

Abstract
Although the medial prefrontal cortex (mPFC) is classically defined by its reciprocal connections with the mediodorsal thalamic nucleus (MD), the nature of information transfer between MD and mPFC is poorly understood. In sensory thalamocortical pathways, thalamic recruitment of feedforward inhibition mediated by fast-spiking, putative parvalbumin-expressing (PV) interneurons is a key feature that enables cortical neurons to represent sensory stimuli with high temporal fidelity. Whether a similar circuit mechanism is in place for the projection from the MD (a higher-order thalamic nucleus that does not receive direct input from the periphery) to the mPFC is unknown. Here we show in mice that inputs from the MD drive disynaptic feedforward inhibition in the dorsal anterior cingulate cortex (dACC) subregion of the mPFC. In particular, we demonstrate that axons arising from MD neurons directly synapse onto and excite PV interneurons that in turn mediate feedforward inhibition of pyramidal neurons in layer 3 of the dACC. This feedforward inhibition in the dACC limits the time window during which pyramidal neurons integrate excitatory synaptic inputs and fire action potentials, but in a manner that allows for greater flexibility than in sensory cortex. These findings provide a foundation for understanding the role of MD-PFC circuit function in cognition.
Keywords: anterior cingulate cortex, feedforward inhibition, medial prefrontal cortex, mediodorsal thalamus, parvalbumin interneuron
Introduction
The medial prefrontal cortex (mPFC) is implicated in many fundamental cognitive processes, including decision making (Kennerley and Walton, 2011), attention, working memory (Bissonette et al., 2013), emotional control (Sotres-Bayon and Quirk, 2010), and social interaction (Li et al., 2014). The mPFC is generally defined by its reciprocal connections with the mediodorsal thalamus (MD) (Rose and Woolsey, 1948; Divac et al., 1993; Heidbreder and Groenewegen, 2003). Unlike the sensory or motor thalamic nuclei, the MD appears to integrate information from mPFC, limbic structures, and the basal ganglia to flexibly guide behavior. MD lesions or chemogenetic inhibition of MD neurons weaken task-related oscillatory coupling between MD and mPFC and produce deficits in behavioral flexibility and working memory (Floresco et al., 1999; Romanides et al., 1999; Parnaudeau et al., 2013, 2015) that resemble deficits caused by PFC lesions (Hunt and Aggleton, 1998; Mitchell and Chakraborty, 2013). Despite rich anatomical and behavioral data linking MD and mPFC function, how information is transferred between the MD and the mPFC remains obscure.
The functional interaction between sensory thalamus and corresponding sensory cortices has been studied extensively (Castro-Alamancos and Connors, 1997; Petersen, 2007). A key feature of thalamocortical interactions is the feedforward inhibition mediated by cortical inhibitory interneurons (INs) (Agmon and Connors, 1991; Swadlow, 2003) that is thought to control the temporal precision of cortical responses to sensory stimuli (Porter et al., 2001; Beierlein et al., 2003; Gabernet et al., 2005; Sun et al., 2006; Cruikshank et al., 2007; Daw et al., 2007). However, it is unclear whether a similar process operates within the MD-mPFC pathway. Anatomical evidence suggests that MD neurons directly innervate INs in the mPFC (Kuroda et al., 1998, 2004), particularly those that express parvalbumin (PV) (Rotaru et al., 2005). Furthermore, in vivo data suggest that MD stimulation exerts powerful inhibitory gating of hippocampal-evoked firing in mPFC pyramidal neurons (Gigg et al., 1994; Floresco and Grace, 2003). Although MD axons are proposed to activate PV INs in the mPFC to mediate feedforward inhibition (Floresco and Grace, 2003), to date this has not been demonstrated.
In the current study, we characterized the MD-mPFC projection using electrophysiological, optogenetic, and rabies virus-assisted circuit-mapping techniques. We report that activation of MD axons in the dorsal anterior cingulate (dACC) subregion of the mPFC drives feedforward inhibition onto layer 3 pyramidal neurons that is mediated by PV INs. This feedforward inhibition limits the “window of opportunity” during which pyramidal neurons integrate excitatory inputs and gates spike output.
Materials and Methods
Animals.
Mice were group housed under a 12 h light-dark cycle (9:00 A.M. to 9:00 P.M. light), with food and water freely available. The PV-Cre (http://jaxmice.jax.org/strain/008069.html), SOM-Cre (http://jaxmice.jax.org/strain/013044.h t m l), PV-Cre;Ai14, and Rosa26-stopflox-tTA (http://jaxmice.jax.org/strain/008600.html) mice were described previously (Hippenmeyer et al., 2005; Madisen et al., 2010; Taniguchi et al., 2011). All mice have been bred onto C57BL/6N background for at least five generations. Both male and female mice were used. All procedures involving animals were approved by the Institute Animal Care and Use Committees of Cold Spring Harbor Laboratory and conducted in accordance to the National Institute of Health guidelines.
Viral vectors.
Adeno-associated virus (AAV) vectors, AAV-CAG-ChR2(H134R)-YFP, AAV-Ef1a-DIO-eNpHR3.0-EYFP, and AAV-TRE-hGFP-TVA-G, were produced as AAV2/9 serotype by the University of North Carolina Vector Core Facilities (Chapel Hill, NC) and have been previously described (Zhang et al., 2007; Miyamichi et al., 2011; Li et al., 2013). The EnvA-pseudotyped, protein G-deleted rabies-EnvA-SAD-ΔG-mCherry virus (Miyamichi et al., 2011) was produced by the Viral Vector Core Facility at Salk Institute. All viral vectors were stored in aliquots at −80°C until use.
Stereotaxic surgery.
Mice aged postnatal day 40 to 45 (P40-P45) were used for all surgeries. Unilateral viral injections were performed using previously described procedures (Li et al., 2013) at the following stereotaxic coordinates: mediodorsal thalamus, −1.58 mm from bregma, 0.44 mm lateral from midline, and 3.20 mm vertical from cortical surface; dorsal mPFC, 1.94 mm from bregma, 0.34 mm lateral from midline, and 0.70 mm vertical from cortical surface. Surgical procedures were standardized to minimize the variability of AAV injections. To ensure minimal leak into surrounding brain areas, injection pipettes remained in the brain for ∼5 min after injection before being slowly withdrawn. The final volume for CAG-ChR2 AAV injected into MD was 0.3–0.35 μl; for AAV-eF1a-DIO-eNpHR3.0-EYFP injected into dorsal mPFC, final volume was 0.5 μl. We waited 3–4 weeks for maximal viral expression. The titer for the viruses was ∼1012 virus particles/ml.
Rabies tracing and histology.
PV-Cre; Rosa26-stopflox-tTA of 2–6 months of age were used for this experiment. The coordinates used for dorsal mPFC injection were 2 mm from bregma, 0.5 mm lateral from midline, and 0.75 mm vertical from cortical surface. The helper virus AAV-TRE-hGFP-TVA-G (0.1–0.3 μl at 1.6 × 1012 particles/ml) was injected first, followed 2 weeks later by injection at the same location with the rabies-EnvA-SAD-ΔG-mCherry (0.5 μl at 2 × 109 particles/ml). Mice were killed 5–7 d after rabies infection for histological examination.
Mice were transcardially perfused with PBS followed by 4% PFA in PBS. Following 24–48 h of postfixation, coronal brain slices (75 μm) were sectioned using a vibratome. We used a standard immunohistochemical procedure, in which the primary antibodies recognizing green fluorescence protein (GFP) (chicken, Aves Laboratories; 1:1000) and red fluorescence protein (RFP) (rabbit, Rockland; 1:1000) were used to enhance the signals for GFP and mCherry, respectively. Sections were counterstained with NeuroTrace Nissl Stain (Invitrogen). Images were acquired with a Zeiss 780 LSM confocal microscope and were viewed using FIJI (ImageJ). Anatomical regions were identified according to the Mouse Brain in Stereotaxic Coordinates by Franklin and Paxinos and the Allen Institute Mouse Brain Atlas.
For imaging of MD viral injection location and axonal labeling with ChR2-YFP, the above procedures were followed and YFP signals were enhanced by immunohistochemical methods with the antibody recognizing GFP followed by goat anti-chicken secondary antibody conjugated with AlexaFluor-488 (Invitrogen; 1:500). Sections were also counterstained with DAPI (0.5 μg/ml).
Electrophysiology.
Mice were anesthetized with isoflurane and decapitated, whereupon brains were quickly removed and immersed in ice-cold dissection buffer (110.0 mm choline chloride, 25.0 mm NaHCO3, 1.25 mm NaH2 PO4, 2.5 mm KCl, 0.5 mm CaCl2, 7.0 mm MgCl2, 25.0 mm glucose, 11.6 mm ascorbic acid, and 3.1 mm pyruvic acid, gassed with 95% O2 and 5% CO2). Coronal slices (300 μm in thickness) containing mPFC were cut in dissection buffer using a HM650 Vibrating Microtome (Thermo Fisher Scientific) and were subsequently transferred to a chamber containing ACSF (118 mm NaCl, 2.5 mm KCl, 26.2 mm NaHCO3, 1 mm NaH2 PO4, 20 mm glucose, 2 mm MgCl2, and 2 mm CaCl2, at 34°C, pH 7.4, gassed with 95% O2 and 5% CO2). After ∼30 min recovery time, slices were transferred to room temperature and were constantly perfused with ACSF.
For each mouse, after mPFC slices were prepared, MD-containing slices were cut at 350 μm and imaged to examine the location and extent of ChR2 expression in the MD. Mice were excluded if the extent of infection was too large and leaked into surrounding brain regions. Rodent MD lacks interneurons; therefore, all ChR2-infected neurons are expected to be relay projection neurons (Kuroda et al., 1998).
The internal solution for voltage-clamp experiments contained 140 mm potassium gluconate, 10 mm HEPES, 2 mm MgCl2, 0.05 mm CaCl2, 4 mm MgATP, 0.4 mm Na3GTP, 10 mm Na2-phosphocreatine, 10 mm BAPTA, and 6 mm QX-314, pH 7.25 (290 mOsm). To determine IPSC reversal potential (EIPSC), IPSCs were recorded at varying holding potentials (20 mV steps) in the presence of CNQX (5 μm) and APV (100 μm) to block AMPA receptors and NMDA receptors, respectively. IPSC amplitude was measured, and a linear regression was used to calculate the best-fit line, and the x-intercept was used as the EIPSC. Under our recording conditions, the EIPSC was ∼−60 mV. We therefore recorded EPSCs at −60 mV holding potential. IPSCs were recorded at 0 mV. The only drug used for the excitation/inhibition ratio experiments was APV (100 μm). For cell-attached and current-clamp experiments, the internal solution consisted of 130 mm potassium gluconate, 5 mm KCl, 10 mm HEPES, 2.5 mm MgCl2, 4 mm Na2ATP, 0.4 mm Na3GTP, 10 mm sodium phophocreatine, and 0.6 mm EGTA, pH 7.2. No drug was applied.
To characterize PV neuron and pyramidal neuron (PN) membrane properties, we measured resting membrane potential (Vrest) immediately after the rupture of the neuronal membrane. Input resistance (Rin) was determined by measuring voltage change in response to a small hyperpolarizing current pulse (−20 pA, 500 ms) at resting potential. Spike threshold was determined by visual inspection as the membrane potential at the point of greatest dV/dt. Current was injected in steps of 10 pA, and the current threshold was calculated as the amount of current injected that evoked the first action potential.
EPSC latency, 20%–80% rise time, and half-width were calculated from either the averaged trace or individual sweeps for each cell using automated procedures in the AxoGraph X 1.5.4 software. ESPC and IPSC onset latency was calculated as the time from stimulation onset to 10% rise time, with EPSC-IPSC delay calculated as the difference. The 10% rise time has been reported to be a more reliable measure of delay to onset, as it minimizes the contribution of EPSC and IPSC rise time differences that are reflected in the time to peak (Mittmann et al., 2005). IPSCs were recorded at 0 mV, which in our experimental conditions did not correspond to the AMPA reversal potential. Consequently, evoked responses recorded at 0 mV were biphasic, consisting of an inward current that rapidly transitioned to an outward current. To accurately detect the onset of the IPSC, we measured the 10% rise time starting from the peak of the inward current (see Fig. 1D). Onset jitter was defined as the SD of the 10% rise onset, measured across individual sweeps per cell.
Figure 1.

Inputs from MD drive excitation and inhibition onto layer 3 dACC pyramidal neurons. A, Schematic of injection into the MD. B, Left, Representative image of a brain section in which the MD was injected with AAV-CAG-ChR2(H134R)-YFP. Right, Image of a brain section from the same mouse showing the dACC area where ChR2-YFP-expressing axons originating from the MD are clearly visible in layers 1, 3, and 5. LHb, Lateral habenula; PVT, paraventricular thalamus; M2, secondary motor cortex; PL, prelimbic cortex; VLO, ventral and lateral orbital cortex. C, Layer 3 pyramidal neurons in dACC were recorded while MD axons were photo-stimulated. D, IPSCs and EPSCs were recorded at 0 mV and −60 mV, respectively, holding potentials. MD axon stimulation elicited a short-latency monosynaptic EPSC followed by a delayed IPSC. Inset, Enlarged view of the circled area, in which the two arrows indicate the onset of EPSC and IPSC. E, Onset latency of light-evoked IPSCs was longer than that of light-evoked EPSCs. ***p < 0.0001. Data are mean ± SEM.
To evoke synaptic transmission by activating ChR2, we used a single-wavelength LED system (λ = 470 nm; www.CoolLED.com) connected to the epifluorescence port of the Olympus BX51 microscope. To restrict the size of the light beam for focal stimulation, a built-in shutter along the light path in the BX51 microscope was used. Light pulses of 0.5 ms triggered by a TTL (transistor-transistor logic) signal from the Clampex software (Molecular Devices) were used to evoke synaptic transmission. Blue light pulses were delivered once every 10 s, and a minimum of 30 trials were collected. In paired-pulse recordings, 2 blue light pulses separated by 100 ms were delivered. In experiments where INs were inhibited while the MD-mPFC pathway was photostimulated, two red LEDs (λ = 625 nm, Luminus, CBT-40 series) were used to activate eNpHR3.0 in PV or somatostatin (SOM) INs. The red LEDs were secured at ∼20° angle oblique to the surface of the bath solution and were simultaneously triggered by a TTL signal from the Clampex software. The light intensity at the sample was ∼0.8 mW/mm2. In each trial, a red light pulse (20 ms in duration) was triggered 5 ms before the onset of the blue light pulse (0.5 ms in duration) to ensure that the INs were sufficiently inhibited before and immediately following MD axon stimulation. In each slice that was recorded from, we verified the efficacy of red LED-triggered hyperpolarization in PV or SOM INs. For the current-clamp experiments in which both ChR2 and eNpHR3.0 were used, a 0.5 ms blue light pulse was delivered once every 10 s in a 30 s trial. At 5 ms before the second blue light pulse, red light illumination was triggered and sustained for 20 ms. L3 PNs were recorded at resting potential, and neurons were excluded if resting potential did not remain stable over the ∼6 min recording session. Blue light stimulation intensity was set such that neuronal responses were near threshold. To determine the effect of eNpHR3.0 activation on subthreshold responses, we excluded trials in which neurons reached threshold and compared average postsynaptic potentials (PSPs) before, during, and after red light illumination.
Data analysis and statistics.
Data analysis was performed using Origin 9.0 (Origin Laboratory) or GraphPad Prism 6.0 (GraphPad Software) software. Data are mean ± SEM unless otherwise noted. Normality was tested using the Shapiro–Wilk normality test to guide the selection of parametric versus nonparametric tests. Statistical analysis was performed using two-tailed Student's t test for comparison of two groups. Neighboring neurons that were recorded sequentially were considered pairs and subjected to a paired t test. For comparisons across more than two groups, data were analyzed using one-way ANOVA followed by Tukey's post hoc analysis to correct for multiple comparisons. For data with non-normal distribution, nonparametric Mann–Whitney or Wilcoxon signed-rank test was used. p < 0.05 was considered significant.
Results
Activation of MD inputs drives feedforward inhibition in the dACC
To delineate the functional connectivity between the MD and the dACC, we injected the MD with the AAV-CAG-ChR2(H134R)-YFP, which expresses the light-sensitive cation channel channelrhodopsin-2 (ChR2) (Zhang et al., 2006) tagged with the yellow fluorescent protein (YFP) (Fig. 1A,B). ChR2-YFP-expressing axon fibers originating from the MD innervate distinct layers (L) of the dACC, including L1, L3, and L5, with the densest innervation in L3 (Fig. 1B). We used blue light pulses (0.5 ms) to stimulate these axons in acute slices while recording, using whole-cell patch-clamp technique, EPSCs, and IPSCs, from L3 PNs in the dACC (Fig. 1C). Photostimulation of MD axons reliably elicited both EPSCs and IPSCs in all L3 PNs recorded (Fig. 1D), with the onset latency of the IPSCs being longer than that of the EPSCs (EPSCs, 5.48 ± 0.12 ms; IPSCs, 10.24 ± 0.26 ms; n = 29 cells/10 animals; t(−20.72) = 28, p < 0.0001, paired t test) (Fig. 1E). The IPSCs were blocked by application of either GABAA receptor antagonist picrotoxin or AMPA/kainate receptor antagonist CNQX (Fig. 2A,B), indicating that they are polysynaptic inhibitory currents.
Figure 2.
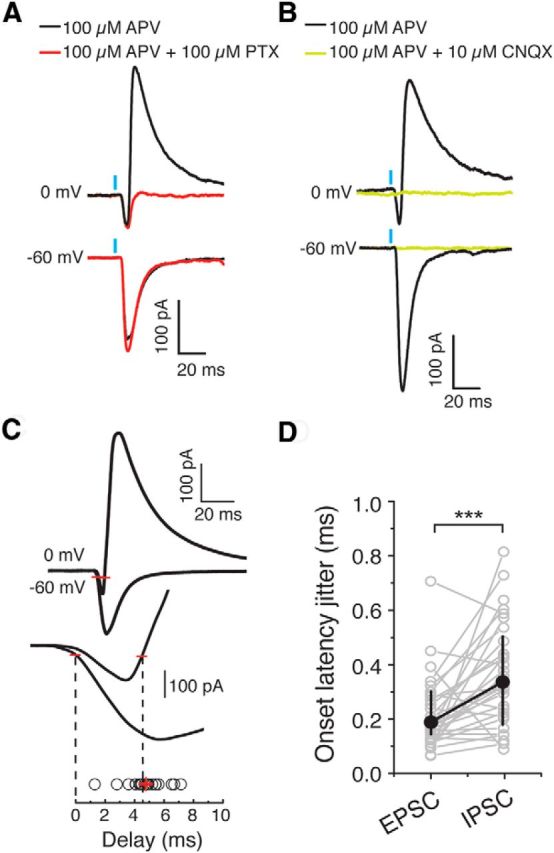
MD-driven disynaptic feedforward inhibition onto pyramidal neurons in the dACC. A, B, MD-evoked outward currents recorded from dACC pyramidal neurons at 0 mV holding potential were blocked by either picrotoxin (PTX) (A) or CNQX (B) in the presence of APV. Similar results were obtained from 5 neurons/5 mice. C, Top, Sample traces of the MD-driven EPSC (inward) and IPSC (outward), for which the 10% rise time was, respectively, marked by two red bars. Bottom, Time period encompassing the two red bars was enlarged to show the onset delay between EPSCs and IPSCs, which was calculated by subtracting the IPSC 10% rise time from the EPSC 10% rise time. Each circle represents the delay measured for one cell. Red error bars indicate mean ± SEM. D, The MD-driven IPSCs exhibit higher-onset jitter than the MD-driven EPSCs. ***p < 0.001 (Wilcoxon signed-rank test). Data are median ± interquartile range.
The estimated delay between the onset of the MD-driven EPSCs and IPSCs onto PNs was 4.76 ± 0.23 ms (n = 29 cells/10 animals; Fig. 2C). The onset jitter of the EPSCs was significantly smaller than that of the IPSCs (EPSCs, 0.24 ± 0.03 ms; IPSCs, 0.38 ± 0.04 ms; n = 29 cells/10 animals; Z = 3.33, p < 0.001, Wilcoxon signed-rank test) (Fig. 2D), as would be expected for monosynaptic versus disynaptic responses. Indeed, the jitter of the MD-driven IPSCs in PNs is similar to what has been previously reported for disynaptic inhibition (Kanichay and Silver, 2008). This observation, together with result from the CNQX experiment (Fig. 2B), suggests that inputs from MD drive disynaptic feedforward inhibition onto L3 PNs in the dACC.
Direct innervation of dACC PV INs by long-range projections from the MD
Fast-spiking, putative PV INs mediate feedforward inhibition in sensory thalamocortical pathways (Sun et al., 2006; Daw et al., 2007). To determine whether PV INs could also mediate feedforward inhibition in the MD-dACC circuit, we first examined whether MD neurons directly innervate PV INs in the dACC. To this end, we used a modified rabies virus system that can trace the monosynaptic inputs onto genetically identified neurons (Miyamichi et al., 2011). In this system, helper virus infection depends on expression of the tetracycline trans-activator (tTA; see Materials and Methods); therefore, we used the PV-Cre;Rosa26-stopflox-tTA mice, in which tTA is selectively expressed in PV neurons. We unilaterally injected the dorsal mPFC in these mice with the helper virus AAV-TRE-hGFP-TVA-G and 2 weeks later injected the same location with the rabies-EnvA-SAD-ΔG-mCherry (Fig. 3A–C). Approximately 5 d later, trans-synaptic labeling was observed as the appearance of red fluorescent rabies-EnvA-SAD-ΔG-mCherry-infected cells. The hGFP/mCherry double-positive cells in the dACC and the prelimbic cortex areas represent the PV starter cells (Fig. 3A,B). This approach revealed that a substantial population of neurons in the MD directly projects onto PV INs in the dorsal mPFC (Fig. 3D,E). In particular, the central and lateral aspects of the MD, which we identified by anterograde tracing to send prominent projections to the dACC (Fig. 1B), contained a high density of neurons back-labeled by the rabies virus (Fig. 3E). Together, these results indicate that the dACC PV INs are directly innervated by neurons in the central and lateral MD, consistent with the previously described topology of MD-mPFC connections (Ray and Price, 1992; Mátyás et al., 2014).
Figure 3.

MD neurons directly innervate mPFC PV interneurons. A, Schematic of the experimental design. B, Left, Representative image of a brain section, in which the helper virus and rabies virus were injected into the dACC. Right, Higher-magnification image of the boxed area. C, Higher-magnification images showing the dACC PV cells infected by the helper virus (left), rabies virus (middle), and both viruses (right). The double-infected (yellow) PV cells represent the starter cells. D, Schematic of retrograde transport of rabies virus into the MD area. E, Representative image of a brain section from the same mouse as that in B, showing the labeling of MD neurons by the retrograde spread of rabies virus expressing mCherry.
dACC PV INs receive direct excitatory synaptic inputs from MD neurons
To determine the functional connectivity between MD neurons and PV INs in the dACC, we injected MD with the AAV-CAG-ChR2(H134R)-YFP at a location that projects to dACC PV INs (Fig. 4A). We used the PV-Cre;Ai14 mice, in which PV INs in the mPFC are readily identified on the basis of their red fluorescence (Fig. 4B). As mentioned above (Fig. 1B), ChR2-YFP-expressing axons arising from infected MD neurons were clearly visible in the dACC, forming axon terminations in L1 and L3, with more diffuse labeling in L5 (Fig. 4B). Interestingly, we observed that there was a marked drop-off in L3 PV INs along the dorsal/ventral axis of mPFC, such that these neurons are much more abundant in the dACC than in the prelimbic cortex (Fig. 4B) (see also Mátyás et al., 2014). This dACC L3 area received the densest MD axonal projections and therefore was the focus of study in subsequent experiments.
Figure 4.
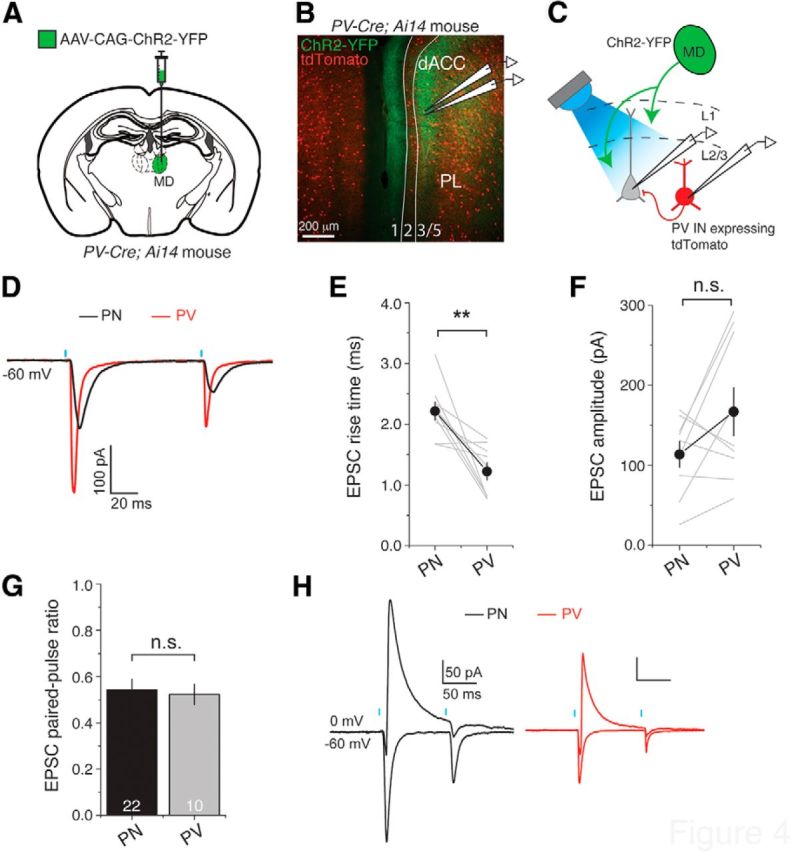
MD neurons directly excite PV interneurons in layer 3 of dACC. A, Schematic of the experimental approach. B, Representative image of a brain section from a PV-Cre;Ai14 mouse, in which the MD was injected with AAV-CAG-ChR2-YFP. The ChR2-YFP-expressing axons originating from the MD terminate in layers 1, 3, and 5 in dACC. PL, Prelimbic cortex. C, Schematic of the recording configuration. D, EPSCs evoked by photostimulation of inputs from the MD were sequentially recorded in pairs of neighboring PV INs and PNs in dACC. E, The rise time of EPSCs was faster in PV INs than in PNs. **p < 0.005 (paired t test). F, G, The amplitude (F) or paired-pulse ratio (100 ms interpulse interval) (G) of EPSCs in PV INs was not significantly different from that in neighboring PNs. n.s., Not significant. Numbers in bar graph indicate the number of cells recorded. H, PV INs also receive feedforward inhibition. The MD-driven feedforward IPSCs onto PV INs or PNs showed strong paired-pulse depression (100 ms interstimulus interval). Data are mean ± SEM.
To confirm that L3 PV INs in the dACC receive direct excitatory synaptic input from the MD, we recorded EPSCs from these neurons as well as neighboring PNs (<50 μm apart) in response to photostimulation of MD axon fibers expressing ChR2 (Fig. 4C). Robust EPSCs were readily recorded from both PV INs and PNs, with the EPSC rise time of PV INs being significantly faster than that of PNs (PV, 1.22 ± 0.14 ms, n = 9 cells/3 animals; PN, 2.21 ± 0.15 ms, n = 9 cells/3 animals; t(8) = 3.89, p < 0.005, paired t test) (Fig. 4D,E). The faster EPSC rise time of PV INs resulted in these neurons reaching peak EPSC amplitude earlier than neighboring PNs (PV INs, 7.7 ± 0.4 ms, n = 9 cells/3 animals; PNs, 11.1 ± 0.4 ms, n = 9 cells/3 animals; t(8) = 8.03, p < 0.001, paired t test), suggesting that MD inputs are capable of evoking action potentials in PV INs within a time frame during which they can shape PN activity driven by thalamocortical excitation. These results demonstrate that L3 PV INs in the dACC receive direct excitatory synaptic inputs from the MD.
Synaptic and cellular properties of PV INs and PNs in the dACC
Next, we compared the synaptic and cellular properties of PV INs with those of PNs in L3 of the dACC using the PV-Cre;Ai14 mice. We found that the amplitude of the MD-driven EPSCs onto PV INs was not significantly different from that onto neighboring PNs (PV INs, 167.2 ± 30.0 pA, n = 9 cells/3 animals; PNs, 113.7 ± 48.8 pA, n = 9 cells/3 animals; t(8) = −1.84, p = 0.10, paired t test) (Fig. 4F). In addition, the MD-driven EPSCs onto both cell types showed strong paired-pulse depression, indicating high presynaptic release probability (PN, 0.54 ± 0.05, n = 22 cells/6 animals; PV, 0.52 ± 0.04, n = 10 cells/3 animals; t(25.82) = 0.32, p > 0.05, t test) (Fig. 4G). Of note, these results differ from findings in the sensory cortices, where thalamic drive onto L4 fast-spiking INs (the putative PV INs) is much stronger and shows a higher degree of paired-pulse depression than that onto PNs in L4, the recipient layer for sensory thalamic projections (Gibson et al., 1999; Beierlein et al., 2003; Gabernet et al., 2005; Cruikshank et al., 2007; Hull et al., 2009; Schiff and Reyes, 2012).
Both PV INs and PNs in the dACC received MD-driven feedforward inhibition that showed strong paired-pulse depression (PNs, 0.07 ± 0.02, n = 13 cells/5 animals; PV INs, 0.15 ± 0.07, n = 6 cells/2 animals; t(5.67) = −1.16, p > 0.05, t test) (Fig. 4H). These results are consistent with sensory thalamocortical transmission, whereby feedforward inhibition decreases dramatically during repeated stimulation, thus increasing the integration window in thalamorecipient neurons in the cortex (Gabernet et al., 2005).
PV INs and PNs in the L3 of dACC had similar resting membrane potentials (PV INs, −74.2 ± 0.9 mV, n = 13 cells/4 animals; PNs, −75.7 ± 0.8 mV, n = 18 cells/4 animals; t(28.87) = −1.22, p > 0.05, t test) and spike threshold (PV INs, −39.4 ± 0.9 mV, n = 13 cells/4 animals; PNs, −39.1 ± 0.6 mV, n = 18 cells/4 animals; U = 120, Z = 0.10, p > 0.05, Mann–Whitney test) (Fig. 5B). There was a trend for PV INs to exhibit lower input resistance (Rin) than nearby PNs (PV INs, 211.3 ± 14.0 MΩ, n = 13 cells/4 animals; PNs, 259.3 ± 20.1 MΩ, n = 18 cells/4 animals; t(28.1) = −1.96, p = 0.06, t test) (Fig. 5C). Lower Rin partly explains why PV INs required larger current injection than PNs to reach spike threshold (PV INs, 142.3 ± 12.7 pA, n = 13 cells/4 animals; PNs, 47.2 ± 5.9 pA, n = 18 cells/4 animals; t(17.16) = −6.81, p < 0.001, t test) (Fig. 5D,E). A similar pattern of intrinsic membrane properties was previously found across fast-spiking interneurons and pyramidal neurons in the barrel cortex (Cruikshank et al., 2007). Finally, both PV INs and PNs were able to fire action potentials in response to optogenetic stimulation of MD axons (Fig. 5F).
Figure 5.

Intrinsic properties of PNs and PV INs in layer 3 of dACC. A, Differential interference contrast image overlaid with fluorescence image showing a tdTomato-expressing PV IN and a neighboring PN recorded in layer 3 of dACC. B, Resting potential (Vrest) and spike threshold (Vthresh) were similar across cell types (Vrest, p > 0.05, t test; Vthresh, p > 0.05, Mann–Whitney U test). C, There was a trend toward PNs exhibiting higher input resistance than PV INs (p = 0.06, t test). D, Representative traces of PV and PN responses (top) to injected currents (bottom). E, PV INs required larger current injection to reach spike threshold than PNs. ***p < 0.001. F, Sample traces showing that PVs and PNs fire action potentials in response to photostimulation (0.5 ms; indicated by blue bars) of ChR2-expressing axons originating from the MD. Similar results were obtained from 5 PV INs and 5 PNs. Threshold data are presented as median ± interquartile range in B; other data are presented as mean ± SEM.
MD-driven feedforward inhibition in the dACC is mediated by PV INs
Several pieces of evidence point to the possibility that PV INs mediate the MD-driven feedforward inhibition in the dACC. First, MD neurons directly project to and make functional synapses onto L3 PV INs in the dACC (Figs. 3 and 4). Second, the kinetics of MD-driven feedforward IPSCs onto dACC PNs were strikingly similar to those evoked by direct photostimulation of local PV INs (rise time: feedforward IPSCs, 2.17 ± 0.18, n = 14 cells/6 animals; PV-mediated IPSCs, 1.76 ± 0.31 ms, n = 5 cells/1 animal; t(−1.13) = 6.75, p > 0.05, t test; decay time: feedforward IPSCs, 29.09 ± 1.27 ms, n = 14 cells/6 animals; PV-mediated IPSCs, 31.22 ± 2.12 ms, n = 5 cells/1 animal; t(7.08) = 0.86, p > 0.05, t test) (Fig. 6A–C).
Figure 6.

Feedforward IPSCs resemble IPSCs directly mediated by PV INs. A, Top, Schematic experimental approach. Bottom, Sample traces of IPSCs recorded in response to MD axon stimulation (feedforward) or direct activation of PV INs (PV-mediated). Traces are normalized and time-aligned to onset. B, Feedforward IPSCs exhibited similar rise time as PV-mediated IPSCs. C, Feedforward and PV-mediated IPSCs exhibited similar 80%–20% decay time. Data are mean ± SEM.
To directly test the hypothesis that PV INs mediate the MD-driven feedforward inhibition, we sought to inhibit them while monitoring the MD-driven synaptic responses in L3 dACC PNs. To achieve this goal, we injected the MD of the PV-Cre mice with AAV-CAG-ChR2(H134R)-YFP and injected the dACC in the same mice with AAV-Ef1a-DIO-eNpHR3.0-EYFP (Fig. 7A). The latter AAV expresses the light-gated chloride pump halorhodopsin (eNpHR3.0) in a Cre-dependent manner, which suppresses neuronal firing in response to red light (Gradinaru et al., 2010). We verified that activation of eNpHR3.0 reversibly inhibited PV IN firing (Fig. 7B) via potent hyperpolarization (41.9 ± 4.8 mV, n = 5 cells/5 animals) of these neurons (Fig. 7C). Inhibition of PV INs dramatically reduced, and in some cases abolished, the MD-driven feedforward inhibitory currents in L3 PNs measured at either 0 mV or −30 mV holding potential (0 mV, n = 11 cells/6 animals, W = −66.00, Z = 2.89, p < 0.001, Wilcoxon matched-pairs signed-rank test) (Fig. 7D,E). This effect was reversible upon cessation of red light illumination (F(1.232,4.93) = 29.88, p < 0.01, one-way repeated-measures ANOVA) (Fig. 7F). Interestingly, inhibition of PV INs prolonged MD-driven EPSCs in PNs, as measured by an increase in the half-width of EPSCs recorded at −30 mV holding potential (light off, 4.37 ± 0.56 ms; light on, 10.72 ± 1.27 ms; n = 10 cells/5 animals, t(−5.12) = 9, p < 0.001, paired t test) (Fig. 7G). In contrast, the peak amplitude of MD-driven EPSCs was not altered by PV IN inhibition (light off, −129.40 ± 26.12 pA; light on, −122.25 ± 30.04 pA; n = 10 cells/5 animals, W = 19.0, Z = −0.82, p > 0.05, Wilcoxon matched-pairs signed rank test) (Fig. 7H).
Figure 7.
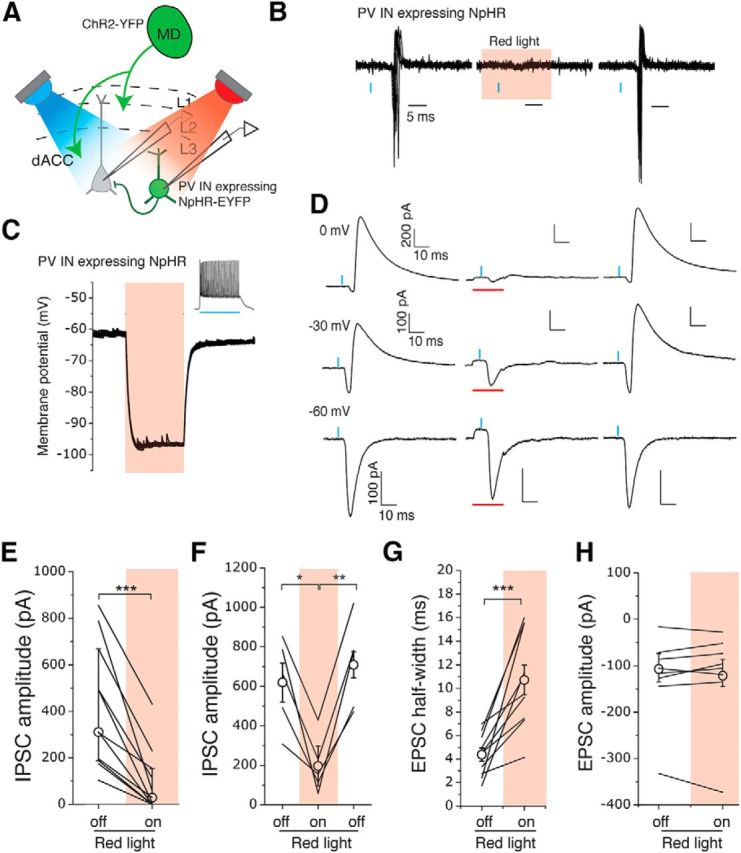
PV INs mediate MD-driven feedforward inhibition in the dACC. A, Schematic of the experimental approach. A blue light (λ = 470 nm) and a red light (λ = 625 nm) were used to activate ChR2 and eNpHR3.0, respectively. B, Sample traces of action potentials recorded in cell-attached mode from a PV IN expressing eNpHR3.0 in response to photo-activation of MD inputs with blue light pulses (blue bars), before, during, and after the presentation of red light (red shading) to activate eNpHR3.0. Red light was triggered 5 ms before the onset of the 0.5 ms blue light pulse and illuminated continuously for 20 ms. C, Photostimulation caused potent hyperpolarization in PV INs expressing eNpHR3.0. Inset, The 500 ms blue light pulse induced fast spiking in an eNpHR3.0-expressing PV IN. D, Sample traces of synaptic currents recorded from layer 3 PNs in the dACC in response to photostimulation (blue bars) of inputs from the MD, before, during, and after the presentation of red light (red bars) to inhibit PV INs. The presentation of red light reversibly suppressed the MD-driven IPSCs recorded at either 0 mV or −30 mV holding potential. E, The peak amplitude of MD-driven IPSCs recorded from PNs at 0 mV was significantly reduced when PV INs were silenced. ***p < 0.001 (Wilcoxon matched-pairs signed-rank test). F, Silencing PV INs reversibly suppressed the MD-driven IPSCs. *p < 0.05 (one-way repeated-measures ANOVA followed by Tukey's test). **p < 0.01 (one-way repeated-measures ANOVA followed by Tukey's test). G, H, Silencing of PV INs increased the half-width (***p < 0.001, paired t test) (G) but did not affect the amplitude (p > 0.05, Wilcoxon matched-pairs signed rank test) (H) of MD-driven EPSCs recorded in PNs at −30 mV holding potential. E, H, Data are median ± interquartile range. All other data are mean ± SEM.
To determine whether the SOM neurons, another major class of inhibitory interneuron, also contribute to the MD-driven feedforward inhibition in the dACC, we repeated the above optogenetic inhibition experiment, with the exception that SOM INs, rather than PV INs, were inhibited. For this purpose, we used the SOM-Cre mice, in which Cre is expressed under the endogenous SOM promoter (Taniguchi et al., 2011) (Fig. 8A).
Figure 8.
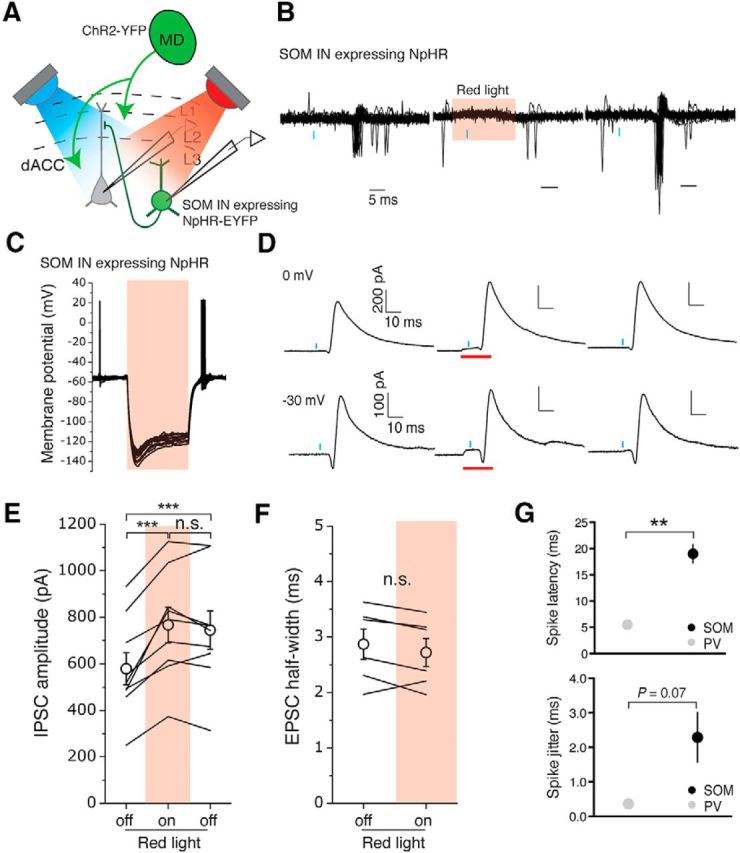
SOM INs do not mediate MD-driven feedforward inhibition in the dACC. A, Schematic of the experimental approach. A blue light (λ = 470 nm) and a red light (λ = 625 nm) were used to activate ChR2 and eNpHR3.0, respectively. B, Sample traces of action potentials recorded in cell-attached mode from a SOM IN expressing eNpHR3.0 in response to photo-activation of MD inputs with blue light pulses (blue bars), before, during, and after the presentation of red light (red shading) to activate eNpHR3.0. Red light was triggered 5 ms before the onset of the 0.5 ms blue light pulse and illuminated continuously for 20 ms. C, Photostimulation caused potent hyperpolarization in SOM INs expressing eNpHR3.0. D, Sample traces of synaptic currents recorded at 0 mV or −30 mV holding potential from layer 3 PNs in the dACC in response to photostimulation (blue bars) of inputs from the MD, before, during, and after the presentation of red light (red bars) to inhibit SOM INs. E, Silencing SOM INs induced a long-lasting increase in the peak amplitude of MD-driven IPSCs recorded in PNs. ***p < 0.001 (one-way repeated-measures ANOVA followed by Tukey's test). n.s., Not significant. F, Silencing SOM INs did not affect the half-width of MD-driven EPSCs recorded in PNs. G, Top, The MD-driven spiking of PV INs and SOM INs (recorded in cell-attached mode) exhibited differences in latency (top) (**p < 0.01 t test) and jitter (bottom) (p = 0.07 t test). Data are mean ± SEM.
Although activation of eNpHR3.0 effectively suppressed SOM IN firing in the dACC (Fig. 8B) and induced potent hyperpolarization (67.25 ± 12.36 mV, n = 5 cells/4 animals) (Fig. 8C), it did not dampen MD-driven feedforward inhibition onto L3 PNs (Fig. 8D); rather, feedforward inhibition was significantly enhanced (F(21.554,12.43) = 32.23, p < 0.0001, n = 9 cells/4 animals; one-way repeated-measures ANOVA) (Fig. 8E). Surprisingly, IPSC amplitude remained higher than baseline even after the red light was turned off (p < 0.001, n = 9 cells/4 animals; one-way repeated-measures ANOVA followed by Tukey's test) (Fig. 8E). Unlike inhibition of PV INs, we found no change in half-width of the MD-driven EPSCs onto PNs when SOM INs were silenced (light off, 2.87 ± 0.27 ms; light on, 2.72 ± 0.25 ms; n = 6 cells/3 animals, t(5) = 1.79, p > 0.05, paired t test) (Fig. 8F).
Like the PV INs, the SOM INs were capable of firing action potentials in response to the stimulation of inputs from the MD. However, the MD-driven spiking of PV INs had significantly shorter latencies than that of SOM INs and exhibited a trend toward lower spike jitter (latency to peak: PV, 5.46 ± 0.13 ms, n = 3 cells/3 animals, SOM, 18.99 ± 1.77 ms, n = 4 cells/3 animals, t(3.03) = 7.64, p < 0.01, t test; jitter: PV, 0.36 ± 0.09, n = 3 cells/3 animals, SOM, 2.29 ± 0.72 ms, n = 4 cells/3 animals, t(3.09) = 2.67, p = 0.07, t test) (Fig. 8G). These results suggest that, under our experimental conditions, SOM IN spiking was driven by local polysynaptic excitation as opposed to direct MD input. This is consistent with the observation that thalamocortical synapses onto SOM INs are weak and facilitate during repetitive activity in vitro (Tan et al., 2008; Xu et al., 2013) and in vivo (Ma et al., 2010). Recent studies further suggest that, at least in visual cortex, SOM INs lack feedforward input and are largely driven by local pyramidal neurons (Adesnik et al., 2012; El-Boustani and Sur, 2014).
Together, these results indicate that the majority of MD-driven disynaptic feedforward inhibition of L3 dACC PNs is mediated by PV INs. Removal of PV-mediated inhibition increased the duration of EPSCs in L3 PNs, suggesting that PV cells normally limit the time window during which PNs are capable of integrating excitatory inputs.
We next asked how PV-mediated feedforward inhibition affects spike output of L3 PNs in the dACC. First, we injected the MD of the PV-Cre;Ai14 mice with AAV-CAG-ChR2(H134R)-YFP and injected the dACC with AAV-Ef1a-DIO-eNpHR3.0-EYFP (Fig. 9A). After confirming that this approach led to specific expression of eNpHR3.0-EYFP in PV INs in the dACC (Fig. 9B), we recorded L3 PNs in whole-cell current-clamp mode at resting potential and measured PSPs in response to light stimulation of MD axons. We found that eNpHR3.0-mediated inhibition of PV INs during a time window encompassing that of MD axon stimulation converted subthreshold PSPs to full-fledged action potentials in 9 of 10 neurons recorded (p < 0.001, F(1.413,11.31) = 21.96, one-way repeated-measures ANOVA, n = 9 cells/5 mice) (Fig. 9C,D). In trials in which MD-driven postsynaptic responses remained subthreshold, inhibition of PV INs significantly and reversibly increased the amplitude of PSPs (p < 0.01, F(1.495,10.47) = 13.07, one-way repeated-measures ANOVA, n = 8 cells/6 mice) (Fig. 9E,F). On average, PV IN silencing increased PSP amplitude 22.2 ± 0.07% above baseline (p < 0.05, post hoc Tukey's test for multiple comparisons) (Fig. 9F) but did not significantly change PSP half-width (p > 0.05, F(1.515,10.60) = 2.10, one-way repeated-measures ANOVA, n = 8 cells/6 mice) (Fig. 9G).
Figure 9.
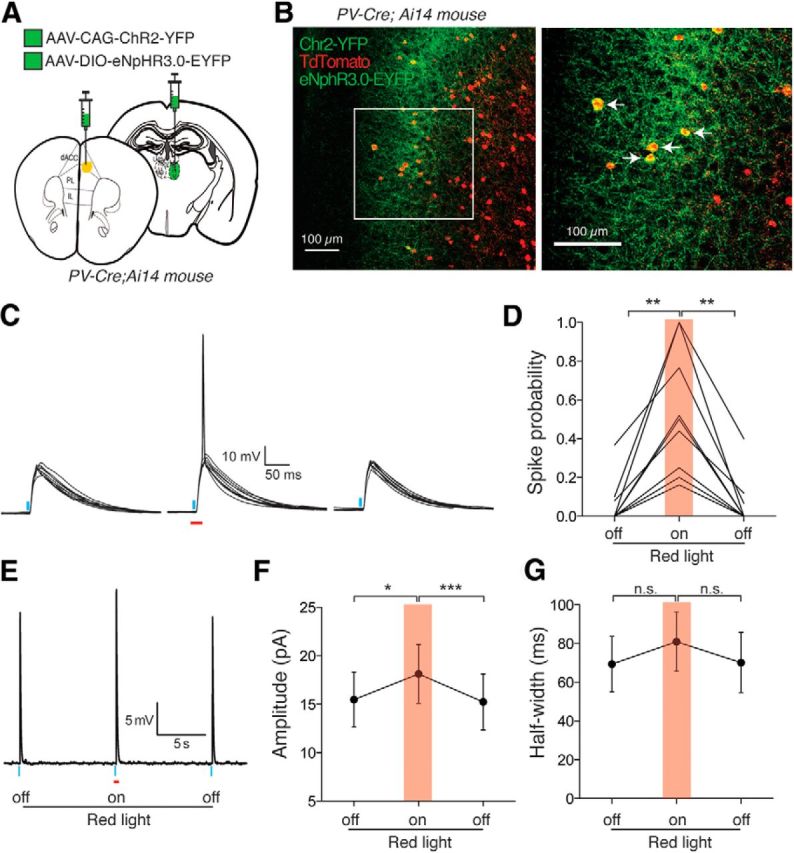
PV IN-mediated feedforward inhibition controls PN spike output in the dACC. A, Schematic of injection. AAV-CAG-ChR2-YFP was injected into MD while AAV-DIO-eNpHR3.0-GFP was injected into dACC of PV-Cre;Ai14 mice in which PV INs are labeled by Tdtomato. B, Labeling of ChR2-YFP+ axons originating from MD, Tdtomato+ PV INs, and eNpHR3.0-EYFP-infected PV INs in L3 of the dACC. Arrows indicate neurons in which eNpHR3.0-EYFP and Tdtomato were coexpressed. C, Sample PSP traces recorded from an L3 PN in response to blue light activation (0.5 ms; blue bars) of MD axons (ChR2+) in the presence or absence of red light illumination (20 ms; red bar) to inhibit local PV INs (eNpHR3.0+). Red light was triggered 5 ms before the onset of the blue light pulse. D, Silencing PV INs reversibly increased ChR2-evoked spike probability in L3 PNs. **p < 0.01 (one-way repeated-measures ANOVA followed by Tukey's test). E, Averaged subthreshold PSP sample trace, showing the effect of red light illumination. F, Silencing PV INs reversibly increased the peak amplitude of ChR2-evoked subthreshold PSPs in L3 PNs. *p < 0.05, ***p < 0.001 (one-way repeated-measures ANOVA followed by Tukey's test). G, Silencing PV INs did not significantly alter the half-width of ChR2-evoked subthreshold PSPs in L3 PNs. n.s., Not significant (one-way repeated-measures ANOVA). Data are mean ± SEM.
Discussion
The organization of the feedforward circuit in sensory thalamocortical systems has been well characterized (Bruno and Simons, 2002; Swadlow, 2002; Isaacson and Scanziani, 2011). It is known, for example, that thalamocortical input engages fast-spiking interneurons, presumably PV INs, in the somatosensory cortex (Bruno and Simons, 2002; Swadlow, 2002; Inoue and Imoto, 2006) and that these neurons mediate thalamocortical feedforward inhibition (Sun et al., 2006; Daw et al., 2007). However, whether the MD (a higher-order thalamic nucleus that does not receive input from the periphery) communicates with the cortex in a similar manner remains unclear.
In this study, we examined feedforward inhibition in the MD-dACC circuit and investigated its underlying mechanisms. By combining electrophysiology, rabies virus-assisted circuit tracing, and optogenetic techniques, we show that MD inputs drive disynaptic feedforward inhibition in the dACC and that MD neurons directly synapse onto and activate PV INs in the dACC. Furthermore, we demonstrate that PV INs in the dACC are the major mediator of this feedforward inhibition, as optogenetic silencing of these neurons greatly suppressed, and in some cases abolished, feedforward inhibition. Silencing local PV INs increased the duration, but not the amplitude, of EPSCs in PNs of dACC, suggesting that PV IN-mediated feedforward inhibition in dACC controls the temporal integration window of PNs, akin to the thalamically recruited inhibition in sensory cortices (Daw et al., 2007; Hull and Scanziani, 2007; Chittajallu and Isaac, 2010; Sotres-Bayon and Quirk, 2010; Isaacson and Scanziani, 2011). Finally, we found that optogenetic silencing of PV INs increased MD-driven spike probability of L3 PNs in the dACC, indicating that PV IN-mediated feedforward inhibition limits the output of PNs in this brain area.
Our results indicate that the SOM INs, which constitute the other major population of cortical inhibitory INs (Rudy et al., 2011), do not mediate thalamocortical feedforward inhibition in the MD-dACC circuit. Instead, MD-dACC feedforward inhibition was enhanced when SOM INs were silenced, suggesting that SOM INs suppress feedforward inhibition under basal conditions. Recent studies indicate that a subpopulation of SOM INs in the somatosensory cortex inhibit fast-spiking INs (Xu et al., 2013). Therefore, it is possible that SOM INs inhibit PV INs in the dACC, and the enhancement of feedforward inhibition upon silencing local SOM INs is caused by the disinhibition of PV INs. We observed SOM INs in the dACC that were spontaneously active (Fig. 8B), raising the possibility that a subset of SOM INs tonically inhibit PV INs. Interestingly, it was shown in visual cortex that in vivo activation of SOM INs suppresses PV IN spiking more strongly than PN spiking during visual processing (Cottam et al., 2013). One explanation for our finding that SOM INs did not contribute to feedforward inhibition in the MD-dACC pathway is that MD-driven spiking in SOM INs occurred at long latencies (∼14 ms after PV spiking) with high jitter, suggesting that SOM INs were driven by local polysynaptic excitation as opposed to direct thalamic input.
We observed several properties of the MD-dACC circuit that are distinct from those of the sensory thalamocortical pathways: (1) the strength of thalamocortical synapses onto PV INs was not significantly stronger than that onto nearby PNs in the MD-dACC circuit; in contrast, the strength of thalamocortical synapses onto fast-spiking INs is typically threefold to fourfold stronger than that onto nearby principal neurons in the sensory pathways (Gibson et al., 1999; Porter et al., 2001; Beierlein et al., 2003; Gabernet et al., 2005; Sun et al., 2006; Cruikshank et al., 2007); (2) in the MD-dACC circuit, the thalamocortical synapses onto PV INs and PNs had similar presynaptic release probability, whereas in the sensory pathways, the thalamocortical synapses onto fast-spiking INs have much higher release probability than those onto PNs (Beierlein et al., 2003; Cruikshank et al., 2010); and (3) although inhibition normally lags behind thalamocortical excitation, the latency from excitation to inhibition onset we observed in the MD-dACC circuit (∼4.8 ms) is longer than what has been reported in the sensory thalamocortical pathways (1–3 ms) (Agmon and Connors, 1991; Pouille and Scanziani, 2001; Gabernet et al., 2005; Cruikshank et al., 2010). These properties, while suggesting weaker feedforward inhibition in the MD-dACC circuit than in the sensory thalamocortical pathways, may allow for greater flexibility or plasticity in the dACC. Nevertheless, how these specific features of feedforward inhibition suit the functional role of the MD-dACC circuit is unclear.
In sensory processing, feedforward inhibition filters out stimulus-evoked thalamocortical responses of cortical neurons for which the stimulus is “nonpreferred” (Wilent and Contreras, 2005) and increases the temporal precision of the cortical operation (Pouille and Scanziani, 2001; Wehr and Zador, 2003). It has been previously reported that GABA blockade broadens spike tuning and reduces stimulus selectivity in auditory (Wang et al., 2000; Chang et al., 2005) and visual (Leventhal et al., 2003; Katzner et al., 2011) cortices. In the PFC, GABAergic interneurons are thought to play an important role in establishing spatial tuning (Rao et al., 2000) and temporal coding (Constantinidis et al., 2002) during an oculomotor working memory task in primates. Notably, inputs from the MD suppress hippocampal-driven activation of neurons in the mPFC, a process that is thought to be important for cognitive functions subserved by the hippocampal-mPFC circuit (Floresco and Grace, 2003). Our findings reveal a mechanism for this MD gating of hippocampal transmission to the mPFC and suggest that this may be a more general mechanism for gating afferent inputs to dACC.
Recent studies have highlighted the role of mPFC PV INs in regulating motivational and aversive behaviors. For example, phasic inhibition of a subpopulation of PV INs in the mPFC correlates with fear expression (Courtin et al., 2014), and chemogenetic inhibition of mPFC PV INs promotes maladaptive behavioral responses to stress (Perova et al., 2015). On the other hand, optogenetic activation of PV INs in the mPFC facilitates extinction of an appetitive operant behavior (Sparta et al., 2014). These findings suggest that reduced activity of PV INs in the mPFC can bias animals toward passive behavioral responses, whereas engagement of these PV INs may be required for flexible, adaptive behaviors. Interestingly, chemogenetic inhibition of MD is reported to impair working memory (Parnaudeau et al., 2013) and a host of flexible goal-directed behaviors (Parnaudeau et al., 2015). The extent to which these behavioral effects can be explained by removal of feedforward drive onto PV INs in the mPFC remains to be explored.
Given that the mPFC exhibits a high degree of recurrent connectivity (Mason et al., 1991; Thomson et al., 1993; Wang et al., 2006) and is extensively interconnected with subcortical structures (Gabbott et al., 2005), it has been proposed that mPFC is more vulnerable to aberrant recurrent excitation if left unchecked, such that subtle changes in mPFC excitation/inhibition balance could broadly dysregulate activity patterns in the brain and promote psychiatric symptoms (Yizhar et al., 2011).
Intriguingly, reduced functional coupling between the PFC and MD (Seidman et al., 1994; Mitelman et al., 2005; Zhou et al., 2007; Schlösser et al., 2008; Minzenberg et al., 2009) and deficits in prefrontal cortical PV INs (Lisman et al., 2008; Lewis et al., 2012) are both observed in schizophrenia, a mental disorder that is marked by cognitive impairment (Lesh et al., 2011). However, the exact nature of MD-PFC circuit dysfunction in schizophrenia remains unclear. Our findings regarding the role of PV INs in mediating feedforward inhibition in the MD-dACC circuit provide mechanistic insight into how thalamofrontal circuits support cognition and may be perturbed in diseases, such as schizophrenia.
Footnotes
This work was supported by National Institutes of Health training grant to K.D. and National Institutes of Health grants to Z.J.H and B.L., Brain and Behavior Research Foundation to Z.J.H. and B.L., and Stanley Family Foundation to Z.J.H. and B.L. We thank Anthony Zador and Ashlan Reid for comments and members of the B.L. laboratory for discussions.
The authors declare no competing financial interests.
References
- Adesnik H, Bruns W, Taniguchi H, Huang ZJ, Scanziani M. A neural circuit for spatial summation in visual cortex. Nature. 2012;490:226–231. doi: 10.1038/nature11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-J. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Gibson JR, Connors BW. Two dynamically distinct inhibitory networks in layer 4 of the neocortex. J Neurophysiol. 2003;90:2987–3000. doi: 10.1152/jn.00283.2003. [DOI] [PubMed] [Google Scholar]
- Bissonette GB, Powell EM, Roesch MR. Neural structures underlying set-shifting: roles of medial prefrontal cortex and anterior cingulate cortex. Behav Brain Res. 2013;250:91–101. doi: 10.1016/j.bbr.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno RM, Simons DJ. Feedforward mechanisms of excitatory and inhibitory cortical receptive fields. J Neurosci. 2002;22:10966–10975. doi: 10.1523/JNEUROSCI.22-24-10966.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Connors BW. Thalamocortical synapses. Prog Neurobiol. 1997;51:581–606. doi: 10.1016/S0301-0082(97)00002-6. [DOI] [PubMed] [Google Scholar]
- Chang EF, Bao S, Imaizumi K, Schreiner CE, Merzenich MM. Development of spectral and temporal response selectivity in the auditory cortex. Proc Natl Acad Sci U S A. 2005;102:16460–16465. doi: 10.1073/pnas.0508239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Isaac JT. Emergence of cortical inhibition by coordinated sensory-driven plasticity at distinct synaptic loci. Nat Neurosci. 2010;13:1240–1248. doi: 10.1038/nn.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinidis C, Williams GV, Goldman-Rakic PS. A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci. 2002;5:175–180. doi: 10.1038/nn799. [DOI] [PubMed] [Google Scholar]
- Cottam JC, Smith SL, Häusser M. Target-specific effects of somatostatin-expressing interneurons on neocortical visual processing. J Neurosci. 2013;33:19567–19578. doi: 10.1523/JNEUROSCI.2624-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtin J, Chaudun F, Rozeske RR, Karalis N, Gonzalez-Campo C, Wurtz H, Abdi A, Baufreton J, Bienvenu TC, Herry C. Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature. 2014;505:92–96. doi: 10.1038/nature12755. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron. 2010;65:230–245. doi: 10.1016/j.neuron.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Ashby MC, Isaac JT. Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat Neurosci. 2007;10:453–461. doi: 10.1038/nn1866. [DOI] [PubMed] [Google Scholar]
- Divac I, Mogensen J, Petrovic-Minic B, Zilles K, Regidor J. Cortical projections of the thalamic mediodorsal nucleus in the rat: definition of the prefrontal cortex. Acta Neurobiol Exp. 1993;53:425–429. [PubMed] [Google Scholar]
- El-Boustani S, Sur M. Response-dependent dynamics of cell-specific inhibition in cortical networks in vivo. Nat Commun. 2014;5:5689. doi: 10.1038/ncomms6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, Grace AA. Gating of hippocampal-evoked activity in prefrontal cortical neurons by inputs from the mediodorsal thalamus and ventral tegmental area. J Neurosci. 2003;23:3930–3943. doi: 10.1523/JNEUROSCI.23-09-03930.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, Braaksma DN, Phillips AG. Thalamic-cortical-striatal circuitry subserves working memory during delayed responding on a radial arm maze. J Neurosci. 1999;19:11061–11071. doi: 10.1523/JNEUROSCI.19-24-11061.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, Scanziani M. Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron. 2005;48:315–327. doi: 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Gigg J, Tan AM, Finch DM. Glutamatergic hippocampal formation projections to prefrontal cortex in the rat are regulated by GABAergic inhibition and show convergence with glutamatergic projections from the limbic thalamus. Hippocampus. 1994;4:189–198. doi: 10.1002/hipo.450040209. [DOI] [PubMed] [Google Scholar]
- Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidbreder CA, Groenewegen HJ. The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci Biobehav Rev. 2003;27:555–579. doi: 10.1016/j.neubiorev.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Scanziani M. It's about time for thalamocortical circuits. Nat Neurosci. 2007;10:400–402. doi: 10.1038/nn0407-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Isaacson JS, Scanziani M. Postsynaptic mechanisms govern the differential excitation of cortical neurons by thalamic inputs. J Neurosci. 2009;29:9127–9136. doi: 10.1523/JNEUROSCI.5971-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt PR, Aggleton JP. Neurotoxic lesions of the dorsomedial thalamus impair the acquisition but not the performance of delayed matching to place by rats: a deficit in shifting response rules. J Neurosci. 1998;18:10045–10052. doi: 10.1523/JNEUROSCI.18-23-10045.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Imoto K. Feedforward inhibitory connections from multiple thalamic cells to multiple regular-spiking cells in layer 4 of the somatosensory cortex. J Neurophysiol. 2006;96:1746–1754. doi: 10.1152/jn.00301.2006. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanichay RT, Silver RA. Synaptic and cellular properties of the feedforward inhibitory circuit within the input layer of the cerebellar cortex. J Neurosci. 2008;28:8955–8967. doi: 10.1523/JNEUROSCI.5469-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzner S, Busse L, Carandini M. GABAA inhibition controls response gain in visual cortex. J Neurosci. 2011;31:5931–5941. doi: 10.1523/JNEUROSCI.5753-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerley SW, Walton ME. Decision making and reward in frontal cortex: complementary evidence from neurophysiological and neuropsychological studies. Behav Neurosci. 2011;125:297–317. doi: 10.1037/a0023575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Yokofujita J, Murakami K. An ultrastructural study of the neural circuit between the prefrontal cortex and the mediodorsal nucleus of the thalamus. Prog Neurobiol. 1998;54:417–458. doi: 10.1016/S0301-0082(97)00070-1. [DOI] [PubMed] [Google Scholar]
- Kuroda M, Yokofujita J, Oda S, Price JL. Synaptic relationships between axon terminals from the mediodorsal thalamic nucleus and gamma-aminobutyric acidergic cortical cells in the prelimbic cortex of the rat. J Comp Neurol. 2004;477:220–234. doi: 10.1002/cne.20249. [DOI] [PubMed] [Google Scholar]
- Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36:316–338. doi: 10.1038/npp.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal AG, Wang Y, Pu M, Zhou Y, Ma Y. GABA and its agonists improved visual cortical function in senescent monkeys. Science. 2003;300:812–815. doi: 10.1126/science.1082874. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, Li B. Experience-dependent modification of a central amygdala fear circuit. Nat Neurosci. 2013;16:332–339. doi: 10.1038/nn.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Mai X, Liu C. The default mode network and social understanding of others: what do brain connectivity studies tell us. Front Hum Neurosci. 2014;8:74. doi: 10.3389/fnhum.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma WP, Liu BH, Li YT, Huang ZJ, Zhang LI, Tao HW. Visual representations by cortical somatostatin inhibitory neurons: selective but with weak and delayed responses. J Neurosci. 2010;30:14371–14379. doi: 10.1523/JNEUROSCI.3248-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason A, Nicoll A, Stratford K. Synaptic transmission between individual pyramidal neurons of the rat visual cortex in vitro. J Neurosci. 1991;11:72–84. doi: 10.1523/JNEUROSCI.11-01-00072.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátyás F, Lee J, Shin HS, Acsády L. The fear circuit of the mouse forebrain: connections between the mediodorsal thalamus, frontal cortices and basolateral amygdala. Eur J Neurosci. 2014;39:1810–1823. doi: 10.1111/ejn.12610. [DOI] [PubMed] [Google Scholar]
- Minzenberg MJ, Laird AR, Thelen S, Carter CS, Glahn DC. Meta-analysis of 41 functional neuroimaging studies of executive function in schizophrenia. Arch Gen Psychiatry. 2009;66:811–822. doi: 10.1001/archgenpsychiatry.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AS, Chakraborty S. What does the mediodorsal thalamus do? Front Syst Neurosci. 2013;7:37. doi: 10.3389/fnsys.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman SA, Byne W, Kemether EM, Hazlett EA, Buchsbaum MS. Metabolic disconnection between the mediodorsal nucleus of the thalamus and cortical Brodmann's areas of the left hemisphere in schizophrenia. J Psychiatry. 2005;162:1733–1735. doi: 10.1176/appi.ajp.162.9.1733. [DOI] [PubMed] [Google Scholar]
- Mittmann W, Koch U, Häusser M. Feed-forward inhibition shapes the spike output of cerebellar Purkinje cells. J Physiol. 2005;563:369–378. doi: 10.1113/jphysiol.2004.075028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamichi K, Amat F, Moussavi F, Wang C, Wickersham I, Wall NR, Taniguchi H, Tasic B, Huang ZJ, He Z, Callaway EM, Horowitz MA, Luo L. Cortical representations of olfactory input by trans-synaptic tracing. Nature. 2011;472:191–196. doi: 10.1038/nature09714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnaudeau S, O'Neill PK, Bolkan SS, Ward RD, Abbas AI, Roth BL, Balsam PD, Gordon JA, Kellendonk C. Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron. 2013;77:1151–1162. doi: 10.1016/j.neuron.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnaudeau S, Taylor K, Bolkan SS, Ward RD, Balsam PD, Kellendonk C. Mediodorsal thalamus hypofunction impairs flexible goal-directed behavior. Biol Psychiatry. 2015;77:445–453. doi: 10.1016/j.biopsych.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perova Z, Delevich K, Li B. Depression of excitatory synapses onto parvalbumin interneurons in the medial prefrontal cortex in susceptibility to stress. J Neurosci. 2015;35:3201–3206. doi: 10.1523/JNEUROSCI.2670-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC. The functional organization of the barrel cortex. Neuron. 2007;56:339–355. doi: 10.1016/j.neuron.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Porter JT, Johnson CK, Agmon A. Diverse types of interneurons generate thalamus-evoked feedforward inhibition in the mouse barrel cortex. J Neurosci. 2001;21:2699–2710. doi: 10.1523/JNEUROSCI.21-08-02699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray JP, Price JL. The organization of the thalamocortical connections of the mediodorsal thalamic nucleus in the rat, related to the ventral forebrain-prefrontal cortex topography. J Comp Neurol. 1992;323:167–197. doi: 10.1002/cne.903230204. [DOI] [PubMed] [Google Scholar]
- Romanides AJ, Duffy P, Kalivas PW. Glutamatergic and dopaminergic afferents to the prefrontal cortex regulate spatial working memory in rats. Neuroscience. 1999;92:97–106. doi: 10.1016/S0306-4522(98)00747-7. [DOI] [PubMed] [Google Scholar]
- Rose JE, Woolsey CN. Structure and relations of limbic cortex and anterior thalamic nuclei in rabbit and cat. J Comp Neurol. 1948;89:279–347. doi: 10.1002/cne.900890307. [DOI] [PubMed] [Google Scholar]
- Rotaru DC, Barrionuevo G, Sesack SR. Mediodorsal thalamic afferents to layer III of the rat prefrontal cortex: synaptic relationships to subclasses of interneurons. J Comp Neurol. 2005;490:220–238. doi: 10.1002/cne.20661. [DOI] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2011;71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff ML, Reyes AD. Characterization of thalamocortical responses of regular-spiking and fast-spiking neurons of the mouse auditory cortex in vitro and in silico. J Neurophysiol. 2012;107:1476–1488. doi: 10.1152/jn.00208.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlösser RG, Koch K, Wagner G, Nenadic I, Roebel M, Schachtzabel C, Axer M, Schultz C, Reichenbach JR, Sauer H. Inefficient executive cognitive control in schizophrenia is preceded by altered functional activation during information encoding: an fMRI study. Neuropsychologia. 2008;46:336–347. doi: 10.1016/j.neuropsychologia.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Seidman LJ, Yurgelun-Todd D, Kremen WS, Woods BT, Goldstein JM, Faraone SV, Tsuang MT. Relationship of prefrontal and temporal lobe MRI measures to neuropsychological performance in chronic schizophrenia. Biol Psychiatry. 1994;35:235–246. doi: 10.1016/0006-3223(94)91254-8. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Quirk GJ. Prefrontal control of fear: more than just extinction. Curr Opin Neurobiol. 2010;20:231–235. doi: 10.1016/j.conb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparta DR, Hovelsø N, Mason AO, Kantak PA, Ung RL, Decot HK, Stuber GD. Activation of prefrontal cortical parvalbumin interneurons facilitates extinction of reward-seeking behavior. J Neurosci. 2014;34:3699–3705. doi: 10.1523/JNEUROSCI.0235-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR, Prince DA. Barrel cortex microcircuits: thalamocortical feedforward inhibition in spiny stellate cells is mediated by a small number of fast-spiking interneurons. J Neurosci. 2006;26:1219–1230. doi: 10.1523/JNEUROSCI.4727-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swadlow HA. Thalamocortical control of feed-forward inhibition in awake somatosensory “barrel” cortex. Philos Trans R Soc Lond B Biol Sci. 2002;357:1717–1727. doi: 10.1098/rstb.2002.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swadlow HA. Fast-spike interneurons and feedforward inhibition in awake sensory neocortex. Cereb Cortex. 2003;13:25–32. doi: 10.1093/cercor/13.1.25. [DOI] [PubMed] [Google Scholar]
- Tan Z, Hu H, Huang ZJ, Agmon A. Robust but delayed thalamocortical activation of dendritic-targeting inhibitory interneurons. Proc Natl Acad Sci U S A. 2008;105:2187–2192. doi: 10.1073/pnas.0710628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM, Deuchars J, West DC. Large, deep layer pyramid-pyramid single axon EPSPs in slices of rat motor cortex display paired pulse and frequency-dependent depression, mediated presynaptically and self-facilitation, mediated postsynaptically. J Neurophysiol. 1993;70:2354–2369. doi: 10.1152/jn.1993.70.6.2354. [DOI] [PubMed] [Google Scholar]
- Wang J, Caspary D, Salvi RJ. GABA-A antagonist causes dramatic expansion of tuning in primary auditory cortex. Neuroreport. 2000;11:1137–1140. doi: 10.1097/00001756-200004070-00045. [DOI] [PubMed] [Google Scholar]
- Wang Y, Markram H, Goodman PH, Berger TK, Ma J, Goldman-Rakic PS. Heterogeneity in the pyramidal network of the medial prefrontal cortex. Nat Neurosci. 2006;9:534–542. doi: 10.1038/nn1670. [DOI] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–446. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- Wilent WB, Contreras D. Dynamics of excitation and inhibition underlying stimulus selectivity in rat somatosensory cortex. Nat Neurosci. 2005;8:1364–1370. doi: 10.1038/nn1545. [DOI] [PubMed] [Google Scholar]
- Xu H, Jeong HY, Tremblay R, Rudy B. Neocortical somatostatin-expressing GABAergic interneurons disinhibit the thalamorecipient layer 4. Neuron. 2013;77:155–167. doi: 10.1016/j.neuron.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Methods. 2006;3:785–792. doi: 10.1038/nmeth936. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, Deisseroth K. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Liang M, Jiang T, Tian L, Liu Y, Liu Z, Liu H, Kuang F. Functional disconnectivity of the dorsolateral prefrontal cortex in first-episode schizophrenia using resting-state fMRI. Neurosci Lett. 2007;417:297–302. doi: 10.1016/j.neulet.2007.02.081. [DOI] [PubMed] [Google Scholar]