70% over a 20 min period before returning back to 5%. Neutral lipids were separated on an Agilent Zorbax XDB-C18 column (i.d., 4.6 × 100 mm) using an isocratic mobile phase chloroform/methanol/0.1 m ammonium acetate (100:100:4) at a flow rate of 300 μl/min. Multiple reaction monitoring transitions were set up for the quantitative analysis of different lipid species and their corresponding internal standards as described previously (Chan et al., 2012). Lipid levels for each sample were calculated relative to the spiked internal standards and then normalized to the total amount of all lipid species measured and presented as the relative mole percentage. Data are presented as mean mole percentage for three samples of each genotype.
Lysosomal, endosomal, and cytosolic vesicle isolation
Two different lysosomal populations preferentially involved in chaperone-mediated autophagy (CMA) or in macroautophagy (MA) were isolated by centrifugation from a light mitochondrial/lysosomal fraction of a pool of two mouse brains in a discontinuous metrizamide density gradient as reported previously (Cuervo et al., 1997). Lysosomal integrity was verified after isolation by measuring β-hexosaminidase latency, and only preparations with >90% intact lysosomes were used.
Cytosolic vesicles and late endosomes as well as a Golgi-enriched fraction were isolated from the same samples as the lysosomes using Percoll gradients as described previously (Castellino and Germain, 1995). Briefly, sample homogenates were centrifuged for 5 min at 2000 × g to spin down unbroken cells, nuclei, plasma membrane, and heavy mitochondria. The supernatant was loaded on a 27% Percoll gradient laid over 2.5 m sucrose cushion and centrifuged for 1 h at 34000 ×g. The band at the interface was enriched in late and early endosomes as well as in Golgi and was further separated on a 10% Percoll gradient by centrifugation at 34000 × g for 1 h. The band above the sucrose cushion corresponds to late endosomes, whereas the band at the interface was enriched for Golgi. A fraction of the supernatant from the first spin of the homogenate was centrifuged at 100,000 × g for 1 h to clear the sample of insoluble components, allowing for the analysis of cytosolic vesicles.
Degradation of substrate proteins by lysosomes in vitro
Intact freshly isolated lysosomal subpopulations isolated as described in the previous section were incubated with a pool of radiolabeled proteins in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer (10 mm MOPS, pH 7.3, 0.3 m sucrose, 1 mm dithiothreitol, and 5.4 μm cysteine) for 30 min at 37°C as described previously (Kaushik and Cuervo, 2009). Reactions were stopped with 20% TCA, filtered through a Millipore Multiscreen Assay System and detected in a liquid scintillation analyzer (PerkinElmer, Wallac). Proteolysis was measured as the percentage of the initial acid-insoluble radioactivity (protein) transformed into acid-soluble radioactivity (peptides and free amino acids). To analyze total proteolytic activity, this assay was performed instead with lysosomes whose membrane had been disrupted by a hypotonic shock and addition of 0.1% Triton X-100.
Binding and uptake of α-synuclein as a CMA substrate by isolated lysosomes
Freshly isolated, intact CMA active lysosomes were incubated with the substrate protein α-synuclein in MOPS buffer at 37°C for 20 min. Where indicated, lysosomes were preincubated with a mixture of protease inhibitors for 10 min at 0°C to inhibit degradation of the substrate inside the lysosomes as described previously (Cuervo et al., 2004). Lysosomes were collected by centrifugation, washed with MOPS buffer, subjected to SDS-PAGE, and immunoblotted for α-synuclein. Binding was calculated from the densitometric analysis as the amount of substrate protein bound to the lysosomal membrane in the absence of protease inhibitors. Uptake was calculated by subtracting the amount of protein associated with lysosomes in the presence (protein bound to the lysosomal membrane and taken up by lysosomes) and absence (protein bound to the lysosomal membrane) of protease inhibitors.
Statistics
Results were analyzed using GraphPad Prism 5.0. A χ2 test was used to determine Mendelian ratios and analyze the tail suspension behavioral test. Behavioral studies were performed blind. Data were first examined for equal variance and then subjected to two-way repeated-measures ANOVA with time and genotype as variables, with Bonferroni's post hoc tests at specific ages. Student's t tests were used for Western blotting analyses. In all studies, n indicates the number of samples per group, and a critical value of p ≤ 0.05 was used. Data are plotted as means ± SEM.
Results
Generation of Atp13a2 null mice
To provide novel insight into the mechanism through which Atp13a2 loss of function—a cause of human Parkinsonism—produces neurotoxicity, we generated Atp13a2 null mice and examined these animals for relevant behavior and histopathological abnormalities. We created Atp13a2 null mice by inserting LoxP sites around exons 2 and 3 to generate Atp13a2-flx mice. Crossing Atp13a2-flx mice to a Rosa-Cre mouse that expresses Cre in the germline resulted in Atp13a2 null mice (Fig. 1A). RT-PCR analysis and cDNA sequencing of RNA derived from these animals confirmed the expected recombination and deletion of Atp13a2 mRNA (Fig. 1B). Atp13a2 protein levels were not examined due to lack of specific Atp13a2 antibodies.
Atp13a2 null mice develop abnormal movements and decreased spontaneous movement
All Atp13a2 genotypes were obtained in Mendelian ratios (84 WT, 150 heterozygous, 87 null mice; total, 321; χ2 = 1.43; df = 2; p = 0.49). Initially indistinguishable from littermate controls, 18-month-old Atp13a2 null mice adopted abnormal clasping positions during tail suspension (Fig. 1C). Whereas wild-type mice reflexively kicked their limbs and rarely exhibited any form of limb clasping, a significant proportion of Atp13a2 null mice clasped their rear limbs, a behavior that was not seen in any mice at 12 months of age. To further characterize the temporal profile of motor abnormalities in these mice, we tested a cohort of 13 null and 16 wild-type littermate male mice every 3 months from 9 to 18 months of age in tests of motor behavior, including the open field, balance beam, and accelerating rotarod. Three null mice died during the study at 6, 12, and 13 months of age. The 12-month-old mouse died of an eye infection; the other two died of unknown causes. In the open field, Atp13a2 null mice displayed decreased spontaneous horizontal movement relative to wild-type littermates (two-way repeated-measures ANOVA, Fgenotype(1,21) = 7.09, p = 0.015; Ftime(3,63) = 1.31, p = 0.28; Finteraction(3,63) = 0.16, p = 0.92; Fig. 1D). No significant difference was observed in the number of rears (two-way repeated-measures ANOVA, Fgenotype(1,21) = 3.69, p = 0.069; Ftime(3,63) = 1.83, p = 0.15; Finteraction(3,63) = 0.054; p = 0.98). No significant differences between Atp13a2 null and littermate control mice were observed in balance beam or Rotarod testing up to 18 months of age (Fig. 1E).
Atp13a2 null mice develop widespread gliosis
The decreased horizontal movements observed in open field testing, in combination with age-related abnormal movements during tail suspension, suggested that Atp13a2 null mice would exhibit neuropathological changes consistent with PD and KRS, which includes neuroinflammation, lipofuscinosis, protein aggregation, and accumulation of α-synuclein. We first examined brains from 18-month-old Atp13a2 null mice and littermate controls, the age of onset of the motor phenotype. We assessed markers of neurodegeneration, lipofuscinosis, and protein aggregation. Aged Atp13a2 null brains exhibited a pronounced increase in gliosis throughout many brain regions, including, but not limited to, cortex, striatum, hippocampus, cerebellum, thalamus, and midbrain (Fig. 2A,B). Gliosis was confirmed by Western blot analysis of microdissected brain tissue, with elevated levels of GFAP protein observed in the cortex, cerebellum, hippocampus, and midbrain of Atp13a2 null brain lysates relative to wild-type controls (Fig. 2C). In contrast to the delayed onset of clasping, gliosis was present in the cortex as early as 1 month of age, though showed the most dramatic increase by 12 months (Fig. 2D). The GFAP-positive percentage of cortex was increased in Atp13a2 null mice at all ages relative to their wild-type littermates, with a fivefold increase in glial immunostaining at 6 months of age, and a 10-fold increase at 12 months of age, consistent with an age-related worsening of gliosis.
Figure 2.

Atp13a2 null CNS tissue exhibits widespread age-dependent gliosis. A, B, Forty micrometer sagittal sections from 18-month-old wild-type (top) or Atp13a2 null (bottom) mouse brains show diffuse GFAP immunoreactivity throughout the brain, including in the cortex, cerebellum, and hippocampus. C, Quantitative Western blot analysis of GFAP expression in the cortex, cerebellum, hippocampus, midbrain, and striatum of 18-month-old mice (5 μg protein loaded/lane; n = 11 wild-type, 10 Atp13a2 null mice). D, Immunohistochemistry for GFAP in the cortex of wild-type and Atp13a2 null mice at the indicated ages. Quantification (right) shows the percentage area of the cortex that is GFAP-positive normalized to wild-type tissue of the same age (n = 4 to 8 animals/genotype at each time point). *p < 0.05. Error bars indicate SEM. Scale bars: B, D, 200 μm.
Increased lipofuscinosis and lysosomal markers in Atp13a2 null mice
A previous study reported increased autofluorescence in the CNS of Atp13a2 null mice suggestive of lipofuscin deposits (Schultheis et al., 2013). Similar to this report, we observed increased autofluorescence (Fig. 3A) in multiple brain regions in Atp13a2 null mice. The autofluorescence was successfully quenched by treatment with 0.3% Sudan Black in 70% ethanol (Fig. 3A, right), allowing us to perform immunofluorescence in tissue from Atp13a2 null brains. Animal models of neuronal ceroid lipofuscinosis with increased autofluorescence and gliosis, including the Tibetan terrier model, exhibit lipofuscinosis by electron microscopy (Koike et al., 2000; Gupta et al., 2001; Katz et al., 2005, 2007; Weimer et al., 2009; Farias et al., 2011). To determine whether similar abnormalities occurred in aged Atp13a2 null mice, we performed electron microscopy on multiple brain regions displaying gliosis and autofluorescence, including cortex, cerebellum, hippocampus, and striatum. Similar to other animal models of NCL, we observed prominent lipofuscin deposits in neurons from all regions displaying autofluorescence and gliosis (Fig. 3B). The lipofuscin deposits consisted of large, electron dense material, frequently containing membranous structures, in close association with large lipid droplets. Lipid droplets typically contain neutral lipids such as triacylglycerols and cholesteryl esters and are ALP substrates (Singh et al., 2009). Lipid droplets were larger and more numerous in Atp13a2 null compared to wild-type littermate tissue (Fig. 3C). To clarify the temporal relationship between lipofuscin and gliosis, we performed an EM analysis of cortex from 1-month-old tissue, when there is gliosis but no autofluorescence. One-month-old Atp13a2 null tissue exhibited lamellated structures, but no difference in lipid droplets or lipofuscin (Fig. 3D). The lamellated structures were similar in appearance to those in a report that identified ultrastructural lamellated structures in muscle and skin biopsies from two siblings with KRS (Malandrini et al., 2013), suggesting that the ultrastructural changes observed in Atp13a2 null mice reflect similar processes to those in patients. Furthermore, our analysis suggests a close association between ultrastructural changes and gliosis, and indicates that abnormalities of lipid homeostasis occur later in the pathogenic cascade.
Figure 3.
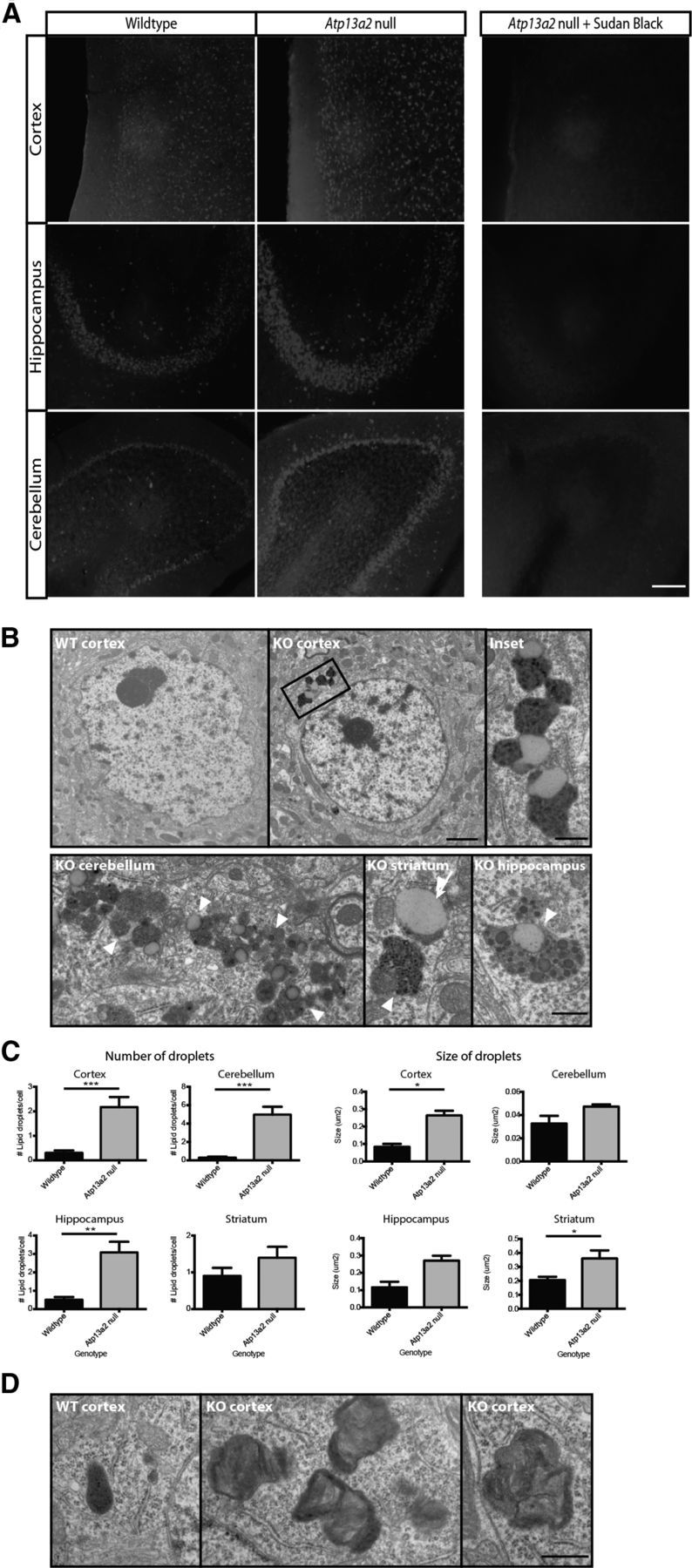
Atp13a2 null neurons accumulate lipofuscin and lipid droplets. A, Autofluorescence in 40 μm sagittal sections from wild-type or Atp13a2 null mice (18 months old). Right, Ten-minute incubation with 0.3% Sudan Black in 70% ethanol quenched autofluorescence. B, Ultrastructural analyses of 12-month-old wild-type (top left) and Atp13a2 null mouse cortex (top middle, right), Purkinje cell layer of the cerebellum (bottom left), striatum (bottom middle), and hippocampus (bottom right). Lipofuscin is indicated with white arrowheads, whereas lipid droplets associated with lipofuscin is indicated with white arrows. C, Quantification of the number and size of lipid droplets associated with lipofuscin in neurons from 12-month-old wild-type and Atp13a2 null mice (n = 40 neurons/genotype). D, Ultrastructural analyses of cortex from 1-month-old wild-type (left) and Atp13a2 null mice (middle, right). Scale bars: A, 200 μm; B, left, middle, 2 μm; B, inset, bottom, D, 500 nm.
In vitro studies suggest that loss of ATP13A2 function causes increases in the number and size of lysosomes (Dehay et al., 2012; Usenovic et al., 2012), potentially as compensation for poor lysosomal function. Similar to these reports, we found increases in immunofluorescent staining (Fig. 4A) and protein levels (B,C) of the lysosomal markers LAMP1 and LAMP2. Bis(monoacylglycero)phosphate (BMP) is a lipid species found exclusively within late endosomes and lysosomes (Kobayashi et al., 1998; Gallala and Sandhoff, 2011) and has been found to accumulate in the lysosomal storage disorder Niemann–Pick type C disease (Chevallier et al., 2008) and Alzheimer's disease (Chan et al., 2012). Lipidomic analysis demonstrated increased levels of total BMP in 18 month Atp13a2 null cortical lipid extracts (Fig. 4D), but no changes in a range of other lipid species (data not shown), consistent with a rather selective perturbation of these organelles. These findings are consistent with the previous in vitro studies and may reflect compensatory changes in the ALP following improper clearance of lysosomal substrates. Increases in LAMP1 were not observed until 6 months of age (Fig. 4E), demonstrating that lysosomal accumulation follows abnormal lipofuscin deposition and gliosis. Although we observed an increase in cortical lipid droplets by ultrastructural analysis, there were no changes to total triacylglycerols and/or cholesteryl esters by lipidomic analysis (triacylglycerols, wild-type mole percentage, 0.049 ± 0.002%; Atp13a2 null mole percentage, 0.053 ± 0.003%; t = 1.085, df = 4, p = 0.34; cholesteryl esters, wild-type mole percentage, 1.635 ± 0.577%; Atp13a2 null mole percentage, 1.694 ± 0.272%; t = 0.092, df = 4, p = 0.93).
Figure 4.

Age-dependent accumulation of lysosomal proteins and lipids in the Atp13a2 null CNS. A, Forty micrometer sections from 18-month-old Atp13a2 null or littermate control mouse brain sections stained for LAMP2 immunofluorescence in cortex and cerebellum. B, Quantitative Western blot analysis of LAMP1 expression in the cortex and cerebellum of 18-month-old mice. Top and bottom bands indicate glycosylated and unglycosylated forms of LAMP1, respectively (20 μg protein loaded/lane; n = 7 wild-type, 6 Atp13a2 null mice). C, Quantitative Western blot analysis of LAMP2 expression in the cortex of 18-month-old-mice (30 μg protein loaded/lane; n = 4 per genotype). D, Total levels of the lysosomal lipid BMP measured in 18-month-old cortical lipid extracts from wild-type and Atp13a2 null animals (n = 3 per genotype). Data are shown as the mean relative mole percentage, which was calculated by normalizing BMP levels to the total moles of all lipid species measured. E, LAMP1 immunofluorescence of cortex from wild-type and Atp13a2 null mice at the indicated ages. Immunofluorescence was quantified as mean fluorescence intensity for images taken from five cortical fields per animal and normalized to wild-type tissue of the same age (n = 4 animals/genotype at each time point). *p < 0.05; ***p < 0.001. Error bars indicate SEM. Scale bars: A, E, 100 μm.
Protein aggregation but no α-synuclein abnormalities in Atp13a2 null mice
The observed lysosomal abnormalities and in vitro studies of ATP13A2 function (Dehay et al., 2012; Usenovic et al., 2012) suggested that proteolysis was disrupted in Atp13a2 null mice, a situation commonly associated with abnormal deposition of aggregated protein. Consistent with this notion, Atp13a2 null tissue exhibited large ubiquitin-positive aggregates (Fig. 5A), most prominently in the cortex and hippocampus, and sparingly in other regions such as the SNpC; such deposits were never observed in wild-type tissue. These ubiquitin-positive inclusions colocalized with immunofluorescence for NeuN, but not GFAP or Iba1, indicating a selective localization within neurons (Fig. 5B). Given the central role of α-synuclein in PD pathogenesis and in vitro data linking ATP13A2 function to α-synuclein homeostasis (Dehay et al., 2012; Usenovic et al., 2012), we next assessed aged Atp13a2 null tissue for changes in α-synuclein levels or aggregation. In contrast to predictions from in vitro studies, no α-synuclein-positive aggregates, changes in α-synuclein levels, or changes in the phosphorylation of synuclein at serine 129 were observed in Atp13a2 null tissue (Fig. 5C–E). Furthermore, α-synuclein antibodies did not colabel the ubiquitin-positive inclusions found in 18-month-old Atp13a2 null mice (data not shown). No difference in α-synuclein solubility was observed in lysates from 18-month-old Atp13a2 null cortex sequentially extracted in buffers of increasing detergent (Fig. 5F; high salt, 1% Triton-X, and 1% SDS), consistent with the immunohistochemical and Western blot analyses.
Figure 5.
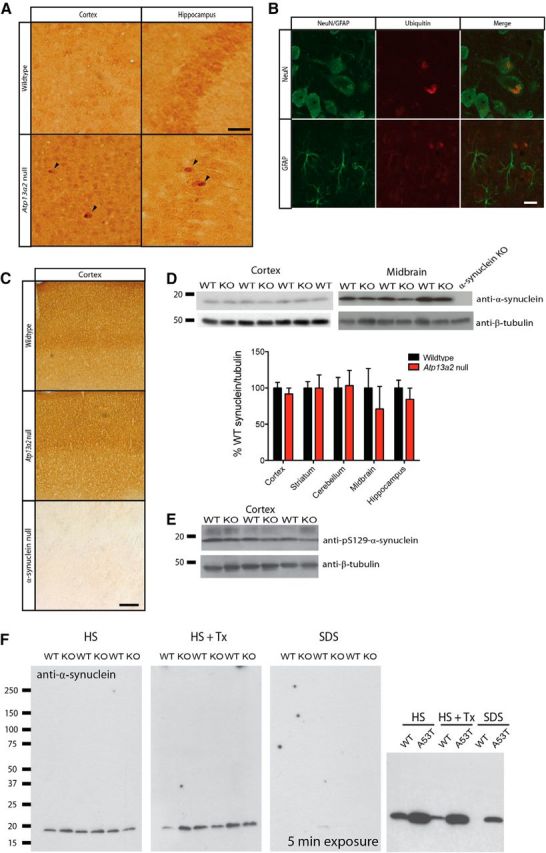
Accumulation of ubiquitin-positive aggregates, but absence of α-synuclein-related pathology in Atp13a2 null mice. A, Forty micrometer sections of cortex and hippocampus from Atp13a2 null or littermate control mouse brains stained for ubiquitin. B, Ubiquitin inclusions (red) from Atp13a2 null mice colocalize with the neuronal marker NeuN, but not GFAP (green). C, Forty micrometer sections from 18-month-old wild-type or Atp13a2 null mouse brains stained for α-synuclein with tissue from α-synuclein null brain shown as a control. D, Quantitative Western blotting of α-synuclein protein levels from multiple brain regions of Atp13a2 null mice (5 μg protein loaded/lane; n = 12 wild-type, 10 Atp13a2 null mice). Error bars indicate SEM. E, Western blotting of pS129-α-synuclein protein from cortex of 18-month-old wild-type and Atp13a2 null mice (30 μg protein loaded/lane). F, Sequential extraction of cortical lysates from 18-month-old wild-type or Atp13a2 null mice in either HS, HS plus 1% Triton X-100 (HS + Tx), or 1% SDS lysis buffer (n = 3 per genotype). Sequential extraction from A53T-synuclein transgenic mice at 6 months of age is shown as a control (far right). Scale bars: A, 200 μm; B, 50 μm; C, 400 μm.
KRS subjects have levodopa-responsive Parkinsonism, so we performed a detailed assessment of the SNpC of aged Atp13a2 null mice for protein aggregation and neurodegeneration. No ubiquitin- or α-synuclein-positive aggregates were observed in dopaminergic cell bodies (data not shown). We conducted unbiased stereology (Tieu et al., 2003) to compare the numbers of SNpC dopaminergic cell bodies between wild-type and Atp13a2 null tissue. We observed no changes in the number of SNpC dopaminergic neurons in the midbrains of aged Atp13a2 null mice (Fig. 6A; TH neurons, wild-type average, 4156 ± 549 neurons/hemisphere; Atp13a2 null average, 4531 ± 817 neurons/hemisphere; n = 7–10 animals/genotype, p = 0.27; Nissl cells, wild-type average, 6892 ± 962 neurons/hemisphere; Atp13a2 null average, 7231 ± 1181 neurons/hemisphere; n = 7–10 animals/genotype, p = 0.52). Similarly, we saw no decrease in the levels of tyrosine hydroxylase in aged Atp13a2 null mice in either the striatum or midbrain by Western blotting (Fig. 6B; wild type, 100 ± 47 a.u.; Atp13a2 null, 80 ± 25 a.u.; n = 4/genotype, p = 0.55) or by fluorescence intensity analysis in the striatum (Fig. 6C; wild type, 100 ± 35 a.u.; Atp13a2 null, 131 ± 31 a.u.; n = 3–4 animals/genotype; t = 1.196, p = 0.93). Although increased GFAP levels were present in microdissected midbrain lysates (Fig. 2C), immunohistochemical analysis of the SNpC did not demonstrate enhanced regional colocalization of TH and GFAP (Fig. 6D). We did observe an increase in LAMP2 immunoreactivity within TH-positive neurons in the SNpC (Fig. 6E), similar to the increased LAMP2 reactivity in other brain regions (Fig. 4). These studies suggest that loss of Atp13a2 function affects the SNpC region to a similar extent as other regions, with no selective degeneration of dopaminergic neurons, similar to other genetic models of PD (Dawson et al., 2010).
Figure 6.
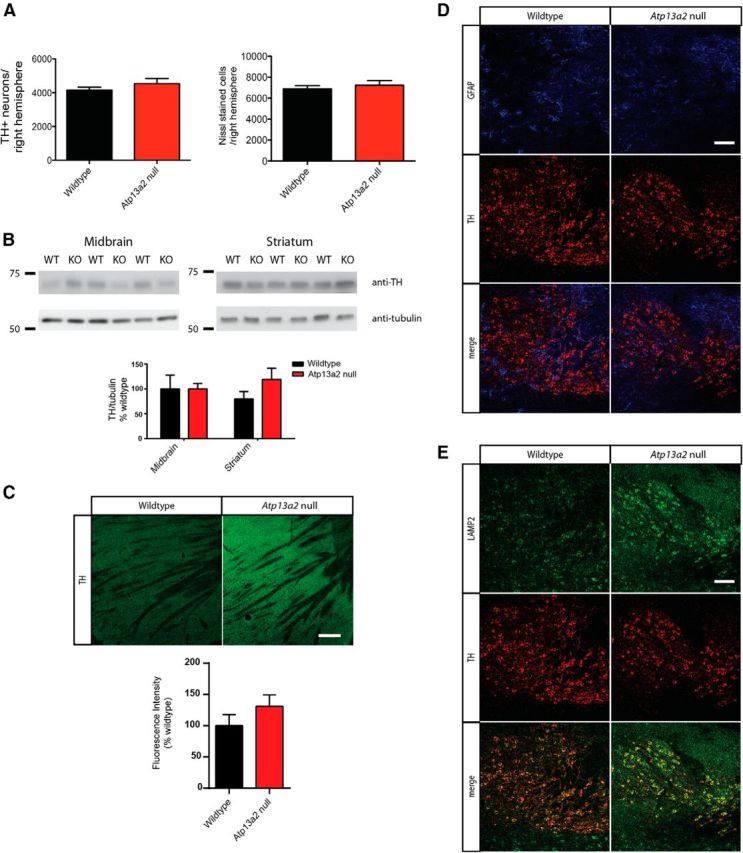
Midbrain dopaminergic neurons do not degenerate in Atp13a2 null mice. A, Stereological analysis of SNpC neurons with TH immunostaining or Nissl staining. The number of dopaminergic neurons in the SNpC was identified by TH immunoreactivity at 18 months of age (n = 10 wild-type, 7 Atp13a2 null mice). B, Semiquantitative Western blotting of TH levels from midbrain and striatum of 18-month-old mice (5 μg protein loaded/lane; n = 12 wild-type, 10 Atp13a2 null mice). C, Immunofluorescence of TH of striata from 18-month-old wild-type or Atp13a2 null mice. D, E, Immunofluorescence of midbrain structures for GFAP (blue), TH (red), or LAMP2 (green) of 18-month-old wild-type or Atp13a2 null mice. Scale bars: C, 20 μm; D, E, 100 μm.
Selective defects in autophagy function in Atp13a2 null mice
The surprising lack of α-synuclein pathology despite clear abnormalities in ALP morphology led us to dissect further the protein processing defects consequent to Atp13a2 loss of function. P62 is an autophagy substrate commonly used as a marker of autophagy function. We observed increased p62 levels in the striatum, cerebellum, and midbrain of Atp13a2 null tissue compared to littermate controls (Fig. 7A). Despite comparable p62 protein levels, the presence of p62-positive protein aggregates in the cortex of Atp13a2 null tissue (Fig. 7B) provided further evidence of defective proteostasis in these mutants. To assess autophagy induction, we examined LC3 levels. Steady-state levels of LC3-II did not differ between Atp13a2 null and wild-type tissue (Fig. 7C), consistent with an absence of marked change in the abundance of autophagic vacuoles by ultrastructural analysis (data not shown). Further biochemical analysis confirmed that the most pronounced changes were observed in the endolysosomal compartments. Maturation of cathepsin D, which occurs as it traffics to more acidic compartments (Zaidi et al., 2008), was impaired in 18-month-old Atp13a2 null cortical and cerebellar lysates, which exhibited higher levels of the immature and intermediate forms and a corresponding decrease in the cleaved, mature form (Fig. 7D). These abnormalities in cathepsin D processing (defined as mature/total cathepsin D) were not observed in 9-month-old Atp13a2 null tissue, indicating that this defect occurs relatively late in the pathogenic cascade, long after the onset of reactive astrocytosis and lysosome accumulation. The defect in cathepsin D maturation is specific, as the levels and maturation of cathepsin B and cathepsin L did not differ from wild-type controls (Fig. 7E). The selective abnormality in cathepsin D processing and the lack of differences between wild-type and Atp13a2 null mice in the analyzed markers of different endocytic compartments (early endosomes, multivesicular bodies, and late endosomes markers; Fig. 7F) raised the possibility that loss of Atp13a2 function may disrupt only a subset of ALP components, pointing to secondary lysosomes (also termed autolysosomes) as the most severely affected compartment.
Figure 7.
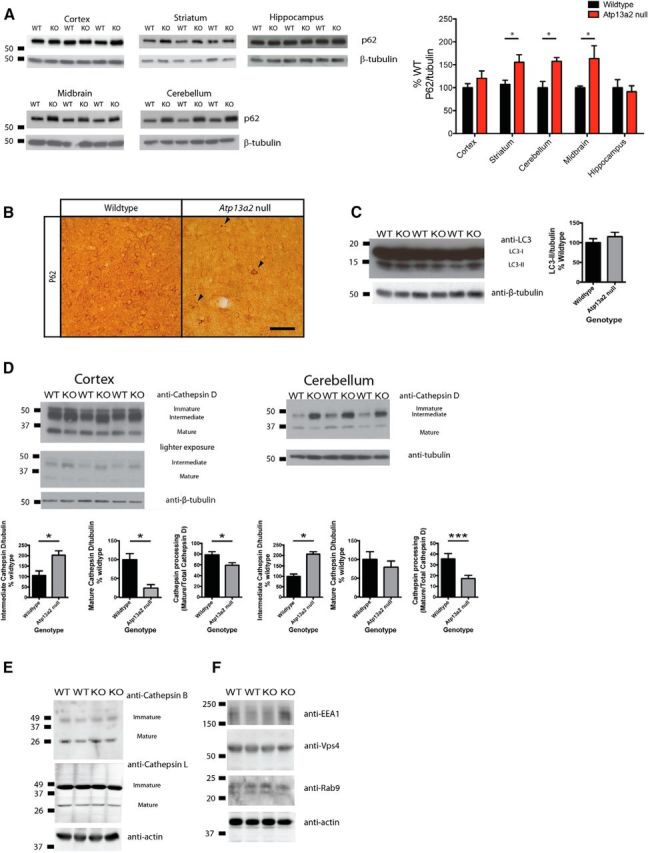
Lysosomal processing of p62 and cathepsin D is abnormal in Atp13a2 null CNS tissue. A, Quantitative Western blotting of p62 levels from 18-month-old mice (5 μg protein loaded/lane; n = 12 wild-type, 10 Atp13a2 null mice). B, P62 immunohistochemistry of 18-month-old wild-type or Atp13a2 null cortex. Scale bar, 100 μm. C, Western blotting of LC3-II from 18-month-old mice; quantitation shown on the right (30 μg protein loaded/lane; n = 7 wild-type, 6 Atp13a2 null mice). D, Quantitative Western blot of cathepsin D levels in the cortex and cerebellum of 18-month-old mice (30 μg protein loaded/lane; n = 7 wild-type, 6 Atp13a2 null mice). Cathepsin D processing was defined as the ratio of mature cathepsin D to total cathepsin D. E, F, Western blotting of cathepsin B and cathepsin L (E) and of markers of early, late, and multivesicular endocytic compartments (F) from whole brain homogenates from 18-month-old mice (n = 4 samples of 2 pooled brains from wild-type and Atp13a2 null mice). *p < 0.05; **p < 0.01. Error bars indicate SEM.
To examine the lysosomal compartment directly, we isolated two subpopulations of lysosomes from whole brains of 18-month-old wild-type and Atp13a2 null mice: those preferentially involved in macroautophagy or CMA (Cuervo et al., 1997; Koga et al., 2010). There was no difference in the membrane integrity of lysosomes from wild-type or Atp13a2 null mice, indicating that the loss of Atp13a2 (a membrane protein) does not render lysosomal membranes more fragile (data not shown). Consistent with the decrease in cathepsin D processing observed in cortical and cerebellar tissue lysates (Fig. 7D), levels of mature cathepsin D were decreased in both populations of lysosomes from Atp13a2 null tissue (Fig. 8A). The abnormality in cathepsin D processing raised the possibility that loss of Atp13a2 may compromise lysosomal acidification, as cathepsin maturation is a pH-dependent process (Zaidi et al., 2008). However, contrary to the increase in the intermediate form of cathepsin D observed in total lysates, levels of this intermediate form were reduced in the two groups of secondary lysosomes from Atp13a2 null mice (Fig. 8A). These findings suggest that rather than a problem of cathepsin processing within lysosomes, the reduced levels in mature cathepsin D were mainly a consequence of compromised trafficking of this cathepsin to lysosomes. Several other findings support the selectivity of the cathepsin D defect and argue against a global disruption of the lysosomal pH gradient in these organelles. We observed no changes in the maturation of other cathepsins (Fig. 7E) and no differences in the total proteolytic activity (Fig. 8B) or the activity of other lysosomal hydrolases (hexosaminidase; Fig. 8C). In addition, in contrast to a previous report of pH neutralization in lysosomes of ATP13A2 mutant fibroblasts (Dehay et al. 2012), we observed no difference in lysosomal pH in Atp13a2 null mouse embryonic fibroblasts as measured by Oregon Green dextran ratio (Fig. 8D; pHwild type 4.30 ± 0.22, n = 174 cells; pHAtp13a2 null 4.26 ± 0.33, n = 154 cells; t = 1.16, df = 326, p = 0.25).
Figure 8.

Isolated lysosomes from Atp13a2 null tissue have decreased cathepsin D levels, but normal proteolytic activity. Lysosomes were isolated from two pooled brains of 18-month-old wild-type or Atp13a2 null mice and probed for ALP proteins and substrates. A, Western blotting of the indicated proteins of lysosomal compartments preferentially related with either CMA or with MA. Protein levels were determined by densitometry (bottom). B, Proteolytic activity of freshly isolated lysosomes. Proteolysis was measured by incubating a pool of radiolabeled cytosolic proteins with the two subpopulations of lysosomes after disruption of their membranes by a hypotonic shock. Results are expressed as the percentage of degradation per microgram of protein and are the average values of triplicate samples from four samples pooled from eight brains of either wild-type or Atp13a2 null mice. C, Hexosaminidase activity in total homogenates and the two subpopulations of lysosomes isolated from the same animals as in B. Values are expressed as percentages of those in wild-type control samples. D, Lysosomal pH, measured by ratiometric imaging following uptake of Oregon Green dextran. The calibration curve (left) was generated by holding wild-type cells at set pH values, and it allowed the conversion of the C2/C1 ratio to pH (right) for wild-type and Atp13a2 null fibroblasts (n = 174 cells for wild-type, 151 cells for Atp13a2 null cells). E, Endogenous levels of α-synuclein and GAPDH in intact lysosomes from brains of wild-type or Atp13a2 null mice. Levels of the indicated proteins were calculated by densitometry (bottom). F, Association of recombinant GST-α-synuclein with CMA active lysosomes previously incubated or not with protease inhibitors (PIs) to determine binding and uptake via CMA. L, Lysosomes incubated without GST-α-synuclein; I, input (1/10 of added protein). Samples correspond to lysosomes isolated from four different preparations. The percentage of monomeric GST-α-synuclein bound and taken up by lysosomes is shown at the right. G, Degradation of a pool of radiolabeled cytosolic proteins by the two subpopulations of lysosomes isolated from brains of wild-type or Atp13a2 null mice was performed as in B but using intact, instead of disrupted, lysosomes. *p < 0.05. Error bars indicate SEM.
In light of the previously described lysosomal degradation of α-synuclein by both macroautophagy and CMA (Webb et al., 2003; Cuervo et al., 2004; Martinez-Vicente et al., 2008; Vogiatzi et al., 2008; Winslow et al., 2010; Ebrahimi-Fakhari et al., 2011; Xilouri et al., 2013) and of the disruption of this process in other PD models, we next analyzed the association of α-synuclein with the isolated lysosomal fractions. Similar amounts of endogenous α-synuclein were detected in wild-type and Atp13a2 null lysosomes, and α-synuclein was more abundant in the subgroup of lysosomes with higher CMA activity (Fig. 8E), as described previously (Martinez-Vicente et al., 2008). Levels of other well-characterized CMA substrate proteins such as GAPDH were also comparable in wild-type and Atp13a2 null lysosomes (Fig. 8D). Moreover, no significant difference was noted in the amount of α-synuclein monomer associated with the macroautophagy population of lysosomes (Fig. 8E; wild type, 100 ± 12.2 a.u.; Atp13a2 null, 132.5 ± 9.2; n = 4 preparations/genotype; t = 2.1, p > 0.05).
To directly analyze the ability of Atp13a2 null lysosomes to take up α-synuclein via CMA, we incubated CMA active lysosomes with GST-tagged α-synuclein (Fig. 8F). While results varied somewhat between material prepared on different days, extensive analysis (n = 12 wild-type, 12 null animals; seven different preparations) demonstrated normal lysosomal uptake of α-synuclein in Atp13a2 null material. These findings are in contrast to changes reported in these measures in models of pathogenic variants of other PD-linked genes, such as α-synuclein and LRRK2 (Martinez-Vicente et al., 2008; Orenstein et al., 2013). The normal ability of Atp13a2 null lysosomes to internalize and degrade a mixture of radiolabeled proteins (Fig. 8G), together with the normal lysosomal levels of both endogenous α-synuclein and GAPDH (Fig. 8E), provide substantial evidence indicating that lysosomal uptake and degradation of protein functions normally in the absence of Atp13a2. These findings highlight further the specificity of the cathepsin D processing defect resulting from Atp13a2 loss of function.
To clarify which compartment within the endolysosomal system was aberrantly processing cathepsin D, we conducted additional analyses of the protein trafficking pathway. We began by pursuing an ultrastructural assessment of the trans-Golgi network (Fig. 9A). A blinded observer quantified the length and number of cisternae/stack of the Golgi in 18-month-old wild-type or Atp13a2 null neurons (n = 40 neurons/genotype). Golgi from Atp13a2 null neurons showed similar structural dimensions to wild-type neurons (Fig. 9A). Furthermore, quantitation of abnormal Golgi shape showed no difference in the proportion of swollen Golgi in Atp13a2 null neurons relative to wild-type controls (wild type, 55 swollen/110 total Golgi; Atp13a2 null, 45 swollen/93 total Golgi; χ2 = 0.10, df = 1, p > 0.05).
Figure 9.

Selective alteration to cathepsin D processing in lysosomes and late endosomes, but not Golgi or cytosolic vesicles. A, Ultrastructural analysis of Golgi from 12-month-old wild-type (left) or Atp13a2 null (right) neurons. Golgi are indicated by black arrows. Quantitation of Golgi length (in nanometers) and numbers of cisternae per stack are shown. Scale bar: 500 nm. B, Subcellular fractions enriched in Golgi, late endosomes, and lysosomes were isolated from five preparations of 10 pooled brains of 18-month-old wild-type or Atp13a2 null mice and probed for cathepsin D, Golgi, and endosome/lysosome markers. Total (B2), mature (B3), and intermediate (B1) cathepsin D were determined by densitometry. Cathepsin D processing (B4) was defined as mature cathepsin D divided by total protein level. C, Cytosolic carrier vesicles were isolated from five preparation of 10 pooled brains of 18-month-old wild-type or Atp13a2 null mice and probed for cathepsin D and LAMP1 as a marker of lysosomes. Total (C2), mature (C3), and intermediate (C1) cathepsin D were determined by densitometry. *p < 0.05.
To further dissect the cathepsin D trafficking defect, we examined cathepsin D processing at each successive step along the protein trafficking pathway, including in isolated Golgi, late endosomes, and lysosomes from aged wild-type and Atp13a2 null brains (using an analysis similar to that performed previously in lysosomes; Fig. 7D). Golgi-enriched fractions derived from Atp13a2 null mouse brains contained comparable levels of the mature and intermediate forms of cathepsin D with decreased levels of the mature form of this protease in this compartment in both groups of animals, suggesting that loss of Atp13a2 does not disrupt cathepsin D processing within the Golgi (Fig. 9B). In contrast, concurrent analysis of the late endosome compartment showed deficits in cathepsin D maturation comparable to that seen in the lysosomes (Fig. 9B), localizing the trafficking deficit between the trans-Golgi network and late endosomes. Cytosolic vesicles trafficking between the Golgi and the late endosomes showed insignificant increases in the levels of intermediate forms of cathepsin D (Fig. 9C). As expected, cathepsin D maturation increased as we analyzed successive compartments in the endolysosomal system, and differences between wild-type and mutants were especially apparent in the cytosolic vesicles and lysosomes. The approximate levels of mature processed cathepsin D in wild-type and mutant mice was as follows: cytosolic vesicles, WT 10%, KO 5%; late endosomes, WT 22%, KO 2%; lysosomes, WT 40%, KO 5%.
Modulation of α-synuclein levels does not affect the onset or extent of histopathology in Atp13a2 null mice
The lack of α-synuclein abnormalities in Atp13a2 null tissue or isolated lysosomes (Figs. 5, 8E,F) suggests that the histopathologic consequences of Atp13a2 loss of function are α-synuclein independent. We tested this possibility by genetically manipulating the levels of α-synuclein in Atp13a2 null mice. We eliminated α-synuclein by generating α-synuclein/Atp13a2 double knockouts using an established knockout line (Dauer et al., 2002). Loss of α-synuclein did not prevent gliosis or accumulation of lysosomes as assessed at 3 months of age, shortly following the onset of these pathologies (Fig. 10A, quantification in B,C). If anything, the double null mice showed a surprising increase in the extent of gliosis, suggesting a worsening of pathology by this one metric. Conversely, we increased α-synuclein levels by at least fourfold in the cortex (Giasson et al., 2002) by ingression of an α-synuclein-overexpressing transgene (Giasson et al., 2002) onto the Atp13a2 null background and examined the onset and severity of pathology at 1 month (before the onset of pathology), 3 months (earliest observed pathology), and 9 months (onset of protein inclusions). This substantial increase in α-synuclein levels did not accelerate the time of onset of gliosis or lysosomal accumulation, as no pathology was observed at 1 month (data not shown), and a similar extent of pathology was observed at 3 months (Fig. 10D). Similarly, severity of histopathology at 9 months was not worse in double mutant compared to Atp13a2 null animals, with an unexpected decrease in the amount of gliosis in double mutant versus Atp13a2 null mice (Fig. 10E, quantified in F,G). These in vivo genetic studies are in accordance with biochemical studies and suggest strongly that the neuropathological changes in Atp13a2 null mice are independent of disruption in α-synuclein homeostasis.
Figure 10.
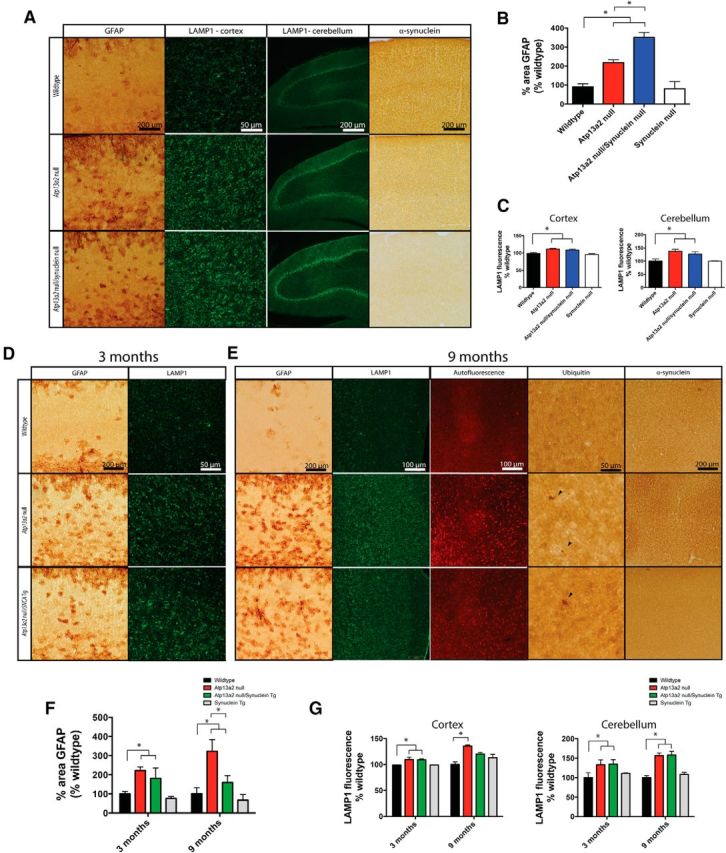
Genetic modulation of α-synuclein levels does not affect the onset or extent of histopathology in Atp13a2 null mice. A, Forty micrometer sections from cortex or cerebellum of 3-month-old wild-type, Atp13a2 null, or Atp13a2 null/SNCA null mice stained for GFAP, LAMP1, or α-synuclein. B, Quantification of percentage area of the cortex positive for GFAP immunoreactivity, as in Figure 2D (one-way ANOVA, F = 33.99, p < 0 .0001; n = 6/genotype). Tukey's multiple comparisons tests were performed to detect differences between individual genotypes. C, Quantification of average fluorescence intensity of the cortex (left) or Purkinje cell layer of the cerebellum (right), as in Figure 4D (one-way ANOVA, Fcortex = 8.24, p = 0.001; Fcerebellum = 5.50, p = 0.008). Tukey's multiple comparisons tests were used to detect differences between individual genotypes. D, E, Forty micrometer sections from cortex of 3-month-old (D) or 9-month-old (E) wild-type, Atp13a2 null, or Atp13a2 null/SNCA transgenic (Tg) mice either unstained or stained for GFAP, LAMP1, ubiquitin, or α-synuclein (n = 9/genotype). F, Quantification of percentage area of the cortex positive for GFAP immunoreactivity, as in Figure 2D (one-way ANOVA, F = 4.71, p = 0.01). Tukey's multiple comparisons tests were used to detect differences between individual genotypes. G, Quantification of fluorescence intensity of the cortex (left) or Purkinje cell layer of the cerebellum (right) as in Figure 4D (one-way ANOVA, Fcortex = 23.20, p < 0.0001; Fcerebellum = 18.03, p = 0.0001). Tukey's multiple comparisons tests were used to detect differences between individual genotypes. *p < 0.05. Scale bars are as indicated in A, D, and E.
Discussion
Considerable attention has focused on the hypothesis that in PD, endolysosomal dysfunction causes neurodegeneration by increasing α-synuclein levels. Our findings dissociate alterations in α-synuclein homeostasis from neuropathological and behavioral abnormalities in a model of endolysosomal dysfunction similar to that causing levodopa-responsive Parkinsonism in humans. Even in the absence of α-synuclein, the neuropathological consequences of Atp13a2 loss of function emerge with the same time course and progress to the same extent as in its presence (or overexpression), emphasizing the α-synuclein independence of these abnormalities. We also show that Atp13a2 deletion causes a quite selective defect in cathepsin D maturation and trafficking to the lysosome, and that lysosomal function per se is normal in these mutants, at least for the several key attributes tested. We further demonstrate that this trafficking defect occurs at a prelysosomal transport step. These findings emphasize that even in the context of a PD-related insult, endolysosomal dysfunction and abnormalities of α-synuclein are not synonymous, and that selective α-synuclein-independent defects in this pathway may contribute pathological phenotypes. Identification of such discrete abnormalities will likely help unravel how disruptions of this pathway can cause diverse neurodegenerative diseases (i.e., endolysosomal defects are also linked to Alzheimer's disease; Nixon 2013). This work highlights the importance of broadly considering the consequences of endolysosomal dysfunction that may contribute to neurodegeneration in PD and related neurodegenerative disorders.
We describe novel molecular, neuropathological, and behavioral features of Atp13a2 loss of function, and outline the broad temporal sequence of these events in vivo—information critical for unraveling the pathogenic cascade leading to neurodegeneration. Atp13a2 null mice develop age-related neuropathological changes, including reactive astrocytosis, lipofuscinosis, protein aggregation, and lysosomal accumulation in multiple brain regions, including the cortex, cerebellum, hippocampus, and striatum. We defined the progression of these changes, which demonstrate the early onset of gliosis (by 1 month of age), followed by onset of lipofuscinosis and autofluorescence by 3 months of age, the accumulation of lysosomal proteins LAMP1, LAMP2, and lysosomal lipid BMP by 6 months, and the aggregation of ubiquitinated proteins and p62 by 12 months. Biochemically, late changes include the aberrant processing of the lysosomal protease cathepsin D. The widespread distribution of these changes, with particularly prominent abnormalities in cortex and cerebellum, are consistent with the clinical features of KRS. Similar to lipid changes seen in Niemann–Pick type C (Chevallier et al., 2008), Alzheimer's disease (Chan et al., 2012), and cathepsin D-deficient mice (Jabs et al., 2008), we describe a rather selective increase in the lipid BMP, suggesting changes to both lysosomal proteins and lipids following Atp13a2 deficiency. While the overall content of triglycerides and cholesterol is comparable between control and Atp13a2-deficient brains, our ultrastructural analysis of the Atp13a2 neurons revealed marked accumulation of lipid content in lysosomes (Fig. 3B). The lack of abnormality observed in other lipids may reflect in part the small relative contribution of neuronal lipids to overall brain lipid content.
Several of our findings are consistent with previous work, including the identification of abnormalities of lysosomal size and number, alterations in protein homeostasis and autophagy, and impaired neuronal viability (Dehay et al., 2012; Usenovic et al., 2012). Our report is the first, however, to demonstrate several additional features of Atp13a2 deletion, including astrocytosis, p62 aggregation, and ultrastructural evidence of lipofuscin and lipid accumulation. Together, these results suggest a model by which loss of Atp13a2 results in endolysosomal pathway dysfunction, characterized by decreased cathepsin D processing and decreased clearance of autophagic substrates including p62 and ubiquitin. Lysosome function itself, however, appears normal in the absence of Atp13a2 function, in terms of pH, ability to uptake and degrade α-synuclein, and proteolytic function generally. The temporal sequence of abnormalities in this KRS model is particularly informative. The accumulation of lamellated structures and glial activation occurs early, months before observable abnormalities in lysosomes and aggregation of p62/ubiquitin. This early accumulation of lamellated structures (also seen in human material; Malandrini et al., 2013) and inflammation, in combination with alterations to protein homeostasis, likely disrupts neuronal health and function before the accumulation of protein aggregates. Considered together, these observations indicate that the observed lysosomal phenotypes (e.g., changes in size and number) are likely to be compensatory responses to a more fundamental, upstream abnormality (as indicated by abnormal cathepsin D processing). The identification of the temporal sequence of these events will enable future work aimed at blocking early abnormalities, which will be required to determine causal relationships.
In contrast to previous work in vivo (Schultheis et al., 2013), we find that Atp13a2 null mice do not develop changes in α-synuclein solubility, despite exhibiting abnormal accumulation of ubiquitinated protein aggregates. We also demonstrate normal lysosomal degradation of α-synuclein, even at 18 months of age. The absence of α-synuclein pathology differs from in vitro studies, which report a modest increase in α-synuclein levels in cells following ATP13A2 knockdown or mutation (Dehay et al., 2012; Usenovic et al., 2012), and is in clear contrast with other cell and mouse models of PD where autophagic-lysosomal dysfunction seems to cause neurotoxicity and elevated α-synuclein levels (Mazzulli et al., 2011; Ahmed et al., 2012; Friedman et al., 2012). These initial findings did not exclude the possibility that soluble toxic forms of α-synuclein not detected by our methods might contribute to the abnormalities in Atp13a2 null mice. To rigorously explore this possibility, we genetically altered α-synuclein levels by crossing Atp13a2 null mice to either mice lacking or overexpressing α-synuclein. In these double mutants, we observed no change in the neuropathology seen in Atp13a2 null mice, even up to 9 months of age. These results—including histopathological, biochemical, and genetic analyses—suggest that abnormalities of α-synuclein homeostasis or folding are not required for the development of gliosis, lipofuscinosis, or neuronal injury resulting from Atp13a2 loss of function.
α-Synuclein aggregation is a frequent byproduct following complete loss of specific lysosomal enzymes such as glucocerebrosidase (Mazzulli et al., 2011) or cathepsin D (Cullen et al., 2009), or direct alterations in the delivery of α-synuclein to lysosomes by autophagic pathways (Webb et al., 2003; Cuervo et al., 2004; Orenstein et al., 2013). Our results suggest that loss of Atp13a2 does not prevent lysosomal targeting or degradation of α-synuclein. In KRS patients, then, the principle insult may result not from α-synuclein, but from either neuroinflammation caused by early astrocytic activation, or from an increased burden within neurons from lipofuscinosis and aggregation of proteins other than α-synuclein. We do find abnormalities of cathepsin D, an enzyme known to degrade α-synuclein (Qiao et al., 2008; Sevlever et al., 2008; Cullen et al., 2009). Complete loss of cathepsin D, as seen in cathepsin D null mice, results in much earlier onset of lipofuscin accumulation (Koike et al., 2000), glial activation, and neurotoxicity (Partanen et al., 2008). Whereas cathepsin D null mice show increased levels of insoluble α-synuclein (Cullen et al., 2009), heterozygous mice do not show changes to α-synuclein levels (Crabtree et al., 2013), consistent with our finding that α-synuclein does not change in the presence of partial loss of lysosomal cathepsin D. Our results do not preclude the possibility that α-synuclein aggregation occurs later in the course of the disease, beyond the 18 month age time point that we examined.
Many aspects of our KRS model more closely resemble the lysosomal storage disorders of NCL than PD. Most strikingly, Atp13a2 null mice develop extensive autofluorescence and lipofuscinosis in the cortex, hippocampus, and cerebellum. This accumulation of granular osmophilic deposits is similar in appearance and regional selectivity to that seen in both the Tibetan terriers with NCL (Riis et al., 1992; Katz et al., 2005, 2007; Farias et al., 2011) and mouse models of NCLs (Koike et al., 2000; Gupta et al., 2001; Weimer et al., 2009). Similar to our findings, NCL mouse models display motor dysfunction, including abnormal clasping (Sleat et al., 2004; Macauley et al., 2009; Weimer et al., 2009). The enhanced vulnerability of cortical, cerebellar, and hippocampal neurons replicates findings in NCL models lacking Ppt1 (Gupta et al., 2001; Bible et al., 2004; Macauley et al., 2009), Tpp1 (Chang et al., 2008), and Cln3 (Weimer et al., 2009). Early gliosis precedes neuronal loss in NCL models (Kopra et al., 2004; Chang et al., 2008), similar to our observations of prominent gliosis in Atp13a2 null mice in mice as young as 1 month old.
Increasingly, genetic, biochemical, and mouse studies emphasize the importance of lysosomal dysfunction in many neurodegenerative diseases, including PD. Mutations in multiple proteins related to the endolysosomal system, including the Gaucher's protein glucocerebrosidase, VPS35, RAB7L1, synaptojanin 1, and ATP13A2, lead to increased risk for familial forms of Parkinsonism. While loss of endogenous Atp13a2 in the mouse more closely resembles neuronal ceroid lipofuscinoses, this does not preclude dopaminergic degeneration in human patients with KRS. Many genetic models of Parkinson disease do not display overt loss of neurons in the SNpC (Dawson et al., 2010), suggesting fundamental differences between neuronal susceptibility in humans and mice. Our findings highlight the importance of defining more clearly how particular alterations to the autophagic-lysosomal system results in neurodegeneration, in the context of NCL, PD, and other neurological diseases.
Footnotes
This work was supported by National Institute of Neurological Diseases and Stroke Grants R01 NS061098 (W.T.D.), R01 NS056049 (G.D.P.), NS038370 (A.M.C.), F31 NS078817 (L.R.K.), and T32 GM007863 (L.R.K.), and by the German Academic Exchange Service (B.S.). We thank the following for help with experiments and/or data analysis: Dr. Xioyan Zhang in the Wang laboratory for Golgi ultrastructural analysis, Brian Gregorka in the Swanson lab for lysosomal pH analysis, and Dr. Eeva-Liisa Eskelinen for interpretation of our electron microscopy images. We thank Abigail Renoux in the Todd lab for help with confocal microscopy, TH analysis, and technical assistance. We thank Dotty Sorensen and Shelley Almburg in the University of Michigan Microscopy and Image Analysis Laboratory, who provided valuable assistance with electron and confocal microscopy.
The authors declare no competing financial interests.
References
- Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, Savitt JM. Development and characterization of a new Parkinson's disease model resulting from impaired autophagy. J Neurosci. 2012;32:16503–16509. doi: 10.1523/JNEUROSCI.0209-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens MI, Brüggemann N, Chana P, Venegas P, Kägi M, Parrao T, Orellana P, Garrido C, Rojas CV, Hauke J, Hahnen E, González R, Seleme N, Fernández V, Schmidt A, Binkofski F, Kömpf D, Kubisch C, Hagenah J, Klein C, Ramirez A. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord. 2010;25:1929–1937. doi: 10.1002/mds.22996. [DOI] [PubMed] [Google Scholar]
- Bible E, Gupta P, Hofmann SL, Cooper JD. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2004;16:346–359. doi: 10.1016/j.nbd.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Brüggemann N, Hagenah J, Reetz K, Schmidt A, Kasten M, Buchmann I, Eckerle S, Bähre M, Münchau A, Djarmati A, van der Vegt J, Siebner H, Binkofski F, Ramirez A, Behrens MI, Klein C. Recessively inherited Parkinsonism: effect of ATP13A2 mutations on the clinical and neuroimaging phenotype. Arch Neurol. 2010;67:1357–1363. doi: 10.1001/archneurol.2010.281. [DOI] [PubMed] [Google Scholar]
- Castellino F, Germain RN. Extensive trafficking of MHC class II-invariant chain complexes in the endocytic pathway and appearance of peptide-loaded class II in multiple compartments. Immunity. 1995;2:73–88. doi: 10.1016/1074-7613(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem. 2012;287:2678–2688. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Cooper JD, Sleat DE, Cheng SH, Dodge JC, Passini MA, Lobel P, Davidson BL. Intraventricular enzyme replacement improves disease phenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. 2008;16:649–656. doi: 10.1038/mt.2008.9. [DOI] [PubMed] [Google Scholar]
- Chevallier J, Chamoun Z, Jiang G, Prestwich G, Sakai N, Matile S, Parton RG, Gruenberg J. Lysobisphosphatidic acid controls endosomal cholesterol levels. J Biol Chem. 2008;283:27871–27880. doi: 10.1074/jbc.M801463200. [DOI] [PubMed] [Google Scholar]
- Crabtree D, Boyer-Guittaut M, Ouyang X, Fineberg N, Zhang J. Dopamine and its metabolites in cathepsin D heterozygous mice before and after MPTP administration. Neurosci Lett. 2013;538:3–8. doi: 10.1016/j.neulet.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem. 1997;272:5606–5615. doi: 10.1074/jbc.272.9.5606. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, Boston H, Saftig P, Woulfe J, Feany MB, Myllykangas L, Schlossmacher MG, Tyynelä J. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2009;2:5. doi: 10.1186/1756-6606-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/S0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109:9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of Loss of P-ty. J Neurosci. 2011;31:14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrick MJ, Yu T, Chung C, Lieberman AP. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet. 2012;21:4876–4887. doi: 10.1093/hmg/dds324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias FH, Zeng R, Johnson GS, Wininger FA, Taylor JF, Schnabel RD, McKay SD, Sanders DN, Lohi H, Seppälä EH, Wade CM, Lindblad-Toh K, O'Brien DP, Katz ML. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis. 2011;42:468–474. doi: 10.1016/j.nbd.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J Neurosci. 2012;32:7585–7593. doi: 10.1523/JNEUROSCI.5809-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallala HD, Sandhoff K. Biological function of the cellular lipid BMP-BMP as a key activator for cholesterol sorting and membrane digestion. Neurochem Res. 2011;36:1594–1600. doi: 10.1007/s11064-010-0337-6. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/S0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci U S A. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabs S, Quitsch A, Käkelä R, Koch B, Tyynelä J, Brade H, Glatzel M, Walkley S, Saftig P, Vanier MT, Braulke T. Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J Neurochem. 2008;106:1415–1425. doi: 10.1111/j.1471-4159.2008.05497.x. [DOI] [PubMed] [Google Scholar]
- Katz ML, Narfström K, Johnson GS, O'Brien DP. Assessment of retinal function and characterization of lysosomal storage body accumulation in the retinas and brains of Tibetan Terriers with ceroid-lipofuscinosis. Am J Vet Res. 2005;66:67–76. doi: 10.2460/ajvr.2005.66.67. [DOI] [PubMed] [Google Scholar]
- Katz ML, Sanders DN, Mooney BP, Johnson GS. Accumulation of glial fibrillary acidic protein and histone H4 in brain storage bodies of Tibetan terriers with hereditary neuronal ceroid lipofuscinosis. J Inherit Metab Dis. 2007;30:952–963. doi: 10.1007/s10545-007-0683-y. [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 2009;452:297–324. doi: 10.1016/S0076-6879(08)03619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392:193–197. doi: 10.1038/32440. [DOI] [PubMed] [Google Scholar]
- Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–3065. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, Shibata M, Yamamoto K, Kominami E, Peters C, von Figura K, Uchiyama Y. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–6906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA. Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes synuclein externalization via exosomes. Hum Mol Genet. 2014;23:2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- Kopra O, Vesa J, von Schantz C, Manninen T, Minye H, Fabritius AL, Rapola J, van Diggelen OP, Saarela J, Jalanko A, Peltonen L. A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Hum Mol Genet. 2004;13:2893–2906. doi: 10.1093/hmg/ddh312. [DOI] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, De Camilli P, Yue Z, Paisán-Ruiz C. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34:1200–1207. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley SL, Wozniak DF, Kielar C, Tan Y, Cooper JD, Sands MS. Cerebellar pathology and motor deficits in the palmitoyl protein thioesterase 1-deficient mouse. Exp Neurol. 2009;217:124–135. doi: 10.1016/j.expneurol.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, MacCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malandrini A, Rubegni A, Battisti C, Berti G, Federico A. Electron-dense lamellated inclusions in 2 siblings with Kufor-Rakeb syndrome. Mov Disord. 2013;28:1751–1752. doi: 10.1002/mds.25470. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16:394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Mehta P, Cooper AA, Veivers D, Heimbach A, Stiller B, Kubisch C, Fung VS, Krainc D Mackay-Sim A, Sue CM. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset Parkinsonism. Hum Mutat. 2011;32:956–964. doi: 10.1002/humu.21527. [DOI] [PubMed] [Google Scholar]
- Partanen S, Haapanen A, Kielar C, Pontikis C, Alexander N, Inkinen T, Saftig P, Gillingwater TH, Cooper JD, Tyynelä J. Synaptic changes in the thalamocortical system of cathepsin D-deficient mice: a model of human congenital neuronal ceroid-lipofuscinosis. J Neuropathol Exp Neurol. 2008;67:16–29. doi: 10.1097/nen.0b013e31815f3899. [DOI] [PubMed] [Google Scholar]
- Podhajska A, Musso A, Trancikova A, Stafa K, Moser R, Sonnay S, Glauser L, Moore DJ. Common pathogenic effects of missense mutations in the P-type ATPase ATP13A2 (PARK9) associated with early-onset Parkinsonism. PLoS One. 2012;7:e39942. doi: 10.1371/journal.pone.0039942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, Xie ZL, Speake LD, Parks R, Crabtree D, Liang Q, Crimmins S, Schneider L, Uchiyama Y, Iwatsubo T, Zhou Y, Peng L, Lu Y, Standaert DG, Walls KC, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, Wu B, Xu F, Erro R, Amboni M, Pappatà S, Quarantelli M, Annesi G, Quattrone A, Chien HF, Barbosa ER, Oostra BA, Barone P, Wang J, Bonifati V. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat. 2013;34:1208–1215. doi: 10.1002/humu.22373. [DOI] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary Parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- Riis RC, Cummings JF, Loew ER, de Lahunta A. Tibetan terrier model of canine ceroid lipofuscinosis. Am J Med Genet. 1992;42:615–621. doi: 10.1002/ajmg.1320420437. [DOI] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, Dickson DW, Mazzulli JR, Bardgett ME, Haik KL, Ekhator O, Chava AK, Howard J, Gannon M, Hoffman E, Chen Y, Prasad V, Linn SC, Tamargo RJ, Westbroek W, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22:2067–2082. doi: 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevlever D, Jiang P, Yen SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry. 2008;47:9678–9687. doi: 10.1021/bi800699v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, Macauley SL, Sidman RL, Shen MM, Zhao Q, Passini MA, Davidson BL, Stewart GR, Lobel P. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R, Przedborski S. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003;112:892–901. doi: 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Krainc D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23:2791–2801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Weimer JM, Benedict JW, Getty AL, Pontikis CC, Lim MJ, Cooper JD, Pearce DA. Cerebellar defects in a mouse model of juvenile neuronal ceroid lipofuscinosis. Brain Res. 2009;1266:93–107. doi: 10.1016/j.brainres.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Hadeed A, al-Din AS, Wreikat AL, Lees AJ. Kufor Rakeb disease: autosomal recessive, levodopa-responsive Parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord. 2005;20:1264–1271. doi: 10.1002/mds.20511. [DOI] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. Alpha-synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöhlke A, Philipp U, Bock P, Beineke A, Lichtner P, Meitinger T, Distl O. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late-onset neuronal ceroid lipofuscinosis in the Tibetan terrier. PLoS Genet. 2011;7:e1002304. doi: 10.1371/journal.pgen.1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, Kirik D, Stefanis L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain. 2013;136:2130–2146. doi: 10.1093/brain/awt131. [DOI] [PubMed] [Google Scholar]
- Zaidi N, Maurer A, Nieke S, Kalbacher H. Cathepsin D: a cellular roadmap. Biochem Biophys Res Commun. 2008;376:5–9. doi: 10.1016/j.bbrc.2008.08.099. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]