

Introduction
Langerhans cell histiocytosis (LCH) is a rare disease that mainly affects children; very rarely it may affect adults. Incidence in adults is around one to two cases per million population.1 It is a disease of unknown aetiology, comprising a group of syndromes involving an abnormal increase in the number of histiocytes.
Recently we came across an adult case of LCH, who presented with diffuse parotidomegaly, and resistant seborrhoeic dermatitis. Diagnosis was clinched on skin biopsy and later HRCT showed characteristic radiological findings even when patient was asymptomatic, prompting us to report the case.
Case report
36 yr old male patient, an old case of alcohol dependence and hypertension was investigated elsewhere for alcoholic liver disease about six months back. This time he presented to Department of oto-rhino-laryngology with bilateral parotidomegaly and enlargement of submandibular glands for about three months and loss of about 5 kg of weight in the last six months. He was referred to dermatology OPD for management of seborrhoeic dermatitis. There was no h/o chronic diarrhoea, fever, chest pain, cough, breathlessness, smoking, urinary symptoms, and pain in the bone and/or swelling/lumps on the skull.
Examination revealed diffuse erythema with greasy scales involving scalp, face, retro auricular region, and both axilla and groins (Fig. 1) some of these lesions were eroded with focal yellowish crusting. Multiple, discrete, scaly, red-brown, papules were seen involving predominantly the seborrhoeic areas of the trunk, most of the nails of both hands and feet showed onychomadesis (Fig. 2). Yellow purulent discharge was present in the ear (Left). Oral hygiene was poor and patient had generalized motility of teeth. There was generalized desquamation of oral mucosa with multiple raw wounds and severe periodontal loss.
Fig. 1.
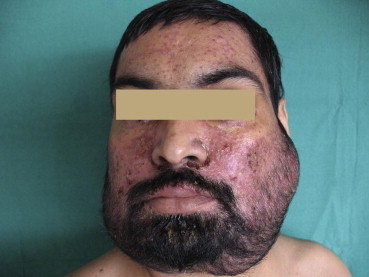
Diffuse erythematous plaques and papules with crusting involving face and scalp.
Fig. 2.
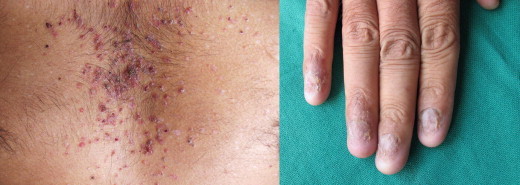
Multiple, discrete, scaly, red-brown, papules over chest and onychomadesis.
Investigations revealed normocytic anaemia with haemoglobin of 11 gm%, with slightly raised ESR. Liver function and renal function tests were normal. Alkaline phosphatase (516 IU/L), GGT (72 IU/L) and serum amylase (98 U/100 ml) were raised. ELISA for HIV, HBsAg and anti HCV antibody was negative.
Patient was treated with tapering doses of oral corticosteroids starting at 30 mg/day and tapered over next three weeks along with topical steroids and antifungals for seborrhoeic dermatitis to which he showed unsatisfactory response.
Skin biopsy from midline of upper back showed mild spongiosis of epidermis, with dense mixed inflammatory infiltrate composed of sheets of histiocytes and lymphocytes in dermis. Many of the histiocytes showed reniform nuclei and nuclear grooving, features associated with LCH (Fig. 3A). Lower dermis showed an inflammatory infiltrate of lymphocytes along with neutrophils and eosinophils. No granulomas were seen. ZN stain was negative for AFB. Immunohistocytochemistry markers CD1a, CD 68 and S100 were positive in the histiocytes (Fig. 3B). FNAC from the parotid glands showed features of inflammatory cystic lesion.
Fig. 3.
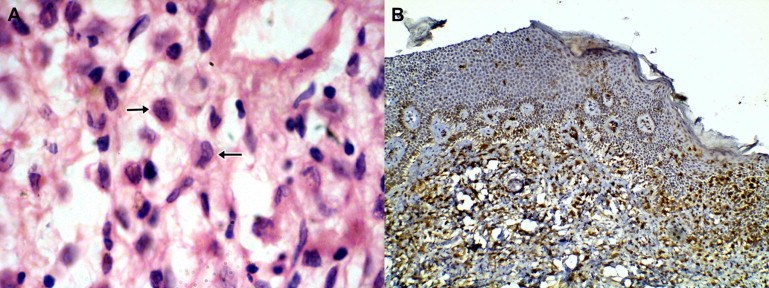
(A) H&E stained section (×400) of skin showing with reniform nuclei (short arrow) and mixed inflammatory infiltrate including eosinophils (long arrow). (B) IHC staining for CD1a (×100) showing diffuse positivity in histiocytes in the dermis.
High Resolution Computed Tomography (HRCT) chest showed multiple thin walled cysts in segments of both upper lobes of variable and bizarre shapes. There was gross involvement of the right middle lobe and lower lobe including the costophrenic angle (Fig. 4). Minimal right apical pneumothorax was also noted. USG abdomen revealed a heterogeneously increased echotexture with multiple hypoechoic areas in both lobes. CT scan abdomen revealed hepatomegaly with multiple oval well defined non enhancing hypodense lesions in both lobes of liver. MRI neck revealed bilateral parotid enlargement with multiple cysts in both the parotid and submandibular glands (Fig. 5). Upper GI endoscopy was normal.
Fig. 4.
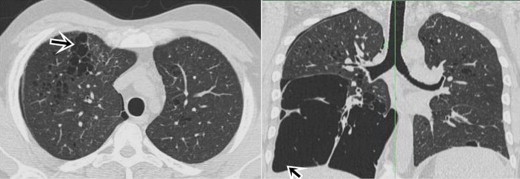
Axial & coronal CT sections show variable sized, bizarre thin walled cysts in upper zones & large cystic changes involving the costophrenic angles.
Fig. 5.
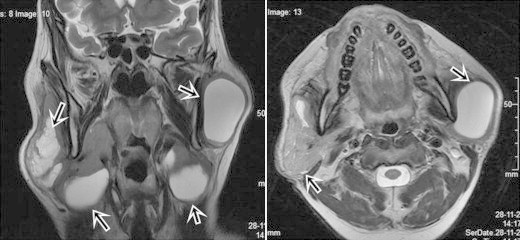
Axial & coronal T2W MRI images show large cystic changes in the parotid & submandibular glands bilaterally.
He was transferred to malignant disease treatment centre for chemotherapy. Initial treatment was prednisolone 40 mg/m2/day orally, weekly reduction after week 4 and IV Vinblastine 6 mg/m2 bolus for six weeks. Patient responded favourably to initial treatment, skin lesions cleared off, swelling of both parotid and submandibular glands has resolved. Patient is presently on continuation treatment consisting of prednisolone 40 mg/m2/day orally for five days in a week every three weeks, IV Vinblastine 6 mg/m2 bolus every three weeks, and 6 mercaptopurine 50 mg/m2 daily orally for 12 months. He is currently on follow-up.
Discussion
Paul Langerhans first described the epidermal dendritic cells in 1868.2 LCH is an accumulation or proliferation of a clonal population of cells bearing the phenotype of a Langerhans cell, these cells being arrested at an early stage of activation and are functionally deficient.3
The diagnosis of LCH must be established histologically. The Writing Group of the Histiocyte Society identified three levels of confidence in the diagnosis of LCH.4
-
1.
A presumptive diagnosis. This is in a patient with disease clinically consistent with LCH and with histology consistent with the diagnosis.
-
2.
A diagnosis. This is established when the histology is consistent with LCH and lesional cells are shown to express S100 and/or α-d-mannosidase activity.
-
3.
A definitive diagnosis. This is established when histology is consistent with a diagnosis of LCH and the lesional cells are shown to express CD1a or to have intracytoplasmic Birbeck granules on electron microscopy.
LCH has varied clinical manifestations and an unpredictable course.5 LCH is traditionally divided into three groups: “unifocal, multifocal unisystem, and multifocal multisystem”.6 Multifocal multisystem LCH, earlier called Letterer-Siwe disease, is a rapidly-progressing disease involving Langerhans cells proliferation in many tissues. It generally affects children under 2 years of age, and has poor prognosis. Though cutaneous LCH responds well to oral or topical steroids, severe disease requires a systemic approach.7 The 5-year survival is only 50% even with aggressive chemotherapy.8 Adult LCH commonly affects smokers and is usually unifocal. Multisystem LCH in adults is rare.3
In multifocal multisystem disease, skin involvement is in the form of recurrent pyoderma like lesions with crusting and scaling, vesicopustular and purpuric eruption occurring in crops over the face, scalp and trunk, resembling seborrhoeic dermatitis. “Intraoral haemorrhage, gingivitis, loose teeth, precocious eruption of teeth and ectopic teeth also may be seen”.9 Lesions may affect a variety of other systems, including liver (20%), spleen (30%), and lymph nodes (50%).
Number of organs involved and the presence of organ dysfunction determines the prognosis of LCH. Single system disease has excellent prognosis, almost 100%, and for multiple system disease survival is around 80%.10
Our case fits into definitive diagnosis category as per Writing Group of the Histiocyte Society.4 However it is unique since it had bilateral parotid and submandibular gland enlargement with large cystic changes which is hitherto unreported in Langerhans cell histiocytosis, and HRCT chest revealing pulmonary cystic changes with rare involvement of the costophrenic angle.11 The patient was asymptomatic with regard to respiratory system. The diagnosis was established only when the treatment resistant skin lesions were biopsied.
The recommendation that skin biopsy should be performed on atypical cases of seborrhoeic dermatitis who do not respond to conventional treatment, keeping this diagnosis in mind, is therefore reiterated.3
Conflicts of interest
All authors have none to declare.
References
- 1.Baumgartner I., von Hochstetter A., Baumert B., Luetolf U., Follath F. Langerhans cell histiocytosis in adults. Med Pediatr Oncol. 1997 Jan;28(1):9–14. doi: 10.1002/(sici)1096-911x(199701)28:1<9::aid-mpo3>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 2.Langerhans P. Über die Nerven der menschlichen Haut. Arch Abl B Pathol. 1868;44:325–337. [Google Scholar]
- 3.Chu T. Langerhans cell histiocytosis. Australas J Dermatol. 2001 Nov;42(4):237–242. doi: 10.1046/j.1440-0960.2001.00527.x. [DOI] [PubMed] [Google Scholar]
- 4.Histiocytosis syndromes in children Writing group of the histiocyte society. Lancet. 1987 Jan 24;1(8526):208–209. [PubMed] [Google Scholar]
- 5.Kusumkumary P., Kumari P.T., Chellam V.G., James F.V., Nair K.M. Langerhans cell histiocytosis in children less than 2 years of age. Indian Pediatr. 1999;36:29–36. [PubMed] [Google Scholar]
- 6.Kumar Abbas, Fausto Aster. 8th ed. Elsevier Saunders; Philadelphia, Penn: 2010. Robbins and Cotran Pathologic Basis of Disease. Ch 13, Disorders of White Blood Cells, 631–632. [Google Scholar]
- 7.Park L., Schiltz C., Korman N. Langerhans cell histiocytosis. J Cutan Med Surg. 2012;16(1):45–49. doi: 10.1177/120347541201600109. [DOI] [PubMed] [Google Scholar]
- 8.Muller J. Langerhans cell histiocytosis in childhood. J Pediatr Sci. 2010;2(3):e28. [Google Scholar]
- 9.Hartmann K.S. Histiocytosis X. A review of 114 cases with oral involvement. Oral Surg Oral Med Oral Pathol. 1980;49:38–54. doi: 10.1016/0030-4220(80)90030-4. [DOI] [PubMed] [Google Scholar]
- 10.Muller J., Garami M., Hauser P. Hungarian experience with Langerhans cell histiocytosis in childhood. Pediatr Hematol Oncol. 2006;23:135–142. doi: 10.1080/08880010500457988. [DOI] [PubMed] [Google Scholar]
- 11.Webb R., Muller N.L., Naidich D.P., editors. Cystic Lung Disease. 4th ed. Lippincott Williams & Wilkins; Philadelphia: 2009. High resolution CT of lung; pp. 368–380. [Chapter 12] [Google Scholar]