

Abstract
Mitochondria are dynamically transported in and out of neuronal processes to maintain neuronal excitability and synaptic function. In higher eukaryotes, the mitochondrial GTPase Miro binds Milton/TRAK adaptor proteins linking microtubule motors to mitochondria. Here we show that Drosophila Miro (dMiro), which has previously been shown to be required for kinesin-driven axonal transport, is also critically required for the dynein-driven distribution of mitochondria into dendrites. In addition, we used the loss-of-function mutations dMiroT25N and dMiroT460N to determine the significance of dMiro's N-terminal and C-terminal GTPase domains, respectively. Expression of dMiroT25N in the absence of endogenous dMiro caused premature lethality and arrested development at a pupal stage. dMiroT25N accumulated mitochondria in the soma of larval motor and sensory neurons, and prevented their kinesin-dependent and dynein-dependent distribution into axons and dendrites, respectively. dMiroT25N mutant mitochondria also were severely fragmented and exhibited reduced kinesin and dynein motility in axons. In contrast, dMiroT460N did not impair viability, mitochondrial size, or the distribution of mitochondria. However, dMiroT460N reduced dynein motility during retrograde mitochondrial transport in axons. Finally, we show that substitutions analogous to the constitutively active Ras-G12V mutation in dMiro's N-terminal and C-terminal GTPase domains cause neomorphic phenotypic effects that are likely unrelated to the normal function of each GTPase domain. Overall, our analysis indicates that dMiro's N-terminal GTPase domain is critically required for viability, mitochondrial size, and the distribution of mitochondria out of the neuronal soma regardless of the employed motor, likely by promoting the transition from a stationary to a motile state.
Keywords: Miro, mitochondria, transport
Introduction
Transport of mitochondria into axons and dendrites is mediated by the microtubule (MT) motors kinesin and dynein, and is critical for sustaining neuronal excitability, synaptic transmission, and survival. Impairments of mitochondrial transport also contribute to a number of neurodegenerative disorders (for review, see Mattson et al., 2008; Zinsmaier et al., 2009; Saxton and Hollenbeck, 2012; Sheng and Cai, 2012; Millecamps and Julien, 2013). However, mechanisms regulating mitochondrial motility out of the neuronal soma remain enigmatic.
The atypical GTPase Miro is an outer mitochondrial membrane protein found in almost all eukaryotes and typically exhibits two GTPase domains and two Ca2+-binding EF-hand domains (Vlahou et al., 2011). Despite this evolutionary conservation, divergent roles have been suggested for Miro in lower and higher eukaryotes. In yeast, Miro is primarily required for mitochondrial homeostasis, facilitating the dissociation of mitochondria from the endoplasmic reticulum (ER) after ER-associated mitochondrial division and phospholipid exchange. In metazoa, Miro is primarily required for mitochondrial transport, although it also has been implicated in mitochondrial homeostasis (for review, see Lee and Lu, 2014).
In flies, Drosophila Miro (dMiro) and Milton are both critically required for transport of mitochondria into axons (Stowers et al., 2002; Guo et al., 2005; Russo et al., 2009). Mechanistically, dMiro recruits cytosolic Milton, which serves as an adaptor linking the heavy chain of conventional kinesin (KHC) to mitochondria (Stowers et al., 2002; Glater et al., 2006). However, whether dMiro and Milton are required for dynein-mediated transport into dendrites remains unclear.
In mammals, mitochondrial motor linkage is mediated by two Miro genes, Miro1 and Miro2, and two Milton-like adaptor genes, trafficking protein kinesin binding 1 (TRAK1) and TRAK2 (i.e., Milton-1/OIP106 and Milton-2/GRIF-1, respectively; Fransson et al., 2003, 2006; Brickley et al., 2005; Koutsopoulos et al., 2010). TRAK1 is preferentially localized to axons, facilitating kinesin-mediated anterograde mitochondrial transport, whereas TRAK2 is abundant in dendrites, facilitating dynein-mediated anterograde transport (van Spronsen et al., 2013). Miro's EF-hand domains control the activity of the Milton/TRAK-motor complex. In Drosophila, Ca2+ activation of the EF hands triggers binding of Miro kinesin's motor domain, disengaging kinesin from MTs, while in mammals it triggers the release of kinesin from the Miro/TRAK complex (MacAskill et al., 2009a; Wang and Schwarz, 2009).
The role of Miro's two GTPase domains for mitochondrial transport has been less clear. Biochemical studies showed that loss of Miro's N-terminal or C-terminal GTPase activity has no effect on the ability of Miro1 and Miro2 to coimmunoprecipitate TRAK proteins (Fransson et al., 2006). Loss of Miro1's N-terminal GTPase activity also had no effect on the mitochondrial recruitment of TRAK2 (MacAskill et al., 2009b). However, a presumed constitutively active mutation of the N-terminal GTPase domain abolished the recruitment of TRAK2 in hippocampal cell cultures and altered the mitochondrial distribution (MacAskill et al., 2009b).
We performed a genetic analysis of analogous mutations in dMiro's two GTPase domains. Our study suggests that only dMiro's N-terminal GTPase domain is critically required for viability, mitochondrial size, and the kinesin- and dynein-dependent distribution of mitochondria into axons and dendrites, respectively.
Materials and Methods
Fly stocks.
Flies were raised on standard cornmeal medium with dry yeast at 24°C. The strain expressing green fluorescent protein (GFP) tagged by an N-terminal mitochondrial localization signal (w1118, P[w+; UAS-mGFP]) was obtained from W. Saxton (University of California Santa Cruz, Santa Cruz, CA). The point mutations dmiroSD32 (29 bp deletion at Y89) and dmiroB682 (W105/stop) prematurely truncate dMiro within the N-terminal GTPase domain and are considered null mutations (Guo et al., 2005; Russo et al., 2009). The motor Gal4 drivers OK6 (Aberle et al., 2002) and elav-C155 (Lin and Goodman, 1994) were used to express all UAS-transgenes in a wild-type (w1118) or a heteroallelic dmiro-null mutant genetic background (dmiroB682/SD32). For expression in sensory neurons, we used the Gal4 drivers 21-7, C380, 477, 2-21, and 5-40 (Grueber et al., 2003; Song et al., 2007; Sanyal, 2009).
For all experiments, strains containing a homozygous UAS-dMiro transgene in a wild-type or heterozygous dmiro-null mutant genetic background (w1118; P[w+, UAS-dMiro-X] or w1118; P[w+, UAS-dMiro-X]; dmiroB682/TM6TbSb) were crossed to strains containing a homozygous Gal4 driver and a UAS-mGFP transgene in a wild-type or heterozygous dmiro-null background (w1118; P[w+, Gal4-driver]; P[w+, UAS-mGFP] or w1118, P[w+, UAS-mGFP], dmirosd32/TM6TbSb) to generate progeny that express a single copy of the UAS transgene driven by the Gal4 driver in the respective genetic background (w1118; P[w+, Gal4-driver]/P[w+, UAS-dMiro-X]; P[w+, UAS-mGFP]/+ or w1118; P[w+, Gal4-driver]/P[w+, UAS-dMiro-X]; dmirosd32, P[w+, UAS-mGFP]/dmiroB682). Progeny of either sex were used for all experiments, except experiments using the C380 Gal driver for which only female progeny were used.
Generation of dMiro mutant transgenes.
The previously generated myc-tagged dMiro cDNA (dMiro-RC; Guo et al., 2005) was used as a template for site-directed mutagenesis (Stratagene QuikChange Mutagenesis) using standard procedures and the following primers: T25N, 5′-GCCGGGGTGGGTGCGACGTCGTTGATTCTGTCTCTG-3′; A20V, 5′-GCTCGTCGGCGACGTCGGGGTGGG-3′; T460N, 5′-AAGGGATCAGGAAAGAATGGAATGTGCAGGGGATTC-3′; K455V, 5′-GTCATGTGATTGGACCAGTGGGATCAGGAAAGACTGG-3′. After verifying the introduced point mutations by DNA sequencing, mutant cDNAs were directionally cloned into a NotI- and KpnI-cleaved pUAST vector (Brand and Perrimon, 1993). Double mutants were generated by fusing cDNAs containing individual mutations in the N- and C-terminal GTPase domain. RsrII- and KpnI-cleaved cDNA fragments encoding the second GTPase domain were excised from pUAST-dMiroK455V and pUAST-dMiroT460N and inserted into RsrII/KpnI-cleaved pUAST-dMiroA20V and pUAST-dMiroT25N. pUAST plasmids were injected into w1118 embryos, generating transgenic animals (Rainbow Transgenic). At least two independent strains were obtained for each transgene. After outcrossing all other chromosomes, strains containing homozygous UAS transgenes on the second chromosome were generated containing a wild-type-like genetic background (w1118) or a dmiro-null mutant background (dmiroB682).
To generate a transgene that expresses only a dMiro-RC transcript under the control of a dmiro promoter, the region including intron 3, the alternative spliced exon 4, and intron 5 was excised from a genomic dmiro transgene and replaced in frame with the respective dMiroRC-specific exon. First, a genomic transgene was generated that contains the dMiro gene from 1975 bp upstream (chromosome 3R:24,039,705, flybase) to 3544 downstream (3R:24,034,186) of its major transcriptional start site (3R:24,037,730). The respective region was PCR amplified with the primers 5′-GCTGAGCCCGATCCCACACATCACC-3′ and 5′-CCTCGACAAGCCAGAGCTGTGTACAGACTC-3′, and cloned into a pUAST vector (pUAST-dMiro). Next, two DNA fragments containing small overlaps were PCR amplified for recombineering. The first fragment contained the 3′ end of the dmiro gene, spanning from the 5′ end of exon 5 to the end of the previously cloned 3′ DNA dmiro gene fragment of pUAST-dMiro [primers 5′-CCTGAGGCAATTCGGCCTGATGACGG-3′ (3R:24,035,001, 5′ end exon 5) and 5′-GTCACACCACAGAAGTAAGGTTCCTTCAC-3′ (in pUAST)]. The second fragment was amplified from a dMiro-RC cDNA in the pUAST-myc-dMiro-RC vector, and contained half of exon 3 just including a unique RsrII site to the 5′ end of exon 5 [primers 5′-GAGCGCCGCATCGATTTGGCC-3′ (3R:24,036,143 within exon 3) and 5′-CCGTCATCAGGCCGAATTGCCTCAGG-3′ (3R:24,035,001; 5′ end exon 5)]. The two PCR fragments were then combined by recombineering with a RsrII/KpnI-cut pUAST-dMiro vector using a GeneArt seamless cloning kit (Invitrogen). After DNA sequence verification, the single-isoform version of genomic dMiro was PCR amplified with primers containing a 5′ NotI or KpnI site (5′-GAGCGGCCGCGCTGAGCCCGATCCCACACATCACC-3′ and 5′-GAGGTACCCCTCGACAAGCCAGAGCTGTGTACAGAC-3′), cleaved with NotI and KpnI, and ligated into a NotI/KpnI-cut pBID vector (Wang et al., 2012). The pBID-genomic-dMiro-RC plasmid was injected into y1, M(vas-int.Dm)ZH-2A w*; M(3xP3-RFP.attP')ZH-22A embryos expressing ϕC31 integrase and containing an attP landing site on chromosome 2 at 22A (Bischof et al., 2007). Four independent lines were obtained and outcrossed to w1118. Two lines were crossed into a dmiro-null mutant background (dmiroB682).
Generation of dMiro antibody.
A N-terminally His-tagged dMiro-RC cDNA fragment (residues 1–590, pET100 TOPO vector) was expressed in BL21 (DE3) Escherichia coli cells. His-tagged dMiro was purified using Ni-TED agarose and standard denaturing conditions (Affymetrix). The purified protein was injected into two guinea pigs using a standard 60 d injection protocol (Cocalico Biologicals). The obtained antisera were screened by Western blots using both fly head protein extracts and bacterially expressed peptides. One of the obtained antiserums showed a positive signal and was retained. The specificity of the polyclonal serum GP5 was verified by the absence of a signal on Western blots and immunostainings using dmiro-null mutations.
Western blot analysis.
Five to 12 larval brains were homogenized in 50 μl of PBS supplemented with 0.2% Triton X-100 (PBS-T) with protease inhibitors (Sigma-Aldrich). Equivalents of two larval brains were separated by 10% SDS-PAGE gel, transferred onto nitrocellulose membranes, and immunostained using primary antibodies in PBS-T at 4°C overnight, followed by a 2 h incubation with HRP-conjugated secondary antibodies at room temperature. Primary antibodies used were guinea pig anti-dMiro (1:50,000), mouse anti-β-tubulin [1:1000; Developmental Studies Hybridoma Bank (DSHB), DSHB #E7; Research Resource Identifier (RRID): AB_2315513], and mouse anti-myc (1:1000; Cell Signaling Technology, #2276; RRID: AB_2314825) in PBS 0.1% Tween, 5% nonfat dry milk. Secondary antibodies used are anti-guinea pig-HRP (1:20,000; Santa Cruz Biotechnology, catalog #sc-2438; RRID: AB_650492) and anti-mouse-HRP (1:20,000; Pierce, catalog #32430; RRID: AB_1185566). Blots were imaged using ChemiDoc XRS (Bio-Rad).
Immunostainings.
Climbing third-instar Drosophila larvae were dissected in HL-6 solution and fixed in 4% paraformaldehyde, pH 7.3, for 1 h, in Bouin's solution for 3 min (for kinesin). After washing in PBS-T, pH 7.3, and blocking with Superblock (Thermo Scientific) or 0.2% BSA for 1 h, the preparation was incubated with primary antibody overnight at 4°C, washed 3× in PBST for 10 min, and incubated with secondary antibody for 2 h at room temperature. Confocal images were acquired within 2 h after final washing. The following antibodies and dilutions were used: anti-HRP-Cy3 at 1:250 (Jackson ImmunoResearch, #123-165-021); anti-GFP-AF488 at 1:500 (Invitrogen, #A21311; RRID: AB_221477); mouse anti-myc at 1:3000 (Cell Signaling Technology, #2276); anti-dynein at 1:250 (DHSB, #2C11-2-s); anti-KHC at 1:50,000 (Cytoskeleton, #AKIN010); mouse anti-Cysteine-string protein (anti-CSP) at 1:200 [DSHB, #DCSP-2 (6D6); RRID: AB_528183]; mouse anti-Bruchpilot (anti-Brp) at 1:400 (DSHB, #NC-82; RRID: AB_528108); RRID: AB_10707921); anti-mouse-Cy3 at 1:500 (Jackson ImmunoResearch, #715-165-150); goat anti-mouse-AF488 at 1:500 (Invitrogen, #A21121; RRID: AB_10053811); goat anti-rabbit-Cy3 at 1:500 (Jackson ImmunoResearch, #111-165-003; RRID: AB_2307387), and goat anti-HRP-AF647 at 1:500 (Jackson ImmunoResearch, #123-605-021; RRID: AB_2338967).
To immunostain sensory neurons in the body wall, dissected larvae were fixed for 15 min in 4% paraformaldehyde, pH 7.3, washed in PBS, and incubated at 4°C overnight in a 1:1000 dilution of anti-GFP Alexa Fluor 488 and a 1:400 dilution of anti-HRP-Cy3 in PBS with 0.2% or 0.5% Triton X-100.
Analysis of mitochondrial transport in axons.
Mitochondria were live imaged in motor neurons of dissected climbing third-instar larvae using HL-6 solution supplemented with 7 mm l-glutamate and 0.6 mm CaCl2, as described previously (Louie et al., 2008; Russo et al., 2009). Briefly, images of 1024 × 280 pixels (∼58.9 × 16.1 μm) were acquired from individual segmental nerves near the ventral nerve cord. A centered region of interest (ROI; 872 × 278 pixels) was photobleached for 180 s followed by the immediate acquisition of 200 images (1024 × 280 pixels, one frame per 1.006 s). The movements of ≤12 clearly labeled mitochondria were tracked if they remained visible in ≥30 consecutive frames using National Institutes of Health ImageJ imaging software (Schneider et al., 2012) and the plug-in MTrackJ (E. Meijering, University Medical Center of Rotterdam, Netherlands) as described previously (Louie et al., 2008). The obtained x–y–t coordinates of mitochondrial movements were analyzed with Java-based software that expands on the Excel-based analysis software described earlier (Louie et al., 2008). An individual movement was defined by a minimal displacement of 0.151 μm/s. Mitochondria undergoing anterograde and retrograde transport were distinguished based on their overall net movement direction before calculating the described parameters as described previously (Russo et al., 2009).
Quantification of motile mitochondrial densities in axons.
Images measuring 1024 × 280 pixels (∼58.9 × 16.1 μm) of mGFP-labeled mitochondria of larval motor axons were obtained at a rate of 1 s−1, pseudocolored in green (time, 0 s) and magenta (time, 120 s), and merged to visualize motile (green and magenta) and stationary (white) mitochondria. Since mitochondria marked green in the merged images were transported out of the ROI during the imaging period and magenta-marked mitochondria were transported into the ROI, only green- and white-marked mitochondria were used to determine the density of motile and stationary mitochondria at time 0. A mitochondrion that was displaced by at least three-quarters of its length was considered to be motile. The number of mitochondria per ROI was normalized to the respective area. The percentage of motile mitochondria was determined as a fraction of the total number of mitochondria in a nerve ROI.
Quantification of mitochondrial densities.
To determine the mitochondrial density at neuromuscular junctions (NMJs) of larval motor neurons (MNs) and dendrites of sensory neurons (SNs), respective confocal images were obtained from dissected third-instar larvae that were immunostained for mGFP-labeled mitochondria and neuronal membranes (anti-HRP). For analysis of NMJs, images were obtained from muscles 6/7 in segment 3-4. The area of a synaptic bouton marked by HRP and the respective area of the bouton occupied by mitochondria marked by mitoGFP was measured using ImageJ software to determine the fraction of the synaptic area occupied by mitochondria. For analysis of dendrites, images of ventral posterior da (vpda) SNs were obtained from segment A2. The length of primary and secondary dendrites and the number of mitochondria in the respective dendrites was measured using ImageJ software. The amount of dendritic mitochondria was normalized to dendritic length.
Tetramethylrhodamine methyl ester perchlorate imaging.
Third-instar larvae were dissected in HL-6 solution supplemented with 7 mm l-glutamic acid in the absence of Ca2+. Larvae were incubated for 25 min in HL-6 solution supplemented with 20 nm tetramethylrhodamine methyl ester perchlorate (TMRM; Invitrogen), rinsed briefly, and incubated for imaging in HL-6 solution containing 5 nm TMRM. Mitochondria at larval NMJs were imaged using a confocal microscope equipped with a multiargon 488 nm laser to detect mGFP and a HeNe 543 nm laser to detect TMRM. To normalize the TMRM signal, mitochondria were then depolarized using 3 μm carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma-Aldrich) in HL-6 solution containing 5 nm TMRM. After 3 min, images of the depolarized mitochondria were obtained using the red and green channels. ImageJ imaging software was used to measure the mean TMRM fluorescence intensity of presynaptic mitochondria in single slices before and after CCCP treatment. TMRM fluorescence was expressed as ΔF(Fbefore CCCP − Fafter CCCP)/Fbefore CCCP.
Statistical analysis.
All data with a non-Gaussian distribution are presented by box-whisker plots showing the median (line), the 25th–75th percentile (box) range, and the 10th–90th percentile (whisker) range. Data with a Gaussian distribution are presented by graph bars showing the mean ± SEM. Data were evaluated for their statistical significance by Mann–Whitney, one-way, or two-way ANOVA testing and the indicated post hoc tests using Prism software (Graphpad Software, www.graphpad.com). N and n indicate number of animals and mitochondria, respectively. p values of <0.05, <0.01, and <0.001 are indicated with one, two, and three asterisks, respectively.
Results
Our previous studies established that dMiro is critically required for the axonal transport of mitochondria in larval MNs of Drosophila (Guo et al., 2005; Russo et al., 2009). To determine whether this function requires the activity of dMiro's two GTPase domains, we generated dMiro cDNA transgenes containing established loss-of-function as well as potential gain-of-function mutations using the Gal4 system for targeted gene expression (Brand and Perrimon, 1993). To abolish GTPase activity, we introduced the amino acid substitutions T25N in the N-terminal GTPase domain of dMiro and T460N in the C-terminal GTPase domain of dMiro (Fig. 1A). Analogous substitutions were previously used in yeast (S19N and S462N) and mammalian Miro (N18 and N432). In yeast, S19N and S462N abolished GTP hydrolysis and failed to rescue mitochondrial defects of yeast Miro-deletion mutants (Kornmann et al., 2011; Koshiba et al., 2011; Murley et al., 2013). In mammals, the substitutions N18 and N432 were regarded as dominant-negative mutations (Fransson et al., 2003, 2006; MacAskill et al., 2009b) due to their similarity to the dominant-negative Ras-S17N mutation (Feig and Cooper, 1988; Feig, 1999; Cantrell, 2002). However, yeast MiroS19N and MiroS462N did not induce dominant phenotypes (Koshiba et al., 2011).
Figure 1.
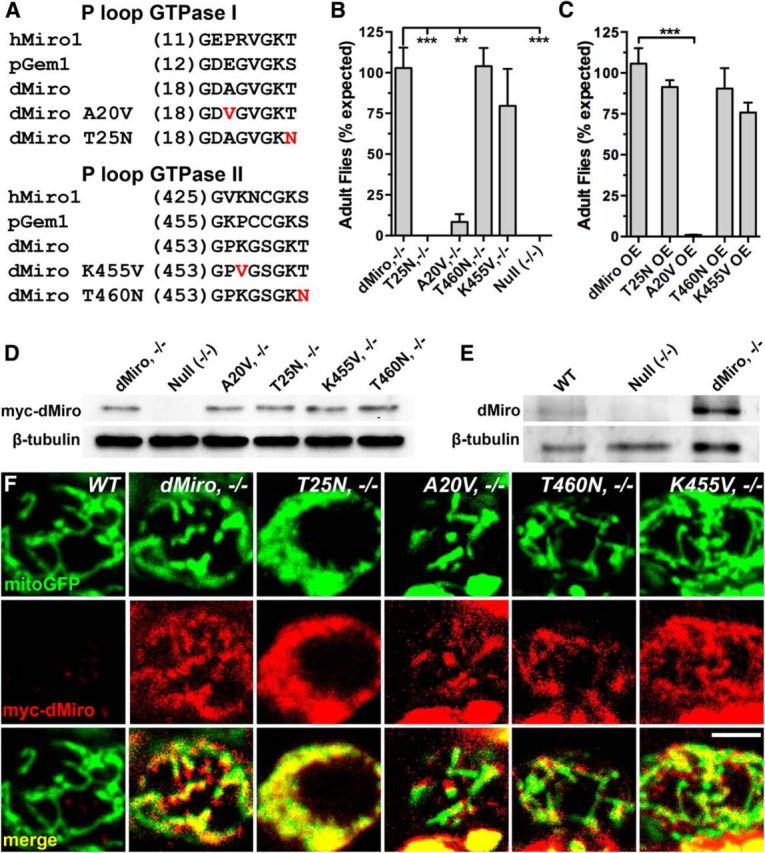
Transgenic expression of GTPase mutant dMiro proteins. A, Amino acid sequence of the P loop in the N-terminal (top) and C-terminal GTPase domain (bottom) of human Miro1 (hMiro1), yeast Miro (pGem1), and dMiro. The point mutations (T25N, T460N, A20V, K455V) introduced into the GTPase domains of myc-tagged dMiro (RC-transcript, FlyBase) are highlighted in red. B, C, Normal myc-dMiro (control) and mutant myc-dMiroA20V, myc-dMiroT25N, myc-dMiroK455V, and myc-dMiroT460N were pan-neuronally expressed in dmiro-null mutants (−/−, B) or (C) overexpressed (OE) in otherwise wild-type animals (w1118) using an elav-Gal4 driver. Viability was assayed daily by counting freshly hatched adult flies. Data represent means ± SEM of three independent matings. Only significant differences between control and mutant genotypes are indicated by asterisks (p < 0.05, N > 3, n > 100; 1-way ANOVA, Tukey's post test). D, E, Protein expression levels of myc-tagged GTPase-mutant dMiro proteins that were transgenically expressed in dmiro-null mutant (−/−) neurons. Immunoblots of larval brain protein extracts were stained with anti-Myc (D) or anti-dMiro (E) antibodies. β-Tubulin was used as loading control to normalize mutant protein levels to control (dMiro, 1.0; dMiroA20V, 0.9; dMiroT25N, 0.8; dMiroK455V, 1.1; dMiroT460N, 0.9; n = 3). Genotypes are indicated. F, Coimmunolocalization of the mitochondrial marker mitoGFP (green, top row) with myc-tagged dMiro proteins (red, middle row) in MN cell bodies of third-instar larvae. Wild-type control (WT) neurons lack transgenic expression of myc-dMiro. Transgenes encoding dMiroA20V, dMiroT25N, dMiroK455V, dMiroT460N, and normal dMiro were coexpressed with a transgene encoding mitoGFP in motor neurons of dmiro-null mutants with an Ok6-Gal4 driver. Scale bar, 2 μm.
We also introduced the amino acid substitutions A20V and K455V into dMiro's N- and C-terminal GTPase domain, respectively. Ectopic expression of analogous mutations in human Miro1 and Miro2 (13V and 427V) altered the structure and distribution of mitochondria in cultured cells. These dominant effects were suggested to be of a constitutively active nature (Fransson et al., 2003, 2006; Saotome et al., 2008; MacAskill et al., 2009b) since both substitutions were modeled after the constitutively active Ras-G12V mutation (Wittinghofer, 1998). However, since the two respective amino acids are divergent among human Miro, yeast Miro, and dMiro (Fig. 1A), the genetic nature of the mutations required further studies.
To verify proper expression of the myc-tagged mutant proteins, we expressed the respective cDNA transgenes in neurons of heteroallelic dmiroB682/SD32-null mutant animals using the pan-neuronal elav-Gal4 driver. Expression levels of both normal and mutant dMiro proteins were comparable among each other (±0.2-fold of control, p > 0.05; Fig. 1D) but significantly higher than levels of endogenous dMiro (Fig. 1D,E). Immunostainings using anti-myc antibodies confirmed a mitochondrial localization of the transgenically expressed GTPase mutant dMiro proteins (Fig. 1F).
dMiro's N-terminal GTPase domain is critical for survival
An interesting aspect of dMiro's role for mitochondrial biology is that only its neuronal function is critical for survival of the animal (Guo et al., 2005; Russo et al., 2009). To determine the significance of dMiro's GTPase domains for viability, we compared the phenotypic effects of expressing normal (control) and GTPase mutant dMiro in heteroallelic dmiro-null mutant neurons (dmirosd32/B682; Guo et al., 2005). This strategy allowed a dual assessment of the mutant protein in regard to the absence of endogenous dMiro activity (null) and its ability to restore normal function compared with normal dMiro. This strategy thus minimized potential complications associated with dominant-negative and/or constitutively active mutations (Feig, 1999; Cantrell, 2002). To evaluate the potential dominant nature of the examined mutations, we also overexpressed normal and mutant dMiro in an otherwise wild-type genetic background (w1118).
Compared with normal dMiro, elav-driven pan-neuronal expression of dMiroT25N in dmiro nulls was lethal during late third-instar and early pupal development and adult escapers were never observed (p < 0.001; Fig. 1B), suggesting that a normal activity of dMiro's N-terminal GTPase domain is critical for survival. In contrast, neuronal overexpression (OE) of dMiroT25N had no significant effect on viability (p = 0.7; Fig. 1C), indicating that dMiroT25N might not be a dominant-negative mutation.
Expression of dMiroA20V in dmiro nulls reduced viability compared with normal dMiro (p < 0.01; Fig. 1B). However, dMiroA20V allowed a limited number of adult survivors, which exhibited crumpled wings, had progressive defects in locomotion, and rarely survived longer than a few days. dMiroA20V OE also significantly impaired viability (p < 0.001; Fig. 1C) to similar levels as dMiroA20V expression in nulls (Fig. 1B,C). This dominant OE effect could not be easily reconciled with the expected effects of a constitutively active mutation since the lethality induced by dMiroA20V expression in nulls was not enhanced by the presence of endogenous dMiro.
Neuronal expression of dMiroT460N and dMiroK455V in dmiro nulls restored a normal viability (p > 0.7; Fig. 1B), indicating a dispensable role of dMiro's C-terminal GTPase domain. In addition, neuronal OE had no significant effects on viability (p > 0.1; Fig. 1C).
dMiro's N-terminal GTPase domain is required for a normal mitochondrial distribution in axons
Null mutations of dMiro cause a severe depletion of mitochondria in axons and dendrites of larval MNs that is accompanied by an accumulation of mitochondria in the soma (Guo et al., 2005; Russo et al., 2009; Fig. 2A). To test whether the two GTPase domains of dMiro are required for a normal distribution of mitochondria, we coexpressed mitoGFP with mutant or normal dMiro protein (control) in larval MNs of heteroallelic dmiro-null mutants using the Ok6-Gal4 driver. Expression of dMiroT25N failed to restore a normal subcellular distribution of mitochondria in MNs, leaving dendrites, axons, and NMJs depleted of mitochondria (p < 0.001; Fig. 2B–G). Instead, mitochondria accumulated in the soma of MNs like in dmiro-null mutants alone (Fig. 2A). The severe depletion of mitochondria at dMiroT25N mutant NMJs was similar to the effects of dmiro-null mutations (p > 0.05; Fig. 2E–G). Like dmiro-null mutants, dMiroT25N mutant synaptic boutons exhibited a significantly reduced size compared with control [p < 0.001; dMiro, 9.0 ± 5.5 (SD), n = 247; dMiroT25N, 5.4 ± 3.2, n = 113; dmiro, 6.9 ± 5.8, n = 113]. Hence, a normal activity of dMiro's N-terminal GTPase domain appears critical for a normal distribution of mitochondria into axons, which primarily requires kinesin-driven transport (Pilling et al., 2006).
Figure 2.

dMiro's N-terminal GTPase domain is required for a normal distribution of mitochondria in motor neurons. A–J, mitoGFP was transgenically coexpressed with normal myc-dMiro (control), myc-dMiroA20V, myc-dMiroT25N, myc-dMiroK455V, or myc-dMiroT460N in MNs of dmiro-null mutants (Null) using an Ok6-Gal4 driver. Mitochondria in larval MNs were visualized by confocal live imaging (A, H–J) or immunostainings using anti-GFP and anti-HRP antibodies (B–G). Graph bars represent mean ± SEM. Box whisker plots represent median (line), 25th–75th percentile (box) range, and 10th–90th percentile (whisker) range. Significant differences among indicated genotypes are indicated by asterisks (1-way ANOVA and Tukey's post-test or Kruskal–Wallis and Dunn's post-test). A, Mitochondria (green) in the soma of larval MNs (arrow) that are located along the midline of the ventral nerve cord (VNC), and axons (arrow) of segmental nerves exiting the VNC. Scale bar, 25 μm. B, Distribution of mitochondria (green) in motor axons (red) of segmental nerves proximal to the VNC. Genotypes are indicated. Arrowheads indicate abnormally elongated mitochondria with thin protrusions that often connect mitochondria of normal width. Scale bar, 5 μm. C, Distribution of presynaptic mitochondria (green) at larval NMJs (red) on muscle 6/7 of the indicated genotypes. Scale bar, 20 μm. D, Overall density of mitochondria in motor axons of proximal segmental nerves (p < 0.01, N > 7). E, Percentage of synaptic boutons containing mitochondria at larval NMJs (p < 0.001, N > 4). F, Mitochondrial area of synaptic boutons (p < 0.05, N > 5, n > 56). G, Mitochondrial area of terminal synaptic boutons (p < 0.05, N > 5, n > 56). H, Length of motile mitochondria in proximal motor axons of the indicated genotypes (p < 0.05, N > 18). I, Number of stationary mitochondria that were longer than 6 μm in proximal motor axons (50-μm-long ROI; p < 0.05, N > 7). J, Normalized TMRM fluorescence (ΔF/F; ΔF = Fpol − Fdepol.) of stationary mitochondria at larval NMJs. Genotypes are indicated (p < 0.05, N > 111).
The impaired dendritic distribution of mitochondria in unipolar MNs of dMiroT25N and dmiro-null mutants (Fig. 2A) is difficult to interpret since the MT organization in the primary neurite is not known, even though it has been resolved for axons and dendrites (Stone et al., 2008). Hence, the defect may be either caused by a primary defect in kinesin-driven transport while traversing the primary neurite, by a dendritic defect in dynein-driven transport, or by a combination of both.
In contrast to dMiroT25N, expression of dMiroT460N in dmiro-null mutant MNs facilitated a normal distribution of mitochondria in the soma, proximal motor axon, and the NMJ compared with dMiro control (p > 0.05; Fig. 2A–G). Notably, expression of normal dMiro in dmiro nulls causes an accumulation of mitochondria in synaptic boutons that is especially pronounced at terminal boutons (Guo et al., 2005; Russo et al., 2009). dMiroT460N did not disrupt this mitochondrial accumulation at terminal boutons (p = 0.9; Fig. 2F,G) and the proportional increase in bouton area (data not shown). Hence, a normal activity of dMiro's C-terminal GTPase domain is likely expendable for the distribution of mitochondria into neuronal processes.
Expression of dMiroA20V and dMiroK455V in dmiro nulls facilitated a normal distribution of mitochondria in axons and NMJs. The mitochondrial density in proximal axons, the amount of synaptic boutons invaded by mitochondria, and the mitochondrial area of synaptic boutons, including terminal boutons, was similar to those of control (p > 0.05; Fig. 2B–G). dMiroA20V and dMiroK455V also had no effect on the average size of synaptic boutons compared with control [p > 0.3; dMiro, 9.0 ± 5.5 (SD), n = 247; dMiroA20V, 11.2 ± 8.1, n = 169; dMiroK455V, 9.4 ± 5.1, n = 60].
To assess potential effects on the distribution of other synaptic cargoes, we examined the synaptic distribution of vesicle-associated CSP (Zinsmaier et al., 1994) and the active zone component Brp (Wagh et al., 2006). The synaptic levels of CSP were reduced in synaptic boutons of dMiroT25N, dMiroA20V, and dmiro-null mutant NMJs from 78 to 84% of control (p < 0.001) while dMiroT460N and dMiroK455V had no significant effect (p > 0.7; Fig. 3A,D). The levels of Brp were decreased in synaptic boutons of dMiroT25N, dMiroA20V, dMiroT460N, and dMiroK455V to various degrees ranging from 54 to 71% of control (p < 0.001; Fig. 3B,E). In addition, the intensity of anti-HRP-stained neuronal membranes was significantly reduced at NMJs of dMiroT25N, dMiroT460N, dMiroK455V, and dmiro-null mutants (p < 0.001) while dMiroA20V had no significant effect (p = 0.99; Fig. 3F).
Figure 3.

Effects of mutations in dMiro's GTPase domains on the distribution of synaptic components. A–H, Normal (control) and GTPase mutant myc-dMiro proteins (A20V, T25N, K455V, or T460N) were expressed in dmiro-null mutant (Null, −/−) neurons using an elav-Gal4 driver. Neuronal membranes, synaptic vesicles, and active zones at larval NMJs on muscle 6/7 were visualized by confocal imaging of immunostainings using anti-HRP, anti-CSP and anti-Brp antibodies, respectively. Asterisks indicate significant differences among indicated genotypes (Kruskal–Wallis, Dunn's post-test). A, B, Distribution of synaptic vesicle-associated CSP (A) and the active-zone component Brp (B) at synaptic boutons of larval NMJs visualized by HRP stainings. Genotypes are indicated. Scale bar, 20 μm. C, Accumulations of CSP (green) and HRP (red) in sensory and motor axons of larval nerves proximal to the VNC. Scale bar, 10 μm. D–F, Quantification of CSP (D), Brp (E), and HRP (F) fluorescence at immunostained synaptic boutons of larval NMJs that were normalized to dMiro control levels. Genotypes are indicated (p < 0.05, n > 56, N > 4). G. Quantification of protein accumulations that were immunopositive for CSP and HRP in sensory and motor axons of larval nerves proximal to the VNC (p > 0.05, N > 10).
Since anti-HRP antibodies detect ≥7 different neuronal proteins in Drosophila (Paschinger et al., 2009), the reduced HRP staining could indicate potential problems in mitochondrial energy supply or protein logistics. To test for a general axonal transport defect, we expressed normal and dMiro mutant proteins in all neurons to ensure a homogenous genotype of sensory and motor axons in the larval nerve. However, we found no evidence for general traffic jams of axonal cargoes (p > 0.05; Fig. 3G), which typically can be visualized by an accumulation of synaptic vesicle precursors and other cargo vesicles transporting neuronal proteins (Gunawardena and Goldstein, 2001; Gunawardena et al., 2003). Overall, the phenotypic effects of the GTPase mutations on neuronal proteins did not correlate with the effects on the axonal distribution of mitochondria. Accordingly, the severe depletion of mitochondria in dMiroT25N mutant axons is not caused by a general defect of the axonal transport machinery.
Mutations in dMiro's N-terminal and C-terminal GTPase domains alter the structure of axonal mitochondria
A number of previous studies reported effects of altered Miro levels or mutations in Miro's GTPase domains on mitochondrial structure from yeast to mammals (Frederick et al., 2004; Fransson et al., 2006; Saotome et al., 2008). In flies, dmiro-null mutations also reduced the average length of axonal mitochondria to less than ∼0.6 μm (Russo et al., 2009). dMiroT25N expression in dmiro-null mutant MNs caused a similar effect and significantly reduced the length of both motile and stationary (data not shown) mitochondria (p < 0.001; Fig. 2B,H), suggesting a potential role of dMiro's N-terminal GTPase domain for maintaining mitochondrial structure.
dMiroA20V, dMiroK455V, and dMiroT460N had no significant effect on the length of motile mitochondria in axons (p = 0.99; Fig. 2B,H). However, both dMiroA20V and dMiroK455V significantly increased the number of stationary mitochondria that exceeded a length of 6 μm (p < 0.01; Fig. 2B,I). In addition, both dMiroA20V and dMiroK455V caused two morphologically different types of elongated axonal mitochondria: one type exhibited a normal width while the second type exhibited extraordinarily thin regions that protruded from sections of normal width and were often connected to other mitochondria of normal width (Fig. 2B, arrowheads). Since this shape is reminiscent of a mitochondrion being pulled apart by opposing forces, it seems possible that this phenotype could be due to an impaired coordination of transport, causing a tug-of-war between kinesin and dynein motors. Alternatively, this abnormal shape could be caused by an impaired dissociation of stationary mitochondria from a “docking site,” or an impaired coordination of mitochondrial fission with transport. Since OE of dMiroA20V and dMiroK455V had similar effects (data not shown), an interpretation of these dominant neomorphic effects is difficult (see Discussion).
Notably, both dMiroA20V and dMiroK455V also induced the formation of abnormally large and swollen mitochondria, which could indicate a deterioration of mitochondrial structure due to a loss of mitochondrial health. To address this possibility, we measured the membrane potential of axonal mitochondria at larval NMJs using the cell-permeant potentiometric fluorescent dye TMRM (Ehrenberg et al., 1988; Plásek and Sigler, 1996). Compared with wild-type control, expression of normal dMiro in dmiro mutants caused a significant increase in the average TMRM fluorescence of mitochondria (p < 0.001; Fig. 2J), which is likely caused by the higher-than-normal expression levels of dMiro (Fig. 1D,E). Expression of dMiroT460N and dMiroK455V caused a similar increase (p > 0.8). However, dMiroA20V mutant mitochondria exhibited normal TMRM fluorescence levels compared with wild type (p = 0.4; Fig. 2J). Hence, the structural effects on mitochondria induced by dMiroA20V and dMiroK455V expression cannot be explained by a deteriorated mitochondrial membrane potential.
Loss of dMiro's N-terminal GTPase activity impairs both kinesin-driven and dynein-driven transport of axonal mitochondria
To directly examine the requirement of dMiro's GTPase domains for axonal transport of mitochondria, we live-imaged mitoGFP-tagged mitochondria in proximal motor axons of larval nerves, as previously described (Louie et al., 2008). This system allows for an unambiguous distinction between kinesin-driven and dynein-driven movements (MT plus and minus end-directed, respectively) because the axons exhibit a uniform MT plus end-out orientation (Pilling et al., 2006; Stone et al., 2008). Of the few mitochondria present in dMiroT25N mutant axons, most were stationary for >2 min and only very few were motile compared with control (p < 0.001; Fig. 4A–C), consistent with a severely reduced flux (rate) of both anterogradely and retrogradely moving mitochondria (p < 0.001; Fig. 4D,E). However, the latter defect could simply be a consequence of the severe depletion of axonal mitochondria.
Figure 4.

dMiro's N-terminal GTPase domain is required for normal motility of axonal mitochondria. A–E, mitoGFP was coexpressed with myc-dMiro (control, dMiro, −/−), myc-dMiroA20V, myc-dMiroT25N, myc-dMiroK455V, or myc-dMiroT460N in MNs of dmiro-null mutants (Null, (−/−)) using an Ok6-Gal4 driver. Confocal time-lapse images visualizing mitochondria in motor axons of the segmental larval nerve exiting the VNC were acquired at a rate of 1 s−1. Asterisks indicate significant differences of indicated mutant genotypes from controls (Kruskal–Wallis and Dunn's post-test). A, Distribution of mitochondria that were motile (green or magenta) or stationary (white) in proximal axons of motor neurons during the 2-min-long imaging period. Scale bar, 5 μm. B, C, Overall density of motile (B) and stationary (C) mitochondria in proximal axons of motor neurons (p < 0.05, N > 7). D, E, Mitochondrial flux of anterograde (D) and retrograde (E) transport in motor axons of proximal larval nerves (p < 0.05, N > 15).
To examine the effects of dMiroT25N on mitochondrial motility, we tracked the transport of mitoGFP-tagged mitochondria in larval motor axons over a 200 s period (Fig. 5A). Transport of mitochondria in a given direction is the net product of opposing movements that are driven by the opposite polarity motors kinesin and dynein, which drive alternating bidirectional movements that are interspersed with short stationary phases (Morris and Hollenbeck, 1993; Hollenbeck, 1996; Zinsmaier et al., 2009; Saxton and Hollenbeck, 2012). Individual movements of a mitochondrion were termed a “run” (Morris and Hollenbeck, 1993; Hollenbeck, 1996; Pilling et al., 2006; Russo et al., 2009), which we further categorized into two types: individual runs (iRuns) that are preceded and followed by a run in the opposite direction, and consecutive runs (cRuns) in one direction. Kinesin motors in particular use on average 4.2 cRuns before a reversal in direction occurs. This phenomenon is described by the term “trip,” which is exclusively defined by a reversal in transport direction, while a run is defined by either a stop or a reversal in direction. Hence, trips can be formed by a single iRun or multiple cRuns.
Figure 5.

Effects of dMiroT25N and dMiroT460N on kinesin-driven motility during anterograde axonal transport of mitochondria. A–I, mitoGFP was coexpressed with myc-dMiro (control, dMiro, −/−), myc-dMiroT25N, or myc-dMiroT460N in motor neurons of dmiro-null mutants [Null (−/−)] using an Ok6-Gal4 driver. Confocal time-lapse images of mitoGFP-tagged mitochondria in motor axons in the segmental larval nerves exiting the VNC were acquired at a rate of 1 s−1 immediately after photobleaching a 50-μm-long ROI to track motions of mitochondria moving into the bleached area. Asterisks indicate significant differences among indicated genotypes (Kruskal–Wallis and Dunn's post-test). A, Each plot shows typical tracks of movements by an individual mitochondrion for 180 s. For comparison, the start of individual tracks was set to zero. Net-positive and net-negative movements represent anterograde and retrograde transport, respectively. B, Net velocity of anterograde mitochondrial transport in motor axons (p < 0.001, N > 20). C, D, Fraction of time mitochondria spent on MT plus end-directed trips (C) and short stops (D) during anterograde transport (p < 0.05, N > 20). E, Average duration of stops of mitochondria during anterograde transport (p < 0.05, N > 18). F, G, Distance of MT plus end-directed cRuns (F) and trips (G) during anterograde mitochondrial transport (p < 0.001, N > 20). H, Number of MT plus end-directed cRuns during anterograde mitochondrial transport (p < 0.01, N > 20). I, Fraction of MT plus end-directed trips that exhibited cRuns (p < 0.05, N > 29).
dMiroT25N severely impaired anterograde mitochondrial transport. Compared with normal dMiro, dMiroT25N expression in dmiro nulls significantly reduced the net velocity of transport (p < 0.001; Fig. 5A,B), even though a few dMiroT25N mutant mitochondria (∼1 of 20) exhibited relatively normal transport (Fig. 5A). Specifically, dMiroT25N reduced the time mitochondria spent on MT plus end-directed, kinesin-driven trips (p < 0.001), the distance of kinesin-driven runs and trips (p < 0.001), the number of kinesin-driven cRuns (p < 0.001), and the percentage of kinesin trips containing >1 run (p < 0.01; Fig. 5C,F–I). In addition, dMiroT25N significantly increased the duration of stops and the time mitochondria spent motionless (p < 0.001; Fig. 5D,E). All of these effects induced by dMiroT25N were statistically indistinguishable from those of dmiro-null mutations (p > 0.05; Fig. 5B–I). The impaired kinesin motility of anterogradely moving mitochondria was not caused by a tug-of-war between kinesin and dynein motors since dMiroT25N had no significant effect on the distance of dynein-driven runs (control, 0.27 ± 0.1 μm; T25N, 0.28 ± 0.14 μm; p = 0.9, Mann–Whitney) and trips (control, 0.30 ± 0.13 μm; T25N, 0.35 ± 0.16 μm; p = 0.14). Together, the severe defects induced by dMiroT25N indicate a critical role of dMiro's N-terminal GTPase domain for kinesin-driven movements driving anterograde transport.
dMiroT25N affected retrograde mitochondrial transport less severely than anterograde transport. dMiroT25N expression in dmiro nulls impaired the net velocity of retrogradely moving mitochondria to a level that was significantly different to both dmiro nulls and normal dMiro (p < 0.05; Fig. 6A). In addition, dMiroT25N impaired the distance of dynein-driven iRuns (data not shown), cRuns, and trips compared with control (p < 0.05; Fig. 6E,F). Like dmiro-null mutations, dMiroT25N also had no effect on the number of dynein-driven cRuns and the percentage of dynein trips containing cRuns (p > 0.05; Fig. 6G,H). In contrast to dmiro nulls, dMiroT25N did not significantly affect the preferential use of MT dynein-driven trips, the time mitochondria spent motionless, and the duration of stops (p > 0.05; Fig. 6B–D). The differential effects of dMiroT25N and dmiro-null mutations on retrograde transport excluded the possibility that T25N disrupts all protein activities of dMiro. Together, these findings indicate an important requirement of dMiro's N-terminal GTPase domain for dynein-driven movements driving retrograde transport.
Figure 6.

Effects of dMiroT25N and dMiroT460N on dynein-driven motility during retrograde axonal transport of mitochondria. A–H, mitoGFP was coexpressed with myc-dMiro (control, dMiro, −/−), myc-dMiroT25N, or myc-dMiroT460N in MNs of dmiro-null mutants [Null (−/−)] using an Ok6-Gal4 driver. Confocal time-lapse images of mitoGFP-tagged mitochondria in motor axons were acquired at a rate of 1 s−1 as described previously. Asterisks indicate significant differences among indicated genotypes (Kruskal–Wallis and Dunn's post-test). A, Net velocity of retrograde axonal mitochondrial transport (p < 0.05, N > 36). B, C, Fraction of time mitochondria spent on MT minus end-directed trips (B) and short stops (C) during retrograde transport (p < 0.05, N > 20). D, Average duration of stops of mitochondria during retrograde transport (p < 0.01, N > 18). E, F, Distance of MT minus end-directed cRuns (E) and trips (F) during retrograde transport (p < 0.05, N > 36). G, Number of MT minus end-directed cRuns during retrograde transport (p > 0.05, N > 40). H, Fraction of MT minus end-directed trips that exhibited cRuns (p > 0.05, N > 29).
Notably, a very small fraction of T25N mutant mitochondria exhibited almost normal anterograde and retrograde transport (Fig. 5A), which might be due to a maternal effect or a second form of mitochondrial motor linkage that is independent of dMiro, as has been suggested for fly and mammalian systems (Glater et al., 2006; Koutsopoulos et al., 2010).
Genetic nature of dMiroT25N
To evaluate the genetic nature of dMiroT25N, we overexpressed dMiroT25N in otherwise wild-type-like larval MNs (w1118). In the case of a dominant-negative mutation, one expects that expression in a wild-type and a null background would induce qualitatively similar phenotypes, even though increased levels of endogenous protein may gradually decrease the phenotypic severity of the mutation (Muller, 1932; Wilkie, 1994). Hence, one expects that dMiroT25N OE would deplete both dendrites and axons of mitochondria, like its expression in dmiro nulls (Fig. 2A). However, this was not the case. dMiroT25N OE in MNs did not deplete axons of mitochondria (Fig. 7A, arrows). In contrast, dMiroT25N OE caused an accumulation of mitochondria at terminal boutons of NMJs compared with wild type (p < 0.001), which was similar to the accumulation induced by normal dMiro OE [p = 0.52; wild type, 3.9 ± 1.6 (SD); dMiroT25N OE, 7.1 ± 1.9; dMiro OE, 8.0 ± 2.7; N > 10]. However, dMiroT25N OE had no gross effect on the distribution of dendritic mitochondria compared with wild type (Fig. 7A, arrowheads). In contrast, dMiro OE depleted dendrites of mitochondria (Fig. 7A). The effects on the mitochondrial distribution induced by dMiroT25N OE were not consistent with those expected by a dominant-negative mutation.
Figure 7.

Comparison of dMiroT25N-induced and dMiroT460N-induced phenotypes in the absence and presence of endogenous dMiro. A–I, myc-dMiro (control), myc-dMiroT25N, or myc-dMiroT460N were coexpressed with mitoGFP in MNs of dmiro-null mutants (−/−) or overexpressed (OE) in MNs of wild-type control (w1118) using an Ok6-Gal4 driver. Confocal time-lapse images of mitoGFP-tagged mitochondria in motor axons were acquired at a rate of 1 s−1. Asterisks indicate significant differences for the pairwise comparison between effects induced by expression in a null and wild-type background (Mann–Whitney) and differences from wild type (WT) and dMiro OE among all indicated genotypes (Kruskal–Wallis and Dunn's post-test). A, Confocal images of mitoGFP-tagged mitochondria (green) in the soma of larval MNs, their dendrites located in the neuropil of the VNC (arrowheads), and axons (arrows) of segmental nerves exiting the VNC. Scale bar, 25 μm. B. Mitochondrial flux of anterograde transport in motor axons of proximal larval nerves (p < 0.05, N > 8). C, Net velocity of anterograde mitochondrial transport in axons (p < 0.05, N > 20). D, Percentage of time mitochondria spent on plus end-directed trips during anterograde transport (p < 0.01, N > 20). E, F, Distance of MT plus end-directed cRuns (E) and trips (F) during anterograde mitochondrial transport (p < 0.001, N > 20). G, Number of MT plus end-directed cRuns during anterograde transport (p < 0.01, N > 20). H, Fraction of MT plus end-directed trips that exhibited cRuns during anterograde transport (p < 0.001, N > 29). I, Number of cRuns per MT plus end-directed trips (p < 0.01, N > 62).
To better test whether dMiroT25N resembles a dominant-negative mutation, we examined to what degree its phenotypic effects on anterograde mitochondrial transport in MN axons are sensitive to the presence of endogenous dMiro. Pairwise comparison showed that the presence of wild-type protein strongly attenuated the severe impairments induced by dMiroT25N expression in dmiro nulls (p < 0.05; Fig. 7B–I). This strong attenuation of dMiroT25N phenotypes is remarkable since dMiroT25N expression levels were much higher than those of endogenous dMiro (Fig. 1D,E). Nevertheless, dMiroT25N OE still caused some dominant effects. Compared with wild-type and dMiro control, dMiroT25N OE reduced the net velocity of transport and distance of kinesin-driven cRuns while it increased the time mitochondria spent on kinesin-driven trips (p < 0.01; Fig. 7C–E). Importantly, the latter was qualitatively the opposite effect of its expression in dmiro nulls. Hence, the phenotypic effects of dMiro OE overall are inconsistent with those of a dominant-negative mutation.
Notably, dMiroT25N OE and dMiro OE had essentially the same effect on number of kinesin-driven cRuns, the percentage of kinesin-driven trips containing cRuns, and the number of cRuns per trip (p < 0.05; Fig. 7G–I), suggesting that these effects are exclusively caused by dMiro OE and do not require the activity of the N-terminal GTPase domain.
dMiro's N-terminal GTPase domain is required for the dynein-dependent distribution of dendritic mitochondria in sensory neurons
The differential effects of dMiroT25N expression in dmiro nulls on kinesin-driven and dynein-driven axonal transport of mitochondria (Figs. 5, 6) could have been caused by an exclusive defect in kinesin motility, since dynein-driven retrograde transport of mitochondria in fly MN axons depends on normal kinesin activity (Pilling et al., 2006). However, it also was possible that dMiroT25N may prevent both kinesin-driven and dynein-driven transport because dMiroT25N failed to properly distribute mitochondria into MN dendrites (Fig. 2A). To better resolve the significance of the N-terminal GTPase domain for dynein-driven transport, we turned to multidendritic SNs.
Mitochondria of multidendritic SNs (class I, vpda; (Grueber et al., 2002) were visualized by transgenic expression of mitoGFP using the 21-7 Gal4 driver (Song et al., 2007). The examined SNs are located in the larval body wall and feature a single axon and a major primary dendrite (arrowheads) that emerges from the soma (arrow) and gives rise to a number of uniquely organized secondary branches that run almost parallel to each other (Fig. 8A). In contrast to mammalian dendrites, larval SN dendrites exhibit a MT minus-end-out organization and require dynein for anterograde transport of mitochondria (Grueber et al., 2002; Rolls et al., 2007; Satoh et al., 2008; Stone et al., 2008; Rolls, 2011).
Figure 8.

Effects of dMiroT25N on the distribution of mitochondria in dendrites of sensory neurons. A–H, mitoGFP was transgenically coexpressed with normal myc-dMiro (control), myc-dMiroT25N, or dmiro promoter-driven genomic transgene expressing only dMiro-PC in vpda SNs of dmiro-null mutants [Null (−/−)] using the indicated 21-7 and C380 Gal4 drivers. Mitochondria in larval SNs were visualized by confocal imaging of immunostained larval body wall preparations using anti-GFP and anti-HRP antibodies. Asterisks indicate significant differences among indicated genotypes (Kruskal–Wallis and Dunn's post-test). A, Mitochondria (green) in dendrites and soma of vpda SNs. Arrows and arrowheads indicate the soma and primary dendrite of the SN. Scale bar, 20 μm. B, Mitochondria (green) in vpda SN axons of segmental nerves proximal to the VNC. Arrowheads denote abnormally small mitochondria of dmiro-null and dMiroT25N mutants. Scale bar, 10 μm. C–E, Density of mitochondria in primary (C), secondary (D), and all (E) dendrites of vpda neurons (p < 0.05, N > 7). F, G, Average length of primary (F) and secondary (G) vpda SN dendrites (p < 0.05, N > 7). F, Cumulative length of vpda SN dendrites (p < 0.05, N > 7).
In wild-type SNs, mitoGFP-tagged mitochondria were found throughout primary and secondary dendrites in irregularly spaced intervals (Fig. 8A). However, SNs of dmiro-null mutants exhibited a severe depletion of dendritic as well as axonal mitochondria that was accompanied by an accumulation of somatic mitochondria (Fig. 8A,B). The few dendritic mitochondria were mainly constrained to proximal regions of the primary dendrite, and rarely present in secondary or tertiary dendrites (Fig. 8A,C–E). The loss of dendritic mitochondria in dmiro-null mutant SNs was associated with a structural impairment of the dendritic tree, reducing the average length of secondary but not primary dendrites (Fig. 8F,G), which together reduced the overall size of the dendritic tree (Fig. 8H). Notably, a similar impairment of the dendritic tree has been observed after knockdown of TRAK2 in cultured hippocampal neurons (van Spronsen et al., 2013).
Expression of normal dMiro in dmiro-null mutant SNs from a transgene containing a dmiro promoter restored the distribution of mitochondria in both axons (data not shown) and dendrites, and even caused a significant increase in the amount of dendritic mitochondria (Fig. 8A,C–E). Hence, this suggests that dMiro is required for both the dynein-driven dendritic and the kinesin-driven axonal distribution of mitochondria.
Notably, 21-7 Gal4-driven expression of normal dMiro in dmiro-null mutant SNs restored the distribution of mitochondria in axons but not dendrites (Fig. 8A,B). This phenomenon mirrored the effects of Ok6 Gal4-driven expression of dMiro in dmiro-null mutant MNs (Fig. 2A). In both cases, the depletion of dendritic mitochondria was caused by a dominant OE effect since both 21-7 and Ok6-driven OE of dMiro induced a severe depletion of mitochondria in dendrites but not axons of otherwise wild-type SNs and MNs (Figs. 9A–C, 7A, respectively). To minimize the effects of dMiro OE on dendritic mitochondria, we tested a number of Gal4 drivers, including 477, 2-21, 5-40, and C380 (Grueber et al., 2003; Song et al., 2007; Sanyal, 2009). Of these, only C380 Gal4-driven expression attenuated the dMiro OE effect and partially restored the dendritic distribution of mitochondria compared with dmiro nulls (p < 0.05) to 45.7% of wild-type levels (p < 0.001; Fig. 8A,C–E).
Figure 9.

Effects of dMiro and dMiroT25N OE on the distribution of mitochondria in dendrites of sensory neurons. A–D, mitoGFP was coexpressed with normal myc-dMiro (dMiro OE) or myc-dMiroT25N (T25N OE) in vpda SNs of wild-type control (WT, w1118) using a 21-7-Gal4 driver. Mitochondria were visualized by confocal imaging of immunostained larval preparations using anti-GFP and anti-HRP antibodies. Asterisks indicate significant differences among indicated genotypes (Kruskal–Wallis and Dunn's post-test). A, Mitochondria (green) in dendrites and soma of vpda SNs. Arrows and arrowheads indicate the soma and primary dendrite of the SN. Scale bar, 20 μm. B, Mitochondria (green) in distal SN axons of the larval nerve and the VNC. Scale bar, 20 μm. C, Density of mitochondria in dendrites of vpda SN neurons (p < 0.01, N > 7). D, Cumulative length of vpda SN dendrites (p < 0.05, N > 7).
To test the requirement of dMiro's N-terminal GTPase domain for distributing mitochondria into dendrites, we expressed dMiroT25N in dmiro-null mutant SNs using both the 21-7 and C380 Gal4 driver. However, C380-driven dMiroT25N expression had essentially no effect on the reduced amount of dendritic mitochondria compared with dmiro-null mutant SNs (p > 0.7), and was significantly different from the partial rescue induced by normal dMiro expression (p < 0.001; Fig. 8A,C–E). 21-7-driven expression caused a similar depletion of dendritic mitochondria (data not shown). Importantly, both 21-7-driven and C380-driven (data not shown) expression of dMiroT25N also failed to restore the distribution of mitochondria into axons of the examined SNs (Fig. 8B), which mirrored the effects of dMiroT25N expression in dmiro-null mutant MNs (Fig. 2B-G). Together these data suggest that dMiro's N-terminal GTPase domain is required for both the kinesin-driven and the dynein-driven distribution of mitochondria into axons and dendrites, respectively.
Notably, dMiroT25N OE disrupted the dominant effect of normal dMiro OE on dendritic mitochondria in both MNs and SNs (Figs. 9A,C, 7A, respectively). In SNs, dMiroT25N OE still caused a slight reduction to 73% of control levels (Fig. 9A,C). The remaining effect was likely a consequence of increased dMiro-mediated kinesin linkage since T25N did not disrupt the increase in kinesin-driven trip motility induced by dMiro OE (Fig. 7F–I). Nevertheless, the effect of dMiroT25N OE was significantly different to the almost complete depletion of dendritic mitochondria induced by dMiro OE (p < 0.001; Fig. 9C). dMiroT25N OE had no effect on the length of the dendritic tree (p = 0.7), which was significantly reduced by normal dMiro OE (p < 0.01; Fig. 9D).
dMiro's C-terminal GTPase domain modulates retrograde mitochondrial transport in axons
Expression of the C-terminal GTPase mutation dMiroT460N in dmiro nulls restored a normal density of motile and stationary mitochondria in proximal axons of MNs (p > 0.9; Fig. 4A–C) and facilitated a normal flux of anterograde mitochondrial transport compared with normal dMiro (p > 0.2; Fig. 4D).
Consistent with a normal axonal mitochondrial distribution, dMiroT460N had no effect on the net velocity, the preferential use of kinesin motors, and the distance of kinesin-driven cRuns and trips compared with normal dMiro (p > 0.3; Fig. 5A–C,F,G). However, dMiroT460N slightly but significantly reduced the distance of kinesin-driven iRuns (data not shown), while it increased the duration of stops and the time mitochondria spent motionless (p < 0.05; Fig. 5D,E). Notably, dMiroT460N increased the number of kinesin-mediated cRuns and the percentage of kinesin trips with cRuns (p < 0.01; Fig. 5H,I). However, the resulting increase in the distance of kinesin-mediated trips was not significantly different from that of control (p > 0.05; Fig. 5G).
Together, the almost perfect rescue of dmiro-null phenotypes by dMiroT460N expression suggest that dMiro's C-terminal GTPase domain may be largely dispensable for anterograde axonal transport of mitochondria. Nevertheless, the dMiroT460N-induced increase of the number of kinesin-driven cRuns and the percentage of trips with cRuns indicated a potential modulatory role (Fig. 5H,I). However, this may not be the case since dMiroT460N OE affected anterograde transport in a paradoxical way. dMiroT460N OE reduced the net velocity of anterograde transport and the distance of kinesin-driven cRuns compared with dMiro OE, wild-type control, and dMiroT460N expression in nulls (p < 0.01; Fig. 7C). In addition, dMiroT460N OE reduced the number of cRuns compared with dMiro OE (p < 0.01; Fig. 7G) to wild-type levels while expression in dmiro nulls caused a significant increase compared with dMiro OE (p < 0.01; Fig. 5H). These paradoxical phenotypic effects of dMiroT460N are inconsistent with a role of the C-terminal GTPase domain modulating kinesin-driven transport. Instead, they may be a consequence of a role of the domain unrelated to transport.
In contrast to anterograde transport, dMiroT460N expression in dmiro nulls significantly impaired retrograde mitochondrial transport. Compared with dMiro control, it reduced the net velocity, the time spent on dynein-mediated trips, and the distance of dynein-mediated trips (p < 0.05; Fig. 6A,B,F). dMiroT460N also caused an increase in the time mitochondria spent on stops (Fig. 6C). All of these effects were significantly less severe than those of dmiro nulls (p < 0.05; Fig. 6A–F). dMiroT460N had no significant effect on the duration of stops, the distance of dynein-driven cRuns, the average number of cRuns, or the fraction of dynein-mediated trips containing cRuns (p > 0.05; Fig. 6D,E,G,H). Together, these data suggest that dMiro's C-terminal GTPase domain modulates retrograde transport, possibly by modulating dynein motility or other aspects of mitochondrial biology.
Expression of dMiroA20V and dMiroK455V causes neomorphic gain-of-function effects
The presumed constitutively active mutations dMiroA20V and dMiroK455V did not facilitate normal mitochondrial transport in axons of MNs, even though they facilitated a normal distribution of mitochondria in axons and NMJs (Fig. 2). Compared with normal dMiro, expression of dMiroA20V and dMiroK455V in dmiro nulls significantly reduced the density of motile (p < 0.001) but not stationary mitochondria in proximal axons (p > 0.05; Fig. 4B,C). Both mutations also severely reduced the flux of anterograde and retrograde axonal transport (p < 0.001; Fig. 4D,E). Furthermore, dMiroA20V and dMiroK455V reduced the net velocity of anterograde transport (p < 0.001), the time mitochondria spent on kinesin-driven trips (p < 0.05), and the distance of kinesin-driven runs and trips to a similar degree (p < 0.001; Fig. 10B–E). Retrograde axonal transport was equally impaired as both dMiroA20V and dMiroK455V reduced the net velocity (p < 0.001) and the distance of dynein-driven runs (p < 0.001) and trips (p < 0.05, data not shown).
Figure 10.

Effects of dMiroA20V and dMiroK455V on kinesin-driven motility during anterograde axonal transport of mitochondria. A–F, myc-dMiro (control), myc-dMiroA20V, or myc-dMiroK455V were coexpressed with mitoGFP in MNs of dmiro-null mutants (−/−) or overexpressed (OE) in MNs of wild-type control (w1118) using an Ok6-Gal4 driver. Confocal time-lapse images of mitoGFP-tagged mitochondria in motor axons were acquired at a rate of 1 s−1. Asterisks indicate significant differences for the pairwise comparison between effects induced by expression in a null and wild-type background (Mann–Whitney), and differences from WT and dMiro OE among all indicated genotypes (Kruskal–Wallis and Dunn's post-test). A, Mitochondrial flux of anterograde transport in motor axons of proximal larval nerves (p < 0.001, N > 6). B, Net velocity of anterograde mitochondrial transport in axons (p < 0.05, n > 29, N > 6). C, Fraction of time mitochondria spent on MT plus end-directed trips during anterograde transport (p < 0.05, n > 29, N > 6). D, E, Distance of MT plus end-directed runs (D) and trips (E) during anterograde mitochondrial transport (p < 0.001, n > 29, N > 6). F, Number of MT plus end-directed cRuns during anterograde transport (p < 0.01, n > 29, N > 6).
Since the effects of dMiroA20V and dMiroK455V on axonal mitochondrial transport were difficult to reconcile with those of the corresponding loss-of-function mutations dMiroT25N and dMiroT460N, we examined their genetic nature by comparing their phenotypic effects in the absence and presence of endogenous dMiro. Pairwise comparison showed that expression of dMiroA20V and dMiroK455V in wild-type and dmiro-null mutants had essentially the same effects on anterograde (p > 0.05; Fig. 10A–F) and retrograde transport of mitochondria (data not shown). Hence, the phenotypic effects of dMiroA20V and dMiroK455V are inconsistent with those of a constitutively active or hypermorphic mutation, whose phenotypes are typically enhanced by increased levels of endogenous wild-type protein. Instead, both mutations exhibited features of a neomorphic gain-of-function mutation (Muller, 1932). Therefore, we refrain from the inherently difficult interpretation of these neomorphic phenotypes since they are often due to the acquisition of a new function that is unrelated to the normal function of the protein (Muller, 1932; Wilkie, 1994).
Mutations in dMiro's C-terminal GTPase domain do not modulate the phenotypic effects of mutations in the N-terminal GTPase domain
Our genetic analysis of dMiroT460N suggests that dMiro's C-terminal GTPase domain is not critical for anterograde mitochondrial transport. Nevertheless, the domain could still cooperate with the N-terminal GTPase domain. To test this, we generated the double-mutant transgenes dMiroT25N-T460N, dMiroT25N-K455V, dMiroA20V-T460N, and dMiroA20V-K455V. The double-mutant proteins were localized properly to mitochondria and their expression levels were slightly higher than those of the respective single-mutant dMiro proteins (1.5–2-fold of dMiro control; data not shown).
Expression of dMiroT25N-T460N and dMiroT25N-K455V in MNs of dmiro-null mutants caused a pronounced accumulation of mitochondria in the soma and a severe depletion in dendrites and axons (Fig. 11A). Proximal axons contained only very few mitochondria while NMJs did not contain >1–3 sporadic mitochondria (Fig. 11B). All of these phenotypic effects were indistinguishable to those of dMiroT25N (p > 0.05; N > 5; data not shown). Hence, the mutations T460N and K455V are not able to modulate the pronounced inhibition of mitochondrial transport that is caused by the T25N-induced inactivity of the N-terminal GTPase domain. These findings do not provide evidence for a cooperative action of dMiro's N-terminal and C-terminal GTPase controlling mitochondrial transport.
Figure 11.

Effects of dMiro GTPase double-mutant proteins on the distribution and transport of mitochondria in axons. A–I, mitoGFP was coexpressed with normal myc-dMiro (control), myc-dMiroA20V, myc-dMiroT25N, myc-dMiroT25N-T460N, myc-dMiroT25N-K455V, myc-dMiroA20V-K455V, or myc-dMiroA20V-T460N in MNs of dmiro-null mutants (−/−) using an Ok6-Gal4 driver. Mitochondria in larval motor neurons were visualized by confocal live imaging or immunostainings using anti-GFP (mitoGFP) and anti-HRP antibodies (neuronal membranes). Asterisks indicate significant differences among indicated genotypes (Kruskal–Wallis and Dunn's post-test). A, Mitochondria (green) in larval MNs in the VNC of dMiroT25N-K455V and dMiroT25N-T460N mutants accumulate in the soma and are absent from axons and dendrites. Scale bar, 30 μm. B, C, Distribution of presynaptic mitochondria (green) at larval NMJs (red) of muscle 6/7. Note the differential sensitivity of mito-GFP-immunostained dMiroA20V-T460N mutant mitochondria (C) to fixation by formaldehyde (FA) and Bouin's fixative. Scale bar, 20 μm. D, Mitochondrial area of synaptic boutons (p < 0.001, n > 98, N > 5). E, Area of synaptic boutons (p < 0.001, n > 98, N > 5). F, Net velocity of anterograde mitochondrial transport in motor axons (p > 0.05, N > 15). G, Fraction of time mitochondria spent on MT plus end-directed trips during anterograde transport (p > 0.05, N > 15). H, I, Distance of MT plus end-directed runs (H) and trips (I) during anterograde mitochondrial transport (p > 0.05, N > 15).
Expression of dMiroA20V-T460N and dMiroA20V-K455V in MNs of dmiro-null mutants facilitated the distribution of mitochondria into axons and NMJs (Fig. 11C). Compared with dMiroA20V, both mutations increased the size of synaptic boutons (p < 0.001; Fig. 11E) and the area occupied by mitochondria, although the latter was only significant for dMiroA20V-T460N (p < 0.01; Fig. 11D). Despite the accumulation of mitochondria at axon terminals, both dMiroA20V-T460N and dMiroA20V-K455V impaired mitochondrial transport in a manner that was indistinguishable from dMiroA20V (p > 0.05; Fig. 11F–I), which is consistent with its proposed neomorphic nature.
Notably, dMiroA20V-T460N expression induced a significant sensitivity of synaptic mitochondria to standard formaldehyde fixation, as it was used throughout this study (Fig. 11C). This problem could be fully eliminated by using Bouin's fixative (Fig. 11C), which visualized a similar amount of synaptic mitochondria as live imaging (data not shown). The cause of this phenomenon is unclear but it could stem from an alteration in the lipid composition of A20V-T460N mutant mitochondrial membranes, assuming that the domains are required for lipid exchange between the ER and mitochondria, as has been demonstrated for yeast Miro (see Discussion; Kornmann et al., 2011).
Discussion
Previous studies demonstrated a critical role of dMiro for the kinesin-mediated distribution of mitochondria into axons (Guo et al., 2005; Russo et al., 2009). Here we extend these studies and show that dMiro is also required for the dynein-dependent distribution of mitochondria into dendrites of SNs. In addition, we used the mutations dMiroT25N and dMiroT460N to determine the biological significance of dMiro's N-terminal and C-terminal GTPase domains. Our analysis suggests a critical neuronal role of dMiro's N-terminal GTPase domain for survival, for normal distribution of mitochondria into axons and dendrites, and for maintaining a normal mitochondrial size. Overall, our analysis indicates that dMiro's N-terminal GTPase domain facilitates the transport of mitochondria by promoting their transition from a stationary to a motile state regardless of the employed motor. In contrast, our analysis of dMiroT460N suggests that dMiro's C-terminal GTPase domain is not essential for viability and the distribution of mitochondria into neuronal processes, although a subtle modulatory role for retrograde mitochondrial transport is indicated. Finally, we show that amino acid substitutions analogous to the constitutively active Ras-G12V mutation in dMiro's N-terminal and C-terminal GTPase domains cause neomorphic phenotypic effects that are likely unrelated to the normal function of both GTPase domains.
Genetic nature of the mutations in dMiro's GTPase domains
The dominant-negative Ras-S17N and constitutively active Ras-G12V mutations served as templates for altering the activities of Miro's two GTPase domains in yeast, flies, and mammals (Fransson et al., 2003, 2006; MacAskill et al., 2009b; Kornmann et al., 2011; Koshiba et al., 2011; Murley et al., 2013). Studies of mammalian Miro overexpressing these GTPase mutant proteins in various cell culture systems reported phenotypic effects that were conceptually difficult to reconcile (Fransson et al., 2003, 2006; MacAskill et al., 2009b). Therefore, we genetically verified the potential dominant nature of the analogous fly mutations.
Expression of the RasS17N analog dMiroT25N in dmiro-null mutants was lethal and severely impaired the distribution and transport of mitochondria in neurons. Remarkably, these effects were strongly suppressed by the presence of endogenous dMiro, suggesting that T25N is essentially a recessive loss-of-function mutation. The recessive nature of dMiroT25N is not a species-specific exception, since the analogous mutation in yeast Miro also had no dominant effects (Koshiba et al., 2011). A potentially recessive nature of the analogous mammalian Miro18N mutation may explain its wild-type-like OE effects on the mitochondrial distribution in cultured hippocampal neurons (MacAskill et al., 2009b). A similar classification could not be done for dMiroT460N.
Expression of the Ras-G12V analog dMiroA20V in dmiro nulls facilitated the distribution of mitochondria into neuronal processes like normal dMiro. However, unlike normal dMiro, dMiroA20V expression in both dmiro nulls and wild-type animals impaired viability and mitochondrial transport to a similar degree, which is a characteristic feature of a neomorphic mutation and inconsistent with a constitutively active mutation (Muller, 1932; Wilkie, 1994). Neomorphic effects are typically not related to normal protein function since they are often due to the acquisition of a toxic protein feature or an unnatural protein interaction (Wilkie, 1994). However, a constitutively active GTPase domain could cause neomorphic effects too. Since GTPase signaling is not meant to operate continuously, a constitutive activity could induce negative-feedback mechanisms counteracting the effects of its chronic activation (Cantrell, 2002).
Remarkably, dMiroK455V had neomorphic effects on mitochondrial transport and structure that were essentially indistinguishable to those of dMiroA20V, and showed no additive effects when combined in a double-mutant protein. The similar effects of dMiroA20V and dMiroK455V contrasted the fundamentally different effects of the corresponding loss of function mutations, dMiroT25N and dMiroT460N, which either prevented or permitted transport, respectively. Hence, the significantly different functional requirements of the GTPase domains make a potential constitutively active nature of dMiroA20V and dMiroK455V unlikely.
How could dMiroA20V and dMiroK455V induce similar dominant phenotypes if only the N-terminal GTPase domain is required for anterograde transport? The neomorphic character of both mutations might provide an explanation. Since both GTPase domains constitute Ras-like domains (Reis et al., 2009; Klosowiak et al., 2013), one can expect that the analogous substitutions A20V and K455V may cause a similar molecular change in each domain. Accordingly, it seems possible that the similar neomorphic effects are caused by the acquisition of either a similar toxic protein feature or a similar unnatural protein interaction, and not by a constitutively active GTPase domain.
dMiro's N-terminal GTPase domain mediates the mobilization of stationary mitochondria
Miro is one of at least two factors that ascertain mitochondrial motor linkage through its association with the Milton/TRAK motor adaptor complex (Fransson et al., 2006; Glater et al., 2006; MacAskill et al., 2009a, 2009b; Wang and Schwarz, 2009; Koutsopoulos et al., 2010; van Spronsen et al., 2013). At least in mammals, polarized mitochondrial transport into either axons or dendrites is achieved by differential interactions of TRAK1 and TRAK2 with kinesin and dynein motors (van Spronsen et al., 2013). The activity of the linked motors is in part controlled by Ca2+ activation of Miro's EF hands arresting transport (MacAskill et al., 2009a; Wang and Schwarz, 2009). However, the significance of Miro's GTPase domains for mitochondrial transport remained unclear.
Previous mammalian studies indicated that Miro's GTPase domains do not control the association of Miro with TRAK proteins. Mutations in the N-terminal (N18) and C-terminal GTPase domain (N432) of Miro1 and Miro2 had no effect on the ability to coprecipitate TRAK1 or TRAK2 (Fransson et al., 2006). In vivo, mycMiro1-N18 also failed to affect the mitochondrial recruitment of cytosolic YFP-TRAK2 (MacAskill et al., 2009b). Our analysis of the MiroN18-analogous fly mutation dMiroT25N revealed that a normal activity of Miro's N-terminal GTPase domain is critical for survival, mitochondrial size, and transport, likely by controlling the transition of mitochondria from a stationary to a motile state.
dMiroT25N expression in the absence of endogenous dMiro caused an accumulation of mitochondria in the soma of MNs and SNs, while axons and dendrites were severely depleted of mitochondria. The few motile mitochondria found in MN axons exhibited defects in both kinesin and dynein motility. Hence, it is unlikely that dMiro's N-terminal GTPase domain resembles a binary switch that controls exclusively kinesin motility or switches mitochondrial motility from a kinesin-driven to a dynein-driven state. Instead, these findings argue that the N-terminal domain controls a fundamentally different mechanism that mediates the transition from a stationary to a motile state regardless of the identity of the subsequently employed MT motor. Accordingly, Miro may mediate mitochondrial transport by providing a membrane anchor linking motors to mitochondria, by controlling the activity of linked motors through its EF-hand domains, and by controlling the transition from a stationary to a motile state through a yet unknown mechanism.
How might dMiro's N-terminal GTPase domain mobilize mitochondria?
The transition of stationary mitochondria to a motile state could be principally achieved by controlling the recruitment of motors to immobile mitochondria, the activity of the linked motors, or the detachment of mitochondria from a stationary site. The first possibility is unlikely since loss of Miro's N-terminal GTPase activity did not inhibit its association with TRAK motor complexes (Fransson et al., 2006; MacAskill et al., 2009b). Instead, the N-terminal domain could control the activity of the linked motors like Miro's EF-hand domains, by either disengaging motors from MTs or triggering their release from Milton/TRAK adaptors. This possibility is supported by the severe motility defects of the few axonal dMiroT25N mutant mitochondria escaping the stationary state. Alternatively, the N-terminal domain could detach mitochondria from a stationary site, which is reminiscent of Miro's role in yeast, where it acts as a negative regulator of a physical ER–mitochondria link (Kornmann et al., 2011; Murley et al., 2013).
The roles of the GTPase domains in yeast Miro are well defined. Yeast mitochondria are physically tethered to the ER by the multiprotein ERMES (endoplasmic reticulum mitochondria encounter structure) complex, which is required for ER-associated phospholipid exchange, mitochondrial division, and the association with actin. Both the N-terminal domain and, to a lesser degree, the C-terminal domain are required for the dissociation of mitochondrial ER contacts and the maintenance of mitochondrial morphology (Boldogh et al., 2003; Frederick et al., 2004; Kornmann et al., 2009, 2011; Toulmay and Prinz, 2012; Voss et al., 2012; Murley et al., 2013).
Metazoan Miro may also dissociate ER–mitochondria contacts, since mammalian Miro1 expressed in yeast is localized to ER–mitochondria interfaces (Kornmann et al., 2011). Accordingly, the accumulation of mitochondria in neuronal somata of dMiroT25N and dmiro-null mutants could be a consequence of abnormal ER–mitochondria interactions downstream of ER-associated mitochondrial division and upstream of mitochondrial dispersion by motor proteins. Like in yeast mutants, dMiroT25N and dmiro-null mutant mitochondria were severely fragmented, which is consistent with a function downstream of mitochondrial division. However, further studies will be needed to elucidate the true nature of the immobilized state induced by the loss of dMiro's N-terminal GTPase activity.
Footnotes
This work was supported by a grant to K.E.Z. from the National Institute of Neurological Disorders and Stroke (R01 NS052664). G.J.R. has been supported by a National Institutes of Health training grant (T32 AG007434). The anti-β-tubulin antibody developed by Michael Klymkowsky, the anti-Brp antibody developed by Alois Hofbauer and Erich Buchner, and the anti-CSP antibody developed by K.E.Z. and Seymour Benzer were obtained from the Developmental Studies Hybridoma Bank, created by the National Institute of Child Health and Human Development of the National Institutes of Health and maintained at the University of Iowa, Department of Biology, Iowa City, IA 52242. We thank Kathryn Cruice and Hojin Seo for technical help and Elliot Imler for critical comments on the manuscript.
The authors declare no competing financial interests.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. Wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/S0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldogh IR, Nowakowski DW, Yang HC, Chung H, Karmon S, Royes P, Pon LA. A protein complex containing Mdm10p, Mdm12p, and Mmm1p links mitochondrial membranes and DNA to the cytoskeleton-based segregation machinery. Mol Biol Cell. 2003;14:4618–4627. doi: 10.1091/mbc.E03-04-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brickley K, Smith MJ, Beck M, Stephenson FA. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem. 2005;280:14723–14732. doi: 10.1074/jbc.M409095200. [DOI] [PubMed] [Google Scholar]
- Cantrell DA. Transgenic analysis of thymocyte signal transduction. Nat Rev Immunol. 2002;2:20–27. doi: 10.1038/nri703. [DOI] [PubMed] [Google Scholar]
- Ehrenberg B, Montana V, Wei MD, Wuskell JP, Loew LM. Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes. Biophys J. 1988;53:785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA. Tools of the trade: use of dominant-inhibitory mutants of Ras-family GTPases. Nat Cell Biol. 1999;1:E25–E27. doi: 10.1038/10018. [DOI] [PubMed] [Google Scholar]
- Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson A, Ruusala A, Aspenström P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278:6495–6502. doi: 10.1074/jbc.M208609200. [DOI] [PubMed] [Google Scholar]
- Fransson S, Ruusala A, Aspenström P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–510. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- Frederick RL, McCaffery JM, Cunningham KW, Okamoto K, Shaw JM. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol. 2004;167:87–98. doi: 10.1083/jcb.200405100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, Jan YN. Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development. 2002;129:2867–2878. doi: 10.1242/dev.129.12.2867. [DOI] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, Jan YN. Different levels of the homeodomain protein cut regulate distinct dendrite branching patterns of Drosophila multidendritic neurons. Cell. 2003;112:805–818. doi: 10.1016/S0092-8674(03)00160-0. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/S0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/S0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. The pattern and mechanisms of mitochondrial transport in axons. Front Biosci. 1996;1:d91–d102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- Klosowiak JL, Focia PJ, Chakravarthy S, Landahl EC, Freymann DM, Rice SE. Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep. 2013;14:968–974. doi: 10.1038/embor.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Osman C, Walter P. The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci U S A. 2011;108:14151–14156. doi: 10.1073/pnas.1111314108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Holman HA, Kubara K, Yasukawa K, Kawabata S, Okamoto K, MacFarlane J, Shaw JM. Structure-function analysis of the yeast mitochondrial Rho GTPase, Gem1p: implications for mitochondrial inheritance. J Biol Chem. 2011;286:354–362. doi: 10.1074/jbc.M110.180034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsopoulos OS, Laine D, Osellame L, Chudakov DM, Parton RG, Frazier AE, Ryan MT. Human Miltons associate with mitochondria and induce microtubule-dependent remodeling of mitochondrial networks. Biochim Biophysic Acta. 2010;1803:564–574. doi: 10.1016/j.bbamcr.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Lee KS, Lu B. The myriad roles of Miro in the nervous system: axonal transport of mitochondria and beyond. Front Cell Neurosci. 2014;8:330. doi: 10.3389/fncel.2014.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DM, Goodman CS. Ectopic and increased expression of fasciclin II alters motor neuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Louie K, Russo GJ, Salkoff DB, Wellington A, Zinsmaier KE. Effects of imaging conditions on mitochondrial transport and length in larval motor axons of Drosophila. Comp Biochem Physiol A Mol Integr Physiol. 2008;151:159–172. doi: 10.1016/j.cbpa.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009a;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Brickley K, Stephenson FA, Kittler JT. GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol Cell Neurosci. 2009b;40:301–312. doi: 10.1016/j.mcn.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–176. doi: 10.1038/nrn3380. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Muller HJ. Further studies on the nature and causes of gene mutations. In: Jones DF, editor. Proceedings of the 6th International Congress of Genetics; Wisconsin: Brooklyn Botanic Gardens; 1932. pp. 213–255. [Google Scholar]
- Murley A, Lackner LL, Osman C, West M, Voeltz GK, Walter P, Nunnari J. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife. 2013;2:e00422. doi: 10.7554/eLife.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschinger K, Rendić D, Wilson IB. Revealing the anti-HRP epitope in Drosophila and Caenorhabditis. Glycoconj J. 2009;26:385–395. doi: 10.1007/s10719-008-9155-3. [DOI] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plásek J, Sigler K. Slow fluorescent indicators of membrane potential: a survey of different approaches to probe response analysis. J Photochem Photobiol B. 1996;33:101–124. doi: 10.1016/1011-1344(96)07283-1. [DOI] [PubMed] [Google Scholar]
- Reis K, Fransson A, Aspenström P. The Miro GTPases: at the heart of the mitochondrial transport machinery. FEBS Lett. 2009;583:1391–1398. doi: 10.1016/j.febslet.2009.04.015. [DOI] [PubMed] [Google Scholar]
- Rolls MM. Neuronal polarity in Drosophila: sorting out axons and dendrites. Dev Neurobiol. 2011;71:419–429. doi: 10.1002/dneu.20836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls MM, Satoh D, Clyne PJ, Henner AL, Uemura T, Doe CQ. Polarity and intracellular compartmentalization of Drosophila neurons. Neural Dev. 2007;2:7. doi: 10.1186/1749-8104-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo GJ, Louie K, Wellington A, Macleod GT, Hu F, Panchumarthi S, Zinsmaier KE. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci. 2009;29:5443–5455. doi: 10.1523/JNEUROSCI.5417-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S. Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr Patterns. 2009;9:371–380. doi: 10.1016/j.gep.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh D, Sato D, Tsuyama T, Saito M, Ohkura H, Rolls MM, Ishikawa F, Uemura T. Spatial control of branching within dendritic arbors by dynein-dependent transport of Rab5-endosomes. Nat Cell Biol. 2008;10:1164–1171. doi: 10.1038/ncb1776. [DOI] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–2104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Onishi M, Jan LY, Jan YN. Peripheral multidendritic sensory neurons are necessary for rhythmic locomotion behavior in Drosophila larvae. Proc Natl Acad Sci U S A. 2007;104:5199–5204. doi: 10.1073/pnas.0700895104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Roegiers F, Rolls MM. Microtubules have opposite orientation in axons and dendrites of Drosophila neurons. Mol Biol Cell. 2008;19:4122–4129. doi: 10.1091/mbc.E07-10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Górska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/S0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Toulmay A, Prinz WA. A conserved membrane-binding domain targets proteins to organelle contact sites. J Cell Sci. 2012;125:49–58. doi: 10.1242/jcs.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77:485–502. doi: 10.1016/j.neuron.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Vlahou G, Eliáš M, von Kleist-Retzow JC, Wiesner RJ, Rivero F. The Ras related GTPase Miro is not required for mitochondrial transport in Dictyostelium discoideum. Eur J Cell Biol. 2011;90:342–355. doi: 10.1016/j.ejcb.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Voss C, Lahiri S, Young BP, Loewen CJ, Prinz WA. ER-shaping proteins facilitate lipid exchange between the ER and mitochondria in S. cerevisiae. J Cell Sci. 2012;125:4791–4799. doi: 10.1242/jcs.105635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Dürrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, Wichmann C, Kittel R, Sigrist SJ, Buchner E. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Wang JW, Beck ES, McCabe BD. A modular toolset for recombination transgenesis and neurogenetic analysis of Drosophila. PLoS One. 2012;7:e42102. doi: 10.1371/journal.pone.0042102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO. The molecular basis of genetic dominance. J Med Genet. 1994;31:89–98. doi: 10.1136/jmg.31.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittinghofer A. Signal transduction via Ras. Biol Chem. 1998;379:933–937. [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Babic M, Russo GJ. Mitochondrial transport dynamics in axons and dendrites. Results Probl Cell Differ. 2009;48:107–139. doi: 10.1007/400_2009_20. [DOI] [PubMed] [Google Scholar]