

Abstract
Psoriasis and psoriatic arthritis represent two paradigmatic conditions characterized by chronic inflammation and possibly autoimmunity, despite the absence of known serum autoantibodies. The two diseases, albeit strongly correlated from clinical, genetic, and epidemiogical standpoints, manifest significant differences in terms of etiology and pathogenetic mechanisms. Nonetheless, Th17 cells appear crucial to both diseases, and IL23 is the cytokine involved in determining the fate of naive CD4+ cells to differentiate into a pathogenic phenotype. This basic experimental observation led to a clear understanding of the immune dysfunction causing psoriasis and psoriatic arthritis but, more importantly, also led to new therapeutic approaches. In recent years, monoclonal antibodies directed to IL12/IL23 (ustekinumab) or IL17 (secukinumab, ixekizumab, brodalumab) are being investigated or have proven to be beneficial for patients with psoriatic disease, thus further supporting the view that Th17 cells play a pivotal role in disease onset and perpetuation. These most recent reports indeed represent significant developments that may allow overcoming the TNFα pathway as the major therapeutic target in chronic inflammation.
Keywords: Psoriasis, Psoriatic arthritis, IL12, IL23, IL17, Inflammation
Introduction
Psoriasis (Pso) and Psoriatic arthritis (PsA) are chronic inflammatory diseases affecting the skin and the spine or peripheral joints [1, 2]. Although Pso and PsA show important differences at immunological and genetic levels, the prevailing view is that they are different manifestations of the same disease, with peculiar penetrance and onset timing. The etiology of both diseases remains largely unknown, but their genetic, immunological, and environmental bases have been recently elucidated. Both Pso and PsA markedly reduce the quality of life of affected people and recent studies have suggested the association of psoriatic disease with other pathological conditions [3–12]. Pso is a chronic relapsing dermatitis, characterized by epidermal hyperplasia, vascular proliferation, and infiltration of leukocytes in the dermis. It usually presents with scaly red plaques on typical areas of the body, such as the scalp, the lumbar and sacral region, and the extensor areas of elbows and knees. PsA is a chronic seronegative arthritis that may be diagnosed in the absence of active Pso in the patient but with a positive history of skin manifestations in one first degree relative [13, 14]. PsA is characterized by enthesitis and/or arthritis involving the distal interphalangeal, axial joints, commonly with active sacroilitis. Despite the obvious association between the two conditions, the mechanisms shared by both diseases remain largely unknown and specific elements point towards a common pathogenesis.
The burden of psoriatic disease is shown by the high prevalence of the disease and its serious impact on the quality of life of affected patients. In fact, Pso is found in 2–3 % of the Caucasian population and approximately 30 % of patients with Pso will develop PsA [15]. Even if the epidemiological profile of PsA is likely to be widely variable across world areas, the prevalence of PsA is generally estimated to be between 0.3 and 1 % of the general population [2]. In approximately 75 % of the cases the skin disease precedes arthritis, while in 15 % of patients Pso appears after PsA and in 10 % the cutaneous and articular involvement are simultaneous [16]. Most frequently, Pso appears around 15–35 years of age, while PsA is highly polymorphic and can occur at any age, in both adults and children, with a peak in the 40–50 years range [17]. Its frequency is similar in both sexes, but men are 3–5 times more likely to manifest a spine involvement [7].
An open issue in both Pso and PsA is that, although current therapies have improved the clinical management, unmet clinical needs still remain. In fact, many patients treated with disease-modifying anti-rheumatic drugs (DMARDs) and TNFα inhibitors may show limited drug tolerance, partial or non-responsiveness, and the long-term use of systemic therapies, such as anti-inflammatory drugs and corticosteroids, is burdened by the risk of serious adverse events. For these reasons, recent efforts in clinical research have focused on the development of new therapies targeting more specifically the pathogenetic mechanisms of psoriatic disease, not only to achieve clinical remission but also to reduce the progression of Pso and PsA. In particular, based on recent insights into the immunopathogenesis of Pso and PsA, the quest for novel therapeutic targets has identified the IL23/Th17 axis as a promising target for specific interventions to inhibit the dysregulated immune system. This major breakthrough is well illustrated by biological therapies targeting IL17A (ixekizumab and secukinumab), IL17R (brodalumab), and IL12/IL23 (ustekinumab) that have been or are being evaluated at advanced stages of development.
Immunopathogenesis of psoriatic disease
Pso and PsA are the two major manifestations of the psoriatic disease spectrum and are believed to share a common pathophysiological pathway characterized by dysfunctional stromal immune cell and cytokine network causing inflammation in the skin, entheses, and joints [18]. The major cytokines involved in Pso and PsA are listed in Table 1. In the earliest phases of research, psoriatic disease was considered to be the result of a Th1 response with IFNγ and IL12 as signature cytokines [19]. In recent years, however, the key role of both the innate immune cells in the affected tissue and adaptive Th17 cells response together with other cytokines (i.e. TNFα, IL23, IL17, IL22) has been clarified. The major contributors to the initiation of the local inflammation and the activation of IL23/Th17 axis appear to be myeloid dendritic cells (mDC) [20]. On the other hand, it has been proposed that IFNα-producing plasmacytoid DC (pDC) [21], LL37 produced by keratinocytes in Pso, mechanical stress [22], and microbial infections [22] contribute to the stimulus that induces DC and other innate immunity cells to trigger inflammation. In clinical terms, these phenomena could explain the wide variability of the disease phenotype and the association with environmental factors. Among the proposed factors, the endogenous antimicrobial peptide LL37 released by keratinocytes plays a relevant role in linking the innate and adaptive immunity in Pso as LL37 aggregates with autogenous DNA and activates toll-like receptor (TLR) nine signaling in pDC, thus resulting in the production by mDC of the pro-inflammatory cytokines TNFα, IL6, IL23, and IFNγ [22]. The local milieu enriched with these cytokines that are produced also by local macrophages further promotes the Th17 polarization of naïve CD4+ Th cells in lymph nodes, triggered by an unknown antigen stimulation [23, 24]. Once activated, Th17 cells migrate to the inflamed tissues, overexpressing the chemokine CCL20 and binding to its receptor CCR6 and perpetuate the immune reaction by attracting neutrophils to the site of inflammation.
Table 1.
| Cytokine | Cellular source | Receptor | Targets | Role in Pso/PsA |
|---|---|---|---|---|
| IL1β | Macrophages, T cells | ILR1 | Keratinocytes, FLS, ECs, OB | Inflammation, osteoclastogenesis, angiogenesis, Th17 amplification |
| IL6 | Macrophages, T cells | IL6R/gp130 | DC, macrophages, T cells | Inflammation, acute phase response |
| IL12 | Th1, monocytes, macrophages | IL12Rβ 1/2 | Th1 differentiation, inflammation, NK activation | |
| IL17A, IL17F | Th17, mast cells, macrophages DCs, NK cells, CD8 T cells | IL17RA/IL17RC | Keratinocytes, FB, OC | Inflammation, neutrophil recruitment, osteoclastogenesis, angiogenesis |
| IL23 | T cells, mast cells, macrophages, DC | IL12Rβ/IL23R | Th17, mDC, OC | Th17 differentiation, mDC activation, osteoclastogenesis |
| IL20 | Monocytes | IL20Rα/β | Th17 | Inflammation |
| IL22 | Th17, NK cells, mast cells | IL22Rα/IL10Rβ | Keratinocytes, FLS | Host defence, Keratinocytes and FB proliferation, inflammation |
| TNFα | Macrophages, mDC, pDC keratinocytes, FB, Th17 | TNFαR1/TNFαR2 | mDc, macrophages, keratinocytes, FLS | Proinflammatory, DC activation, immune cells recruitment, angiogenesis Keratinocyte proliferation, osteoclastogenesis, Th17 amplification |
| IFNα | pDC | IFNαR | mDC | Initation of immune response |
| IFNγ | Macrophages, mDC, Th1, Th17 | IFNγR | mDC, macrophages | Th1 differentiation, inflammation |
| TGFβ | Macrophages, mDC | TGFβR1/TGFβR2 | Keratinocytes, FLS, T cells | Keratinocyte proliferation, inflammation, Th17 differentiation, angiogenesis |
| RANKL | T cells, FB, FLS, OB | RANK | Synovial lining layer | Osteoclastogenesis |
| OPG | OB, FLS | RANKL decoy R | EC below synovial lining | Osteoclastogenesis inhibition |
DC dendritic cell, FB fibroblasts, FLS fibroblast like synoviocytes, OB osteoblasts, OC osteoclasts, EC endothelial cells, pDC plasmacytoid dencritic cells, NK natural killer cells, OPG osteoprotegerin
In Pso, the effector cytokines IL17, IL22, and IL20 produced by Th17 cells activate the STAT3 signaling in keratinocytes, inducing their hyperproliferation and the expression of proinflammatory cytokines, growth factors, antimicrobial peptides, and matrix metalloproteinases that cause chronic skin and articular inflammation [24]. In PsA, Th1- and Th17-driven inflammation is more likely to exert a pro-erosive influence and cause the ultimate damage to the involved joints. In fact, proinflammatory cytokines (i.e. IL23, TNFα, and IL17) activate the NFkB pathway in fibroblasts and synovial cells and induce RANKL and M-CSF expression that promotes osteoclastogenesis and bone absorption, thus leading to destructive arthritis [25, 26]. In this scenario, it is unclear whether other factors such as osteoprotegerin and RANKL levels are increased in PsA [26]. Hyperplasia of synovial lining cells and mononuclear cell infiltration (macrophages, neutrophils, T cells) characterize PsA synovitis and could result from these inflammatory components [27].
The IL23/Th17 axis
A schematic representation of the IL23/Th17 axis is illustrated in Fig. 1. Th17 cells are involved in the pathogenesis of various autoimmune and inflammatory disorders, such as rheumatoid arthritis (RA), Crohn’s disease (CD), asthma, and experimental autoimmune encephalitis (EAE) as a model of multiple sclerosis [27, 28]. IL23 is the upstream cytokine crucial for the development and maintenance of a pathogenic Th17 cell phenotype from naive CD4+ T cells, together with TGFβ and IL6 [23]. IL23 produced by DC and macrophages binds the IL23R on activated Th17 cells and enhances the expression of the orphan nuclear receptor γ-t (RORγ-t), the major transcription factor of Th17 polarization [29]. RORγ-t then further interacts with IL6 induced-STAT3 signaling to stimulate the production of the effector cytokines IL17A, IL17F, IL21, and IL22. Once recruited to the site of inflammation via CCR6-CCL20 chemotaxis, Th17 cells exert their function via cytokines (i.e. IL17, IL21, IL22) that activate signaling pathways on different cells [30]. A recent breakthrough in understanding the pathogenesis of Pso has been made with studies that successfully showed the interaction between the IL23/Th17 axis, keratinocytes, and synovial cells [31]. One downstream cytokine induced by IL23 acting on Th17 cells is indeed IL17 [32] that takes part to mechanisms of delayed immune response to recruit monocytes and neutrophils at sites of inflammation, causing destructive tissue damage [33]. A well-known effect of IL17 is the pro-inflammatory ability to induce airway modifications that has been observed in asthma. Additional functions of IL17 have been related to induction of cytokines and prostaglandins that are involved in autoimmune and inflammatory diseases such as RA and allograft rejection. Other groups also identified an IL23-independent pathway for IL17 production, mainly mediated by TGFβ and IL6 [34], but IL23 is still required to promote survival and proliferation of the T cells that produce IL17 [34].
Fig. 1.
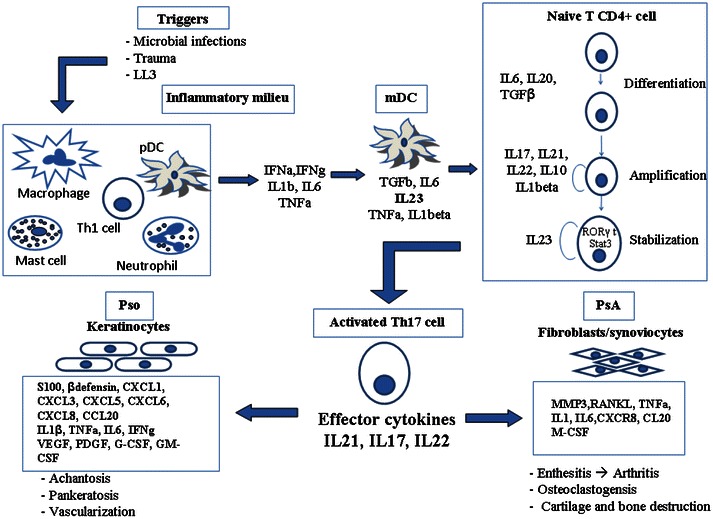
A schematic representation of the Th17 pathway and its involvement in psoriatic disease
A recent study on Th17 development identified serum glucocorticoid kinase-1 (SGK1) as an essential node in the induction of pathogenic Th17 cells. SGK1 is a serine-threonine kinase that is expressed by differentiating Th17 subsets under IL23R signaling and is critical for Th17 stabilization. The experimental study used an elegant approach to demonstrate that SGK1 expression is induced and maintained by exposure to IL23 under Th17 differentiating conditions and is not induced by other T cell subsets (Th0, Th1, Th2). Nonetheless, it was suggested that SGK1 acts by deactivating Foxo1, a direct inhibitor of IL23R expression, thus enhancing the expression of IL23R and stabilizing Th17 phenotype. Interestingly, SGK1 was also reported to act as a salt-sensing kinase, mediator for sodium homeostasis, by phosphorylating epithelial sodium channels (ENaC). A high sodium diet thus enhances the expression of SGK1 and increases Th17 differentiation in vitro and in vivo, thus accelerating the development of autoimmunity. These data raise the issue of whether high sodium salt intake in western diet is responsible for an increased pathogenic Th17 response and increase in autoimmune diseases [35].
The IL23/Th17 axis in Pso and PsA
The first evidence of a major T cell involvement in Pso was provided by the significant clinical response of patients treated with earlier T-cells suppressor agents, such as cyclosporine and alefacept [36, 37], and later costimulation inhibitors such as abatacept [38]. Moreover, psoriatic plaque samples show a marked infiltration of activated T cells which directly correlate with psoriasis area score index (PASI). Similar to cutaneous psoriatic eruptions, significant infiltration of the inflamed synovial membrane by activated CD4+ T cells is also observed in PsA, but the involvement of Th17 axis in PsA remains unclear [39]. Nonetheless, signature cytokines of Th17 cells, such as IL17 and IL22, are highly expressed in skin lesions, joints, and serum of patients with psoriatic disease and correlate with disease severity [40]. As shown in Table 2, the critical role of the Th17 cytokines in Pso is further supported by data from genomewide association studies (GWAS) that identified Pso susceptibility genes among those involved in IL23/Th17 axis, such as IL23R, IL12B, IL23A, TRAF3IP2, and Act1, which regulate IL17 production and IL17-mediated signaling [41, 42]. From a genetic standpoint, however, one concern is related to the difficulty to dissect associations with Pso from those more pertinent to PsA. Finally, compelling evidence of the critical role of IL23/Th17 axis in Pso pathogenesis is provided by clinical studies with new biological agents, as will be discussed further in a dedicated section.
Table 2.
Susceptibility genes involved in Pso and PsA from GWA studies
| Gene | Name | Chromosome | Pathway | Other diseases | References |
|---|---|---|---|---|---|
| IL23A | IL23 p19 | 12q13 | IL23 | [42] | |
| IL12B | IL12/IL23 p40 | 5q33 | IL23 | IBD, PsA, AS, MS | [42, 53, 84–86] |
| IL23R | IL23R | 1p31 | IL23 | IBD, PsA, AS | [42, 53, 87, 88] |
| TYK2 | Tyrosine kinase 2 | 19p13 | IL23 | IBD, SLE, MS? | [89–92] |
| TRAF3IP2 | TNF receptor-associated factor 3 interacting protein 2 | 6q21 | NFKB, IL17 | PsA, RA, CD | [92–96] |
| TNFA IP3 | TNFα-induced protein3 | 6q23 | NFKB | PsA, SLE, RA, CD | [42, 95, 97, 98] |
| TNIP1 | TNFAIP3-interacting protein 1 | 5q33 | NFKB | PsA, SLE, SSc | [42, 99, 100] |
| DEFB4 | Defensin β 4 | 16q12 | IL17 | [101] |
IBD inflammatory bowel disease, PsA psoriatic arthritis, AS ankylosing spondylitis, MS multiple sclerosis, CD celiac disease, RA rheumatoid arthritis, SLE systemic lupus erythematous, SSc systemic sclerosis
IL23 is a member of the IL12 family of cytokines. It is a heterodimeric cytokine composed of the same IL12 subunit called p40 paired with its unique subunit p19 [43]. IL23 is mainly produced by mDC, and in Pso it is upregulated and responsible for the priming of the Th17 response [23], playing a key role in the pathogenesis of skin lesions [44]. The presence of abundant IL23+ DC as well as the elevated mRNA expression of both IL23 subunits (IL23p19 and IL23p40) in psoriatic lesions supports this hypothesis. The role of IL23 in the induction of Pso is confirmed also experimentally by the effect of injecting IL23 in the mouse skin, resulting in epidermal hyperplasia and inflammatory cellular infiltration similar to human Pso, mediated by TNF-α, IL22, IL17A, and IL17F [45]. Studies on experimental models of arthritis have also demonstrated that IL23 affects osteoclastogenesis via an IL17-independent mechanism [46]. The use of therapies such as IL23 inhibitors is associated with reductions of the inflammation, with therapeutic functions similar to those already known from anti-TNFα agents used in PsA like etanercept [47].
IL17 and IL22 are the main effector cytokines of the Th17 polarization and are of primary importance in the protection against bacterial and fungal infections [20]. IL17 is part of the IL17 cytokine family, which consists of six members: IL17A, IL17B, IL17C, IL17D, IL17E/IL25, and IL17F (Table 3). Because of their structural and functional similarities and the fact that they are both produced by Th17 cells, IL17A and IL-17F have been most thoroughly studied and characterized. IL17A and IL17F are most closely related, sharing 55 % homology with each other. Both are active as homodimers, and recently, IL17A/IL17F heterodimers have been identified, and their function is currently being studied [48]. In addition to Th17, other sources of IL17 are mast cells [22], cytotoxic CD8+ T cells, natural killer cells, neutrophils, and macrophages [49]. IL17 induces the expression of pro-inflammatory cytokines and chemokines via IL17R, which recruits neutrophils to inflammatory sites. IL17 is especially relevant in the protection of mucocutaneous surfaces against Candida albicans infection. Recent genetic studies demonstrated that rare human genetic diseases associated with chronic mucocutaneous candidiasis are linked to genetic defects in IL17 signaling, either through autosomal IL17RA or IL17F mutations (i.e. chronic mucocutaneous candidiasis) [50] or high titers of neutralizing antibodies against IL17 cytokines (i.e. autoimmune polyendocrine syndrome type 1, APS-I, secondary to mutations in the AIRE gene) and mutation in STAT3 signaling (i.e. hyper-IgE syndrome) [51]. Thus, an impaired immunity and a deficient Th17 response are responsible for chronic mucocutaneous candidiasis among individuals not only with primary but also acquired immunodeficiency, such as HIV/AIDS, chemotherapy-induced neutropenia, and those assuming chronic systemic corticosteroid therapies or drugs targeting the IL23/Th17/IL17 pathway [52].
Table 3.
| Member | Alternative names | Receptor | Main functions |
|---|---|---|---|
| IL17A | CTLA-8 | IL17RA, IL17RC | Neutrophils recruitment, host defence, autoimmunity |
| IL17B | CX1, NERF | IL17RB | Proinflammatory activity? |
| IL17C | CX2 | IL17RE | Proinflammatory activity? |
| IL17D | IL27, IL27A | Unknown | Proinflammatory activity? |
| IL17E | IL25 | IL17RB, IL17RA | Stimulates Th2 response, suppresses Th17 response |
| IL17F | ML-1 | IL17RA, IL17RC | Neutrophil recruitment, host defence, autoimmunity |
IL22 is a member of the IL10 family of cytokines, which also includes IL19, IL20, and IL24 [22]. IL22 activates IL22R and IL10R2 signaling and induces the release of antimicrobial peptides and matrix metalloproteinases from fibroblasts, endothelial cells, epithelial cells, osteoblasts, macrophages, and DC [20, 21]. An increasing body of evidence suggests that IL17 and IL22 are also important in Pso pathogenesis based on the observation that serum levels of IL17 and IL22 are elevated compared with healthy controls and correlate with disease severity [53, 54]. Moreover, IL17 and IL22 induce the expression of antimicrobial peptides in Pso, such as psoriasin (S100A7), koebnerisin (S100A15), b-defensin 2, S100A8, and S100A9 [55] that can act synergistically as chemoattractants to amplify inflammation [56]. The expression of the neutrophil chemoattractants CXCL1, CXCL3, CXCL5, CXCL6, CXCL8, and the chemokine CCL20 is also upregulated by IL17 [37] and likely promotes skin inflammation. In particular, IL17 exerts a pro-inflammatory activity, and IL22 mainly acts on keratinocytes via STAT3 activation [40]. In Pso, IL22 also induces the proliferation and hyperplasia of keratinocytes, mediates the production of antimicrobial peptides, and alters differentiation of keratinocytes’ leading to hypogranulosis, achantosis, and pankeratosis [57]. In the case of PsA, experimental evidence supporting a role of IL17A in PsA is limited compared with Pso or RA. Th17 cells are increased in the peripheral blood of patients with PsA, and they exhibit a highly differentiated and polyfunctional phenotype suggesting that they play a specific role in the disease [58]. IL17A—producing cells, including Th17 cells and c-Kit—positive mast cells, are increased in the synovial fluid during the course of active PsA [4, 59], and synovial fibroblasts produce higher levels of IL6, CXCL8, and MMP-3 in response to IL17A compared with cells from osteoarthritis patients, an effect that can be blocked by an anti-IL17RA monoclonal antibody. IL17 influences all critical events of inflammation which include induction of adhesion molecules such as ICAM-1, upregulation of chemokines (i.e. IL6, IL8), and angiogenesis. In vitro studies demonstrated that IL17 can induce the expression of RANKL and influence osteoclastogenesis in PsA [47, 60].
New therapeutic targets in Pso and PsA
IL23 targeting (briakinumab and ustekinumab)
Briakinumab and ustekinumab are two fully human monoclonal antibody (mAb), directed at the p40 subunit shared by IL12 and IL23 (Table 4).
Table 4.
| Biologic | Target | Route | Pso status/trial | PsA status/trial |
|---|---|---|---|---|
| Ustekinumab | Human monoclonal antibody against the p40 subunit of IL-12 and IL-23 | SC | FDA-approved | FDA-approved |
| Secukinumab (AIN457) | Human monoclonal antibody directed against IL17A | IV | Phase III: SIGNATURE | Phase III: FUTURE 1 |
| Brodalumab (AMG-827) | Human monoclonal antibody directed against IL17R | IV | Phase III AMAGINE-1,-2,-3 | |
| Ixekinumab (LY2439821) | Humanized monoclonal antibody directed against IL17A | IV | Phase III: UNCOVER-1,-2,-3 | Phase III: SPIRIT-P1 |
Ustekinumab was approved by the FDA for Pso and PsA after demonstration of its efficacy and safety profile in phase III clinical trials. Two placebo-controlled trials, PHOENIX 1 and PHOENIX 2, in fact, showed that ustekinumab was able to ameliorate Pso plaques, pruritus, and nail psoriasis [31]. More importantly, the head-to-head ACCEPT [Active Comparator (CNTO 1275/Enbrel) Psoriasis Trial] study was an active comparator randomized phase III trial against etanercept in the treatment of Pso, where 903 patients with moderate-to-severe Pso received subcutaneous injections of either 45 or 90 mg of ustekinumab (at weeks 0 and 4) or high-dose etanercept (50 mg twice weekly for 12 weeks). At the primary endpoint of 12 weeks, the proportions of patients achieving a PASI75 response in the ustekinumab 45 mg group, ustekinumab 90 mg group, and etanercept 50 mg group were 68, 74 and 57 %, respectively (P = 0.01 and P < 0.001, respectively) [61]. In all trials, ustekinumab at the dosage of 45 or 90 mg significantly improved clinical symptoms of Pso compared with placebo or etanercept. Ustekinumab was generally well tolerated, as most adverse events were mild and did not require treatment adjustment, and the incidence of serious AE was 2 % or less for all treatment arms. In a pooled analysis of data from phase II and III clinical studies investigating the incidence of major adverse cardiovascular events (myocardial infarction, stroke or cardiovascular death) with ustekinumab treatment in patients with moderate to severe Pso, this adverse event was reported in 0.3 % of ustekinumab-treated patients [62]. Nonetheless, ustekinumab may increase the risk of infections or reactivate latent infections [63, 64]. Ustekinumab was approved in 2009 in the US [64] for the treatment of adult patients with moderate-to-severe plaque Pso, while in the European Union [63] ustekinumab is approved for patients who failed to respond to, have a contraindication to, or are intolerant to other systemic therapies or phototherapy for Pso. In the case of PsA, the Food and Drug Administration (FDA) and EU approved ustekinumab in 2013 for treatment of adults with active PsA who have not responded adequately to previous DMARDs. The approval was supported by findings from two phase III multicenter, randomized, double-blind, placebo-controlled trials in naive subjects with active PsA (PSUMMIT1) and in patients previously treated with biological anti-TNFα agents (PSUMMIT II) [65]. These studies compared the efficacy and safety of subcutaneously administered ustekinumab 45 or 90 mg at weeks 0, 4, and then every 12 weeks and included 927 patients diagnosed with active PsA who had at least five tender and five swollen joints and C-reactive protein levels of at least 0.3 mg/dL. PSUMMIT II also included 180 patients with previous exposure to at least one TNFα inhibitor. Results from PSUMMIT I demonstrated that at week 24, 42 and 50 % of patients receiving ustekinumab 45 and 90 mg, respectively, achieved ACR20, the primary endpoint [65]. In PSUMMIT II, 44 % of patients receiving ustekinumab 45 mg and 44 % of patients receiving ustekinumab 90 mg achieved ACR20 at week 24. Additionally, ustekinumab improved soft tissue components of the disease, including dactylitis, enthesitis, and skin involvement as measured by PASI75 [65]. For the treatment of PsA, ustekinumab is administered as a 45-mg subcutaneous injection at weeks 0 and 4, and then every 12 weeks. For patients with co-existent moderate to severe plaque Pso weighing more than 220 lbs (100 kg) the recommended dose is 90 mg subcutaneous injection at weeks 0 and 4, and then every 12 weeks [66]. On the other hand, briakinumab first showed its efficacy in one phase II and different phase III studies for the treatment of patients with moderate-to-severe chronic plaque Pso [67]. In a phase III, 12-week study, 347 patients were randomized in a 2:2:1 ratio to receive 200 mg briakinumab at weeks 0 and 4 followed by 100 mg briakinumab at week 8 (n = 138); 50 mg of etanercept twice weekly 3–4 days apart at weeks 0–11 (n = 141); or placebo injections matching active treatment (n = 68). Briakinumab showed an efficacy superior to both placebo and etanercept at 12 weeks. At the primary end point of week 12, the proportion of patients achieving a Physician’s Global Assessment (PGA) of 0/1 in the briakinumab group, etanercept group, and placebo group were 71, 39.7 and 2.9 %, respectively (p < 0,001). Furthermore, at 12 weeks the proportion of patients receiving a PASI75 response in the briakinumab group, etanercept group, and placebo group was 81.9, 56.0, and 7.4 %, respectively (p < 0,001). Serious adverse event rates were reported in four patients receiving briakinumab (2.9 %), one patient receiving etanercept (0.7 %), and one placebo-treated patient (1.5 %) [67], and, due to adverse cardiovascular effects, briakinumab was ultimately withdrawn [68].
IL17 targeting (secukinumab, ixekizumab, brodalumab)
Three main molecules target IL17 and are being studied for psoriatic disease (Table 4). Response rates with IL17 pathway inhibition vary and these different degrees of response suggest that IL17-mediated pathways play a role in both diseases, but to different, yet to be defined, extents. IL17 inhibitors include secukinumab, a highly selective, fully human IgG1 k mAb directed against the IL-17A cytokine; ixekizumab, a humanized IgG4 anti-IL-17A mAb; and brodalumab, a fully human IgG2 anti-IL-17RA mAb.
Secukinumab (AIN457) is an anti-IL-17A ligand monoclonal antibody approved by the FDA for the treatment of Pso, RA, chronic non-infectious uveitis, and CD. A phase I study in 36 patients with Pso demonstrated that a single dose of secukinumab at 3 mg/kg significantly reduced disease severity (mean PASI score) by 63 % at week 12 versus 9 % for placebo. Clinical responses were associated with reduction in epidermal hyperplasia, IL17-producing cells, and the gene expression of various cytokines and chemokines (e.g., IL17A, IL21, IL22, CCL20, KRT16, and DEFB4) [69]. Data from three phase II studies reported 12-week PASI75 response rates of 81 % in patients with Pso treated with subcutaneous secukinumab at 150 mg (three or four doses) and 83 % in patients treated with intravenous secukinumab [70]. The safety and efficacy of secukinumab in PsA were evaluated in a double-blind, placebo-controlled phase IIA study, in which 42 patients were randomized to treatment with two doses of subcutaneous secukinumab at 10 mg/kg, given 3 weeks apart, or placebo [35]. The ACR20 response rates at week 6 were the primary endpoint and were 39 % with secukinumab compared with 23 % for placebo, failing to reach a statistical significance. The ACR20 response at weeks 12 and 28 for secukinumab was observed in 39 and 43 %, and for placebo, 15 and 18 % of patients, respectively. FUTURE 1, a Phase III randomized, double-blind, placebo-controlled, multicenter study to assess the efficacy and safety of secukinumab in patients with active PsA who were intolerant to or had an inadequate response to anti-inflammatory drugs, DMARDs and/or TNFα inhibitor therapy is ongoing.
Ixekizumab (LY2439821) is a humanized monoclonal antibody directed at IL17A that is being investigated for the treatment of Pso and RA [4]. Recent data from a phase II study in Pso showed week 12 PASI75 response rates of 77, 83, and 82 % following administration of subcutaneous ixekinumab at 25, 75, and 150 mg (weeks 0, 2, 4, 8, and 12), respectively, versus 7.7 % for placebo. Importantly, up to 72 % of patients in the active-treatment groups achieved clear or almost clear Pso status according to Physician’s Global Assessment [71]. Ixekizumab is undergoing evaluation as a treatment for moderate-to-severe Pso also in three phase III studies (UNCOVER-1, -2 and -3) to be completed between February 2018 and April 2019. In detail, UNCOVER-1 is a multicenter, randomized, double-blind, placebo-controlled study characterized by a dosing period followed by a randomized maintenance dosing period and a long-term extension period, to evaluate the efficacy and safety of ixekizumab in patients with moderate-to-severe plaque Pso. UNCOVER-2 is a multicenter, randomized, double-blind, placebo-controlled study in which a comparison of efficacy and safety of ixekizumab is performed with etanercept and placebo in patients with moderate-to-severe plaque Pso [72]. UNCOVER-3 is a 12-week multicenter, randomized, double-blind, placebo-controlled study comparing the efficacy and safety of ixekizumab to etanercept and placebo in patients with moderate-to-severe plaque Pso with a long-term extension period [73]. On the other hand, SPIRIT-P1 is a phase III study of Ixekizumab that is currently enrolling patients with active PsA in a multicenter, randomized, double-blind, active, and placebo-controlled 24-week study followed by long-term evaluation of efficacy and safety of ixekizumab in biologic DMARD-naive patients with active PsA [74].
Finally, brodalumab (AMG 827) is an IL17RA fully human monoclonal antibody that is under investigation for a number of inflammatory conditions, including Pso, PsA, and asthma. A phase I proof-of-concept study in patients with Pso demonstrated significant improvement in the major clinical parameters with 7/8 and 5/8 patients achieving PASI75 by week 6 in the high (700 mg intravenous) and moderate (350 mg subcutaneous) single-dose groups. Improvement in histopathological parameters included reduction in epidermal thickness, Ki-67 [75], and keratin-16 gene expression, and infiltrating leukocyte subsets. In a phase II dose-ranging study, brodalumab (70, 140, or 210 mg at weeks 0, 1, and 2, then every other week or 280 mg monthly) produced dose-dependent improvement in PASI and Physician’s Global Assessment responses [76]. Significant mean PASI improvements were observed at week 12 in 45, 86, 86, and 76 % in the brodalumab 70-, 140-, 210-, and 280-mg groups, respectively, compared with 16 % in the placebo group. PASI75 responses were observed in 33, 77, 82, and 67 % of patients, respectively, in the brodalumab groups and 0 % in the placebo group. Brodalumab is currently undergoing evaluation in three Phase III studies, AMAGINE-1, -2, and -3. In particular, AMAGINE-1 evaluates the efficacy, safety, and effect of withdrawal and retreatment with brodalumab in subjects with moderate-to-severe plaque Pso, with a completion date estimated in March 2014 [77]. AMAGINE-2 and -3 are two Phase III studies to evaluate the efficacy and safety of induction and maintenance regimens of brodalumab compared with placebo and ustekinumab in subjects with moderate-to-severe plaque Pso [78, 79]. AMAGINE-2 is currently enrolling patients, while AMAGINE -3 is ongoing and the estimated completion date is September 2014.
Final remarks and future directions
Recent findings strongly support the view that the Th17 pathway plays a predominant role in Pso and PsA pathogenesis, similar to multiple sclerosis, and animal models should contribute to unravel new details on this promising field of research and potential therapeutic implications. New therapies targeting the pivotal IL23 and the effector IL17 have been developed with promising results. In particular, successful data have been observed by blocking IL23 and IL12 with ustekinumab, later approved for treatment of Pso and most recently PsA, and by blocking IL17 with monoclonal antibodies in phase III trials. Overcoming TNFα as a unique target for Pso and PsA and identifying new pathways to tackle chronic inflammation are mandatory priorities to reduce the risk of partial or temporary response. Furthermore, the development of safer and more effective drugs has opened the way to more selective interventions that arrest the disease progression by blocking the basis of the pathological mechanism of the psoriatic disease. Of note, comorbidities are also expected to mitigate from the new treatments and this advantage should be considered in the approval process of new molecules. However, the pharmacological research into the Th17 pathway maintains numerous promising developments and the axis is a solid horizon for future solutions. In this scenario, possibly more than in any other field of biomedical research, a conjunct effort of clinicians and basic scientists is a necessary step to achieve significant results. Finally, additional and sometimes unsuspected factors, such as dietary salt intake or weight changes, should be taken into account for future trials as determinants of clinical response.
Conflict of interest
Beatrice Marinoni, Angela Ceribelli, Marco S. Massarotti and Carlo Selmi declare they have no conflict of interest.
Human and animal rights
No animal or human studies were carried out by the authors for this article.
Informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (2005). Informed consent was obtained from all patients for being included in the study.
References
- 1.Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64 Suppl 2:ii14–ii17. doi: 10.1136/ard.2004.032482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64 Suppl 2:ii18–ii23. doi: 10.1136/ard.2004.033217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottlieb A, Menter A, Mendelsohn A, Shen YK, Li S, Guzzo C, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet. 2009;373:633–640. doi: 10.1016/S0140-6736(09)60140-9. [DOI] [PubMed] [Google Scholar]
- 4.Raychaudhuri SP, Raychaudhuri SK, Genovese MC. IL-17 receptor and its functional significance in psoriatic arthritis. Mol Cell Biochem. 2012;359:419–429. doi: 10.1007/s11010-011-1036-6. [DOI] [PubMed] [Google Scholar]
- 5.Gottlieb AB, Dann F. Comorbidities in patients with psoriasis. Am J Med. 2009;122(1150):e1–e9. doi: 10.1016/j.amjmed.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Sommer DM, Jenisch S, Suchan M, Christophers E, Weichenthal M. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch Dermatol Res. 2006;298:321–328. doi: 10.1007/s00403-006-0703-z. [DOI] [PubMed] [Google Scholar]
- 7.Horreau C, Pouplard C, Brenaut E, Barnetche T, Misery L, Cribier B, et al. Cardiovascular morbidity and mortality in psoriasis and psoriatic arthritis: a systematic literature review. J Eur Acad Dermatol Venereol. 2013;27(Suppl 3):12–29. doi: 10.1111/jdv.12163. [DOI] [PubMed] [Google Scholar]
- 8.Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA. 2006;296:1735–1741. doi: 10.1001/jama.296.14.1735. [DOI] [PubMed] [Google Scholar]
- 9.Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB, Gelfand JM. Prevalence of cardiovascular risk factors in patients with psoriasis. J Am Acad Dermatol. 2006;55:829–835. doi: 10.1016/j.jaad.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 10.Ramonda R, Lo Nigro A, Modesti V, Nalotto L, Musacchio E, Iaccarino L, et al. Atherosclerosis in psoriatic arthritis. Autoimmun Rev. 2011;10:773–778. doi: 10.1016/j.autrev.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Johnston A, Arnadottir S, Gudjonsson JE, Aphale A, Sigmarsdottir AA, Gunnarsson SI, et al. Obesity in psoriasis: leptin and resistin as mediators of cutaneous inflammation. Br J Dermatol. 2008;159:342–350. doi: 10.1111/j.1365-2133.2008.08655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gisondi P, Targher G, Zoppini G, Girolomoni G. Non-alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol. 2009;51:758–764. doi: 10.1016/j.jhep.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 13.van den Berg R, van Gaalen F, van der Helm-van Mil A, Huizinga T, van der Heijde D. Performance of classification criteria for peripheral spondyloarthritis and psoriatic arthritis in the Leiden Early Arthritis cohort. Ann Rheum Dis. 2012;71:1366–1369. doi: 10.1136/annrheumdis-2011-201081. [DOI] [PubMed] [Google Scholar]
- 14.Helliwell PS, Mease PJ, FitzGerald O, Taylor WJ, van der Heijde D. Peripheral spondyloarthritis and psoriatic arthritis; overlaps and distinctions: a report from the GRAPPA 2012 annual meeting. J Rheumatol. 2013;40:1446–1449. doi: 10.3899/jrheum.130460. [DOI] [PubMed] [Google Scholar]
- 15.Alamanos Y, Voulgari PV, Drosos AA. Incidence and prevalence of psoriatic arthritis: a systematic review. J Rheumatol. 2008;35:1354–1358. [PubMed] [Google Scholar]
- 16.Ceponis A, Kavanaugh A. Treatment of psoriatic arthritis with biological agents. Semin Cutan Med Surg. 2010;29(1):56–62. doi: 10.1016/j.sder.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Gelfand J, Gladman D, Mease P, et al. Epidemiology of psoriatic arthritis in the US population. J Am Acad Dermatol. 2005;53:573. doi: 10.1016/j.jaad.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 18.Scarpa R, Del Puente A, di Girolamo C, della Valle G, Lubrano E, Oriente P. Interplay between environmental factors, articular involvement, and HLA-B27 in patients with psoriatic arthritis. Ann Rheum Dis. 1992;51:78–79. doi: 10.1136/ard.51.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130:1373–1383. doi: 10.1038/jid.2009.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339–1350. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- 21.Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. 2005;202:135–143. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prinz JC. Psoriasis vulgaris—a sterile antibacterial skin reaction mediated by cross-reactive T cells? An immunological view of the pathophysiology of psoriasis. Clin Exp Dermatol. 2001;26:326–332. doi: 10.1046/j.1365-2230.2001.00831.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125–130. doi: 10.1084/jem.20030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaba LC, Krueger JG, Lowes MA. Resident and “inflammatory” dendritic cells in human skin. J Invest Dermatol. 2009;129:302–308. doi: 10.1038/jid.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori G, Cantatore FP, Brunetti G, Oranger A, Colaianni G, Quarta L, et al. Synovial fluid fibroblasts and lymphocytes support the osteoclastogenesis in human psoriatic arthritis. Ann N Y Acad Sci. 2007;1117:159–164. doi: 10.1196/annals.1402.013. [DOI] [PubMed] [Google Scholar]
- 26.Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003;111:821–831. doi: 10.1172/JCI200316069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 28.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Investig. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 30.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeilding N, Szapary P, Brodmerkel C, Benson J, Plotnick M, Zhou H, et al. Development of the IL-12/23 antagonist ustekinumab in psoriasis: past, present, and future perspectives. Ann NY Acad Sci. 2011;1222:30–39. doi: 10.1111/j.1749-6632.2011.05963.x. [DOI] [PubMed] [Google Scholar]
- 32.Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. J Leukoc Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- 33.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 34.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 35.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraan MC, van Kuijk AW, Dinant HJ, Goedkoop AY, Smeets TJ, de Rie MA, et al. Alefacept treatment in psoriatic arthritis: reduction of the effector T cell population in peripheral blood and synovial tissue is associated with improvement of clinical signs of arthritis. Arthr Rheum. 2002;46:2776–2784. doi: 10.1002/art.10543. [DOI] [PubMed] [Google Scholar]
- 37.Colombo MD, Cassano N, Bellia G, Vena GA. Cyclosporine regimens in plaque psoriasis: an overview with special emphasis on dose, duration, and old and new treatment approaches. Sci World J. 2013;2013:805705. doi: 10.1155/2013/805705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mease P, Genovese MC, Gladstein G, Kivitz AJ, Ritchlin C, Tak PP, et al. Abatacept in the treatment of patients with psoriatic arthritis: results of a six-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. Arthr Rheum. 2011;63:939–948. doi: 10.1002/art.30176. [DOI] [PubMed] [Google Scholar]
- 39.Yoo IS, Lee JH, Song ST, Kim JH, Lee HJ, Kang SW. T-helper 17 cells: the driving force of psoriasis and psoriatic arthritis. Int J Rheum Dis. 2012;15:531–537. doi: 10.1111/j.1756-185X.2012.01813.x. [DOI] [PubMed] [Google Scholar]
- 40.Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suarez-Farinas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159:1092–1102. doi: 10.1111/j.1365-2133.2008.08769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duffin KC, Chandran V, Gladman DD, Krueger GG, Elder JT, Rahman P. Genetics of psoriasis and psoriatic arthritis: update and future direction. J Rheumatol. 2008;35:1449–1453. [PMC free article] [PubMed] [Google Scholar]
- 42.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/S1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 44.Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol. 2006;176:1908–1915. doi: 10.4049/jimmunol.176.3.1908. [DOI] [PubMed] [Google Scholar]
- 45.Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2577–2587. doi: 10.1084/jem.20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maeda S, Hayami Y, Naniwa T, Ueda R. The Th17/IL-23 axis and natural immunity in psoriatic arthritis. Int J Rheumatol. 2012;2012:539683. doi: 10.1155/2012/539683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yago T, Nanke Y, Kawamoto M, Furuya T, Kobashigawa T, Kamatani N, et al. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthr Res Ther. 2007;9:R96. doi: 10.1186/ar2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 49.Ogilvie AL, Luftl M, Antoni C, Schuler G, Kalden JR, Lorenz HM. Leukocyte infiltration and mRNA expression of IL-20, IL-8 and TNF-R P60 in psoriatic skin is driven by TNF-alpha. Int J Immunopathol Pharmacol. 2006;19:271–278. doi: 10.1177/039463200601900204. [DOI] [PubMed] [Google Scholar]
- 50.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huppler AR, Bishu S, Gaffen SL. Mucocutaneous candidiasis: the IL-17 pathway and implications for targeted immunotherapy. Arthr Res Ther. 2012;14:217. doi: 10.1186/ar3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol. 2010;185:5453–5462. doi: 10.4049/jimmunol.1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caproni M, Antiga E, Melani L, Volpi W, Del Bianco E, Fabbri P. Serum levels of IL-17 and IL-22 are reduced by etanercept, but not by acitretin, in patients with psoriasis: a randomized-controlled trial. J Clin Immunol. 2009;29:210–214. doi: 10.1007/s10875-008-9233-0. [DOI] [PubMed] [Google Scholar]
- 55.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207–1211. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 56.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 58.Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthr Rheum. 2008;58:2307–2317. doi: 10.1002/art.23655. [DOI] [PubMed] [Google Scholar]
- 59.Noordenbos T, Yeremenko N, Gofita I, van de Sande M, Tak PP, Canete JD, et al. Interleukin-17-positive mast cells contribute to synovial inflammation in spondylarthritis. Arthr Rheum. 2012;64:99–109. doi: 10.1002/art.33396. [DOI] [PubMed] [Google Scholar]
- 60.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Investig. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. 2010;362:118–128. doi: 10.1056/NEJMoa0810652. [DOI] [PubMed] [Google Scholar]
- 62.Reich K, Langley RG, Lebwohl M, Szapary P, Guzzo C, Yeilding N, et al. Cardiovascular safety of ustekinumab in patients with moderate to severe psoriasis: results of integrated analyses of data from phase II and III clinical studies. Br J Dermatol. 2011;164:862–872. doi: 10.1111/j.1365-2133.2011.10257.x. [DOI] [PubMed] [Google Scholar]
- 63.Agency EM. Stelara European public assessment report
- 64.Inc. COB. STELARA (ustekinumab). Prescribing Information
- 65.McInnes IB, Kavanaugh A, Gottlieb AB, Puig L, Rahman P, Ritchlin C, Brodmerkel C, Li S, Wang Y, Mendelsohn AM, Doyle MK, PSUMMIT 1 Study Group Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet. 2013;382(9894):780–789. doi: 10.1016/S0140-6736(13)60594-2. [DOI] [PubMed] [Google Scholar]
- 66.Gottlieb A, Narang K. Ustekinumab in the treatment of psoriatic arthritis: latest findings and clinical potential. Ther Adv Musculoskelet Dis. 2013;5:277–285. doi: 10.1177/1759720X13501021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gottlieb AB, Leonardi C, Kerdel F, Mehlis S, Olds M, Williams DA. Efficacy and safety of briakinumab vs. etanercept and placebo in patients with moderate to severe chronic plaque psoriasis. Br J Dermatol. 2011;165:652–660. doi: 10.1111/j.1365-2133.2011.10418.x. [DOI] [PubMed] [Google Scholar]
- 68.Traczewski P, Rudnicka L. Briakinumab for the treatment of plaque psoriasis. BioDrugs. 2012;26:9–20. doi: 10.2165/11595940-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 69.Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2:52ra72. doi: 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
- 70.Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthr Rheum. 2010;62:929–939. doi: 10.1002/art.27334. [DOI] [PubMed] [Google Scholar]
- 71.Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190–1199. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- 72.ClinicalTrials.gov. UNCOVER-2: Identifier:NCT01597245
- 73.ClinicalTrials.gov. UNCOVER-3: NCT01646177
- 74.ClinicalTrials.gov. SPIRIT-P1: Identifier:NCT01695239
- 75.Gerdes J, Schwab U, Lemke H, Stein H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer. 1983;31:13–20. doi: 10.1002/ijc.2910310104. [DOI] [PubMed] [Google Scholar]
- 76.Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG, Kricorian G, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366:1181–1189. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 77.ClinicalTrials.gov. AMAGINE-1: NCT01708590
- 78.ClinicalTrials.gov. AMAGINE-2: NCT01708603
- 79.ClinicalTrials.gov. AMAGINE-3: NCT01708629
- 80.van Kuijk AW, Reinders-Blankert P, Smeets TJ, Dijkmans BA, Tak PP. Detailed analysis of the cell infiltrate and the expression of mediators of synovial inflammation and joint destruction in the synovium of patients with psoriatic arthritis: implications for treatment. Ann Rheum Dis. 2006;65:1551–1557. doi: 10.1136/ard.2005.050963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han G, Williams CA, Salter K, Garl PJ, Li AG, Wang XJ. A role for TGFbeta signaling in the pathogenesis of psoriasis. J Invest Dermatol. 2010;130:371–377. doi: 10.1038/jid.2009.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamamoto T. Angiogenic and inflammatory properties of psoriatic arthritis. ISRN Dermatol. 2013;2013:630620. doi: 10.1155/2013/630620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Partsch G, Wagner E, Leeb BF, Broll H, Dunky A, Smolen JS. T cell derived cytokines in psoriatic arthritis synovial fluids. Ann Rheum Dis. 1998;57:691–693. doi: 10.1136/ard.57.11.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nair RP, Ruether A, Stuart PE, Jenisch S, Tejasvi T, Hiremagalore R, et al. Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. J Invest Dermatol. 2008;128:1653–1661. doi: 10.1038/sj.jid.5701255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fisher SA, Tremelling M, Anderson CA, Gwilliam R, Bumpstead S, Prescott NJ, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet. 2008;40:710–712. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patsopoulos NA, Esposito F, Reischl J, Lehr S, Bauer D, Heubach J, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Filer C, Ho P, Smith RL, Griffiths C, Young HS, Worthington J, et al. Investigation of association of the IL12B and IL23R genes with psoriatic arthritis. Arthr Rheum. 2008;58:3705–3709. doi: 10.1002/art.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ban M, Goris A, Lorentzen AR, Baker A, Mihalova T, Ingram G, et al. Replication analysis identifies TYK2 as a multiple sclerosis susceptibility factor. Eur J Hum Genet. 2009;17:1309–1313. doi: 10.1038/ejhg.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ellinghaus E, Ellinghaus D, Stuart PE, Nair RP, Debrus S, Raelson JV, et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gregersen PK, Amos CI, Lee AT, Lu Y, Remmers EF, Kastner DL, et al. REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet. 2009;41:820–823. doi: 10.1038/ng.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trynka G, Zhernakova A, Romanos J, Franke L, Hunt KA, Turner G, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut. 2009;58:1078–1083. doi: 10.1136/gut.2008.169052. [DOI] [PubMed] [Google Scholar]
- 96.Ellinghaus E, Stuart PE, Ellinghaus D, Nair RP, Debrus S, Raelson JV, et al. Genome-wide meta-analysis of psoriatic arthritis identifies susceptibility locus at REL. J Invest Dermatol. 2012;132:1133–1140. doi: 10.1038/jid.2011.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thomson W, Barton A, Ke X, Eyre S, Hinks A, Bowes J, et al. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Allanore Y, Saad M, Dieude P, Avouac J, Distler JH, Amouyel P, et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011;7:e1002091. doi: 10.1371/journal.pgen.1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, Rodijk-Olthuis D, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pappu R, Ramirez-Carrozzi V, Ota N, Ouyang W, Hu Y. The IL-17 family cytokines in immunity and disease. J Clin Immunol. 2010;30:185–195. doi: 10.1007/s10875-010-9369-6. [DOI] [PubMed] [Google Scholar]
- 104.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/S1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]