

Multiple technologies and knowledge bases including human genome sequence (1), microarrays (2), and human HapMaps (3) became available beginning in the late-1990s and they represented dramatic scientific and technological advances that could provide a wonderful opportunity to systematically explore how the genome and proteome of the human body respond to serious, potentially lethal injuries. Many investigators in trauma research recognized that top scientists in other medical fields, such as cancer and diabetes, would utilize these technological advances in their fields of interest. There was also a consensus among the trauma investigators that one tremendous obstacle to further advances in the field of injury research was an insufficient systematic understanding of the human response to potentially lethal injuries. It just felt right that injury research also should have this very important opportunity to advance our field.
Why Study Injury Genomics and Proteomics
If you think about it, the majority of NIH funding goes to study human diseases that are not only chronic in nature but even more important, the initiating features are not known and the disease is never cured, only treated. A few examples of these diseases include rheumatoid arthritis, cancer, and atherosclerosis.
Trauma, as a disease, is very different in that our patients are often young, previously healthy, and the disease's beginning is known precisely. Furthermore, it is generally believed that the genome and proteome rapidly change dramatically very soon after injury but it is also thought the genome and proteome return to a more normal pattern after some time interval. These temporal characteristics are shared with another disease state, serious and potentially lethal infection. If these new genomic technologies were used to study the human genome in injured patients who had a high risk of death, then this new knowledge also might be applicable to other disease processes like serious, potentially lethal infection. Possibly by understanding the genomic effects of these stresses (serious injury and infection), new predictive tools and drug targets may be found to mitigate and improve upon the natural human responses to injury and by extension to serious infection.
The Clinical Paradigm
When the Glue Grant program began 15 years ago, the generally accepted clinical paradigm (Figure 1) described an initial phase of multiple organ dysfunction, which is associated with a Systemic Inflammatory Response Syndrome (SIRS) followed by a compensatory response, Compensatory Anti-inflammatory Response Syndrome (CARS), in which phenotypic markers of immunosuppression can be easily demonstrated (4). If for example, devitalized tissues remained or infections occurred, then the second phase of multiple organ dysfunction, CARS had an even higher rate of mortality. The Glue Grant investigators were interested to systematically explore the molecular basis for these phenotypes, in blood, which is the only tissue that can be routinely biopsied. The data that we will explore today comes from the circulating white blood cells (WBCs) and therefore as can be anticipated, the findings are dominated by the host's immune response (5). Genomic data from skin, fat and muscle are included in the Glue Grant datasets but time precludes presentation of these findings.
Figure 1.
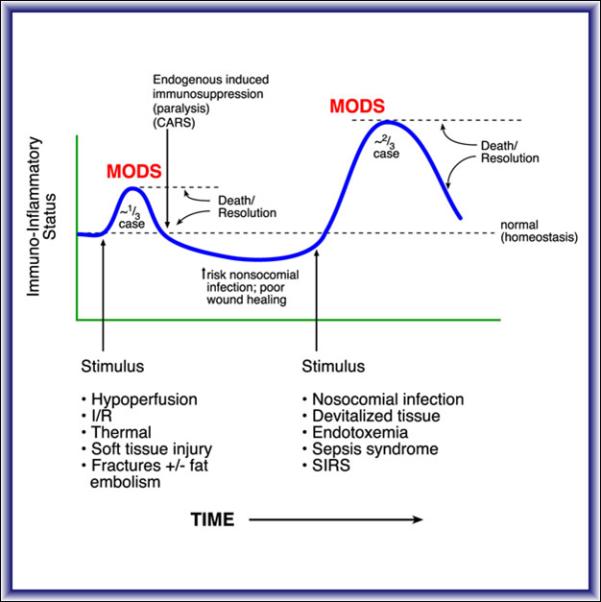
Clinical paradigm for Multiple Organ Dysfunction (MOD). Patients initially present with SIRS during resuscitation and an early MOD syndrome with the potential for an early death. A compensatory period of CARS follows for those patients who survive the early inflammatory insult, but they are subject to a later “second hit” from a nosocomial infection, endotoxemia, or the persistence of devitalized tissue. A late, more severe MOD syndrome can develop with a higher risk of death. Substantial contributions to illustrate this concept came from Ron Maier and Linc Moldawer.
New Technologies
The Glue Grant datasets include gene expression data from microarrays as well as large-scale proteomics from high throughput mass spectroscopy (6-12) as well as functional assays from multiplexed Fluorescence Activated Cell Sorting (FACS) (13-14). In these datasets, it becomes clear that technologies for genomics are far more advanced than for proteomics with one reason being genomics is a 4-letter alphabet (four different nucleic acids for both RNA and DNA), whereas, proteomics is a 20-letter alphabet (20 different amino acids in human tissues). Unfortunately, many of the complex pathophysiological features, for which we seek an understanding, lie in the complexity of proteomics for which multiplexed technology is not currently available.
During the conduct of the study, many new technologies were developed by the Glue Grant investigators including microfluidics for cellular separations (neutrophils, T-cells, and monocytes) (15-24), RNA isolation in critical care patients (25-26), and algorithms in computational biology (27-35) that were necessary for use in critically ill patients. Notwithstanding the technological advances in bioinformatics and computational biology expertly led by Wenzhong Xiao, the extraction of biological knowledge from the petabytes of data generated by the Glue Grant program continues to be a challenge.
Although there may be 20,000 or more human genes, each does not necessarily operate independently, but these molecules operate in small groupings or collections known as modules. Furthermore, within these modules, molecules tend to behave in concert implying that if one knows how a member of the module behaves, then the behavior of the other molecules can be predicted. The advantage of this concept is that the numbers problem can be reduced from understanding the random response of more than 20,000 molecules to a smaller, more manageable number of molecular responses (e.g. a few thousand). Taken together, physiology or pathophysiology that is important to clinicians and scientists is created (e.g., secretion and phagocytosis). Perhaps based upon a better understanding of the genomics, we might learn that these later processes are driven by only a handful of genes. If this turns out to be true, then a very limited number of coordinated interventions could ultimately control these pathophysiologies, thereby creating a human response to injury that is more optimal for survival and recovery.
The Glue Grant Clinical Program
The Glue Grant program was funded for ten years and it remained active with an additional two-year no-cost extension. It was a program with 22 U.S. academic medical centers that truly represented a nationwide experience. With respect to the trauma clinical sites, the vast majority of the patients came from four Level 1 trauma centers: Harborview Medical Center-University of Washington (Ronald Maier and Joseph Cuschieri), Denver Health Medical Center-University of Colorado (Ernest Moore and Jeffrey Johnson), Parkland Memorial Hospital-University of Texas Southwestern Medical Center (Joseph Minei), and Presbyterian University Hospital-U Pittsburgh (Tim Billiar and Jason Sperry). A great deal of the success of the trauma component of the Glue Grant program is owed to Ron Maier, who was the principal investigator for trauma. David Herndon (UTMB and Shriners Hospital-Galveston), who was the principal investigator for burns, led the burn centers that also included Parkland Memorial Hospital-University of Texas Southwestern Medical Center (Brett Arnoldo), Loyola University Medical Center (Richard Gamelli), and Harborview Medical Center-University of Washington (Nicole Gibran and Matthew Klein).
The clinical databases for the Glue Grant program described at www.gluegrant.org/ and www.igenomed.org/mgh are extensive and include data from more than 2,800 trauma and burn patients as well as normal human volunteers with more than 1,200 distinct data fields. This is a very large database that includes not only data from patients and normal human volunteers, but also 76,000 tissue samples and more than 5,000 microarrays organized and archived by principal investigator Lyle (Linc) Moldawer. The first half of the program used what was state-of-the-art at the time microarrays (Affymetrix U133 Plus 2) but during the second half of the program and lead by Wenzhong Xiao and Michael Mindrinos, the program created new microarrays in collaboration with Affymetrix (36). These are currently marketed as GeneChip® Human Transcriptome Array 2.0 (HTA 2.0) and GeneChip® Mouse Transcriptome Assay 1.0 (MTA 1.0) (www.affymetrix.com). As originally designed, these arrays contain >6.9 million features that encode gene exons, exon-exon junction, coding single nucleotide polymorphisms (SNPs) and drug metabolism enzymes and drug transporters (DMET) variations, noncoding functional RNA (f-ncRNA), non-coding antisense expression (as-ncRNA), unannotated transcribed units (UTUs). HTA 2.0 is an array design that covers both coding transcripts and non-coding transcripts. Seventy percent of the probes cover exons for coding transcripts, and the remaining 30% of probes on the array cover exon-exon splice junctions and non-coding transcripts. Although not commercially available yet, the human array also has been annotated for macaque monkeys, which makes these new microarrays useful for genomic studies in man, mouse, and monkey.
In addition to the new arrays, many computational tools were created by the investigators within the program because computational tools at that time were inadequate to accomplish the goals of the Glue Grant. Examples of the new bioinformatics tools include the development of knowledge-based network analysis in collaboration with Ingenuity, Inc. (27), (Extraction of Differential Gene Expression (EDGE) (28,37,38), Surrogate Variable Analysis (SVA) (39), Time Course Analysis of Variance (TANOVA) (32), and Junction and Exon Toolkits for Transcriptome Analysis (JETTA) (40,41), and Significance Analysis for Large-scale Proteomics Studies (SALPS) (42). Together with the HTA described above, these bioinformatics tools have been excellent additions to the field with thousands of downloads significantly contributing to human systems biology.
The First Clinical Study
Of the 1,637 trauma patients over age 16 recruited into the study, one out of ten (167 patients) underwent blood sampling for gene expression using the U133 Plus 2 microarrays. WBC gene expression was determined within 12 hours and 1, 4, 7, 14, and 28 days after their serious blunt trauma. All trauma patients had evidence of shock secondary to hemorrhage demonstrated by a base deficit, a reliable recorded blood pressure <90 mm Hg, and blood given within the first six hours of admission (5,43,44). The patients were treated using ten standard operating procedures (SOPs) that were developed by consensus prior to enrollment and these included SOPs for nutrition, resuscitation, and pulmonary management, among others all of which were published in the Journal of Trauma and Acute Care Surgery (45-55). Consensus SOPs for the treatment of patients with burn injury were also published (56). Even in these American College of Surgeons-verified Level 1 trauma centers, compliance with these SOPs for trauma patients was 10–40% when evaluated by external audit (43,44). However, compared to the general literature for compliance, these compliance rates should be considered to be excellent.
If we intended to recruit comparable patients today, one important change in the entry criteria must be considered. Today, clinicians tolerate much lower hemoglobin concentrations than a decade ago and therefore, the requirement for a blood transfusion within six hours should be extended to 24 hours or the requirement omitted altogether.
In the Glue Grant program using these inclusion criteria together with the SOPs, the patients had an average Injury Severity Score (ISS) of 32; an average incidence of multiple organ failure (MOF) of 35%; an average length of stay (LOS) of 14 days; and nearly ten days of mechanical ventilation (5,43,44). The majority of patients were discharged within two weeks, which is a measure of the high quality of care provided in these Level 1 trauma centers to these patients despite the serious extent of their injuries. The average mortality over the study was 16%, but this annual mortality rate declined from 22% to 11% over the last five years (44).
In the 1,637 trauma patients, the MOF scores decreased over time after injury as shown in Figure 2 (upper panel) (44). The clinical determination of MOF in the first 48 hours is nearly impossible to make for many reasons. These patients are typically either in surgery or otherwise heavily sedated receiving mechanical ventilation in the intensive care unit. Purple represents no MOF; red indicates MOF; and black represents patient deaths. The patients in the upper portion of the figure did not demonstrate MOF. In the mid-portions of the figure, patients exhibited MOF that resolved by five to seven days post-injury and this improvement allowed their hospital discharge relatively early. A smaller portion of patients exhibited relatively severe MOF for two or three weeks but improved enough for discharge between 14 – 28 days of admission. In the lower portion of the figure, a substantial number of patients demonstrated MOF that had not resolved by 28 days and out of the 1,637 patients, the remaining ones died at some time between 48 hours and 28 days after injury.
Figure 2.


Time to recovery and infections associated with severe blunt trauma. (Upper panel) Shown here is time to recovery (TTR) from multiple organ failure (MOF). Heat map of the MOF scores over time after injury up to 28 days for each of the 1,637 massive injured patients. From top to bottom, patients are ordered according to the TTR days. Source: Cuschieri et al.44 © 2012 Lippincott Williams & Wilkins. Reproduced with permission from Wolters Kluwer Health. (Lower panel) Day of onset and frequency of multiple organ failure (MOF), nosocomial infection (NI), and death. Source: Minei et al.43 © 2012 Lippincott Williams & Wilkins. Reproduced with permission from Wolters Kluwer Health.
The frequency for MOF, nosocomial infections, and death after admission are shown in Figure 2 (lower panel) (43). Patients who died within the first two days, which included the vast majority of deaths, were not included in the analysis. Of those alive after two days, subsequent deaths occurred at a very low and declining rate. The MOF frequency was greatest when it was first feasible to determine MOF and its frequency declined precipitously over the next few days. As shown in the panel, MOF did not show a secondary spike in frequency or occurrence. Nosocomial infections began to appear after two days, but the frequency maximum was five to ten days post-injury potentially associated with CARS. To summarize, these 1,637 patients did not show a delayed increase in MOF rates or deaths over the 28 days (Figure 2).
Mortality rates (relative to APACHE, TRISS, and ISS) were compared to the only national, publically-available dataset for trauma, the National Trauma Data Bank (NTDB) in Figure 3 (44). The survival rates for the Glue Grant patients (solid lines) remained higher even when corrected for APACHE, TRISS, or ISS compared to patients in the NTDB database (dashed lines). These improved survival rates were attributed to the SOPs together with the associated external monitoring and audits (44).
Figure 3.




Mortality. Patients divided into quintiles in Panels B, C, and D based underlying score or injury severity. Panel A shows mortality over the entire study period. Observed (solid lines) versus expected (dashed line) outcome for Panel B) mortality by TRISS (P < 0.001), Panel C) mortality by APACHE II (P < 0.001), and Panel D) mortality by NTDB (P < 0.001). Source: Cuschieri et al.44 © 2012 Lippincott Williams & Wilkins. Reproduced with permission from Wolters Kluwer Health.
The Genomic Response after Massive Blunt Trauma
In the circulating white cells in those 167 patients studied when compared to those of the 35 normal volunteers, more than 80% of the WBC genes changed significantly during the 28 days post-injury (5). The term, “genomic storm” has been applied to this dramatic reorganization of the human genome after massive injury. There have been no reports of any other in vivo or in vitro model systems or disease conditions in which a comparable massive genomic effect has been described (5).
Analysis of >80% of the genome simultaneously is not feasible. Using a false discovery rate (FDR) adjusted probability of <0.001 and a 1.5-fold difference, 10,001 genes changed significantly. Restricting the analysis only to those genes changing more than two-fold FDR of <0.001), 5,136 genes are under consideration. Each of the 5,136 genes is shown in Figure 4B over time after injury with those same genes from the normal volunteers shown to the far left of the heat map (5).
Figure 4.

Organ injury and genomic changes associated with severe blunt trauma. (A) The presence and severity of organ injury is represented by colors from blue (least severe) to red (most severe). Black indicates death. (B) K-means clustering of the genes into 30 clusters based on patterns of expression over time. Red indicates increased and blue indicates decreased expression relative to the mean (white). 5,136 genes were differentially expressed between patients and controls (ctrl; FDR <0.001 and at least twofold change). (C and D) Summary of the canonical pathways most affected by trauma. The graph shows the −log10 (p value) of the enrichment of the pathway. © 2011 Xiao et al.5 Originally published in Journal of Experimental Medicine.2011 Nov;208(13):2581-2590. doi: 10.1084/jem.20111354.
The genes are clustered into 30 groups by similarity to temporal patterns (Figure 4B) (5). In cluster 1 (uppermost cluster), the genes are not normally expressed as shown in the normal volunteers but they are shown to be up-regulated as early as 12 hours post-injury. This cluster contains many of those genes that are familiar to us all including IL-1β and TNFα. The genes in cluster 1 are also seen to recover to the pre-injury levels of expression within a day or so; other clusters take longer (days or weeks) before the genes begin to recover to their pre-injury expression levels. IL-10 would be an example of this latter category of genes. Some genes remain highly expressed even 28 days post-injury. As shown in the lower half of Figure 4B, many genes that are highly expressed in the normal volunteers are very significantly down-regulated post-injury. In fact, there are more genes down-regulated relative to normal volunteers than those up-regulated. The vast majority of the expression changes (both down and up-regulation) occur within the first 24 hours post-injury with the exception of only a relatively small proportion of the 5,136 genes with a delayed maximum change in regulation occurring days post-injury. They are an interesting subset of genes near the middle of the heat map, many of which control immunoglobulin responses for example (5).
The ten most significantly regulated pathways (p<10−16) are shown in Figure 4C and D (5). Of the most up-regulated pathways in the circulating WBCs, eight out of the ten are arguably components of the innate immune system, which is consistent with the massive innate immune SIRS response seen after injury. Of those most down-regulated pathways, nine out of the ten are included in the adaptive immune system (5). Many of these down-regulated pathways involve antigen processing. This latter finding might be interesting if one considers that 40% of the new peptides and proteins that appear in the plasma after these injuries are thought to be exclusively intracellular molecules (57).
In the case of patients with complicated clinical trajectories (to be described later), there is likely a persistence of cellular necrosis, apoptosis, and autophagy in many damaged and other tissues after trauma. This cellular debris likely enters the circulation as many unprocessed, self-antigens. There are multiple publications that describe at least a few of these as examples of normally intracellular molecules that are found in the plasma of injured patients (58,59). To have competent antigen-processing pathways after serious injury might not be beneficial, if it only leads to a delayed onset of autoimmune diseases weeks post-injury. It might turn out that restricting or limiting the antigen processing potential of so many self-antigens is a survival benefit.
How Well Do These Genomics of Trauma Mimic Those of Burn Injury?
In the Glue Grant Program, we have three human conditions in which to compare genomic responses: major blunt trauma, burns ≥20% Total Body Surface Area, and endotoxemia in healthy volunteers. The endotoxin studies, performed at UMDNJ by Steve Lowry and Steve Calvano in the first few years of the program (28), were extremely enlightening and guided us to optimize our subsequent genomic studies in patients with trauma and burn injuries. Comparing those genes that change two-fold between the WBCs after burn injury to those after blunt trauma, Pearson's correlation coefficients (R2) is 0.91 (5). Pearson's correlation coefficients can be interpreted to indicate that 91% of the variation in gene expression can be explained by injury mechanism alone (FIGURE 5 upper left) (5,60). The rank sum correlations were also published in the supplemental material and they showed the same pattern although rank sum correlations do not allow quantitative interpretation (60). Additionally, the single dose of endotoxin, which generates an inflammatory reaction mimicking a flu-like syndrome in an otherwise healthy college student, results in an R2 of 0.45. This suggests that the transient stimulation by a single low dose of endotoxin mimics change in 45% of the genes that are seen to respond in very serious injury.
Figure 5.

Correlations of the gene changes among human burns, trauma, and endotoxin and the corresponding mouse models. Scatter plots and Pearson correlations (R2) of the log twofold changes of 4,918 human genes responsive to trauma, burns, or endotoxemia (FDR < 0.001; fold change ≥ 2) and their murine orthologs in the murine models. As shown in the upper left, the genomic responses to human trauma and burns are highly correlated (R2 = 0.91). In contrast, as shown in the lower right, the murine models correlate poorly with each other (R2 = 0.00–0.13) and almost randomly with the corresponding human conditions (R2 = 0.00–0.09). Similar results were seen with rank correlation. © 2013 Seok et al.60 Originally published in Proceedings of the National Academy of Sciences of the United States of America. 2013 Feb;110(9):3507-12. doi: 10.1073/pnas.1222878110.
To be more specific comparing burns with trauma, there were 2,066 genes that are up-regulated and 3,042 that were down-regulated. There were only 28 genes (out of the 5,036 genes) that behaved differently between burns and trauma (5). This level of reproducibility in the human genomic response between burns and trauma is very surprising given that they are two conditions with very different time scales. Although a few trauma patients may still be affected many months after their injuries, many or most burn patients have lingering effects even a year or more. Both burns and trauma are severe stresses that create a very reproducible genomic response. Perhaps, other severe stresses (sepsis and major infections) also might demonstrate comparable genomic responses.
The Toll-like Receptor Responses
As examples, the genomic responses for TLR receptors can be compared among the three human conditions of trauma, burns, and endotoxemia (5). These receptors are molecular pattern receptors, which recognize and respond to chemical patterns that may be either intrinsic (e.g. self-antigens) or extrinsic (e.g. infectious agents). These receptors are an important component of the innate immune system that is immediately available but relatively non-specific in nature. Persistent up-regulation of many Toll-like receptors (TLR1, TLR2, TLR4, TLR5, TLR8, TLR 9, and TLR10) in all three conditions is consistent with the SIRS phenotype that characterizes many of our patients. TLR3, which recognizes double stranded RNA (ds-RNA), and TLR7, which recognizes single stranded (ss-RNA) in endosomes, are both down-regulated. These are remarkable similarities of TLR responses among the three human conditions especially when the comparable genomic responses among murine models of trauma, burns, and endotoxemia not only fail to mimic one another, but also fail to mimic those same responses in patients with injuries.
The Two Contrasting Patient Cohorts
To better understand these genomic responses in patients, let us compare two different patients cohorts: uncomplicated patients, who are simply responding to the severe injury, versus complicated patients, who are not only responding to the injury but in addition, suffering from complications (e.g. nosocomial infections and MOF) (5,43). An example of an uncomplicated patient might be a 20-year old who crashes his BMW into a tree. His injuries include a concussion, multiple left rib fractures with pulmonary contusion, splenic fracture, and a left femoral fracture. Depending upon the severities of each of these injuries, he is at risk of hemorrhagic shock. He arrives at a Level 1 trauma center within an hour, is taken to surgery for the fractured spleen and open reduction and internal fixation of the femur fracture. With supportive care, the cerebral and pulmonary contusions resolve quickly and the patient is discharged after only five to seven days of hospitalization. An example of a complicated patient might be a 55 year-old old, who sustains comparable, but slightly more severe individual organ injuries. He undergoes the same operation, but fails to resolve the pulmonary and cerebral contusions as rapidly. He remains sedated, intubated and ventilated with moderate pulmonary failure, tachycardia, ileus, and other signs of sepsis even five or more days postoperatively.
These two cohorts are not distinguishable by cytokine or WBCs panels. In the Glue Grant study, dozens of cytokine plasma concentrations were compared between the two cohorts with only IL-6, IL1-RA, IL-8, and MCP1 being significantly different. When we checked, we found no differences in WBCs parameters between the two cohorts.
For patients with MOF, a new term, time to recovery (TTR), was created by the Glue Grant investigators (35,43,44). TTR not only reflects that an organ failed but it has a dynamic component to include the time that transpired before the organ recovered. Therefore the term TTR has both a frequency component as well as a temporal or dynamic influence. The patients can be categorized using TTR as uncomplicated (TTR <5 days), intermediate (TTR 5–14 days), and complicated (TTR >14 days) (35).
The Genomics of Two Patient Cohorts
Before the Glue Grant studies, the gene expression profiles in the circulating WBCs of the two contrasting patient cohorts would have been expected to be very different (FIGURE 6). The uncomplicated patient is simply responding to serious blunt trauma and the complicated patient is not only responding to the blunt trauma but also to medical complications. Of the 167 patients for whom there were genomic profiles, 55 patients were uncomplicated, 41 patients were complicated, and the remaining 71 were intermediate (35). Of the 5,136 genes that were two-fold different between patients compared to normal volunteers, the gene expressions can be presented based upon the patient cohort: 55 uncomplicated patients (Figure 6, left panel) and 41 complicated patients (Figure 6, right panel). These two heat maps appear quite similar with very few visual differences, if any.
Figure 6.

Comparisons of the time-course changes between complicated and uncomplicated patients. This is a subset of the data from Figure 4B of the K-means clustering of the genes based upon patterns of gene expression over time in the complicated and uncomplicated patient cohorts. To the left are the 55 uncomplicated patients and to the right are the 41 complicated patients. Red indicates increased and blue indicates decreased expression relative to the mean (white).
A statistical approach is required to identify differences. Of the 5,136 genes that were different by two-fold between patients and normal volunteers, there were 2,391 genes that were statistically different at any single time point comparing uncomplicated and complicated patients. Using a cutoff of more than two-fold, there were only 1,201 genes that were statistically different at one time point (FDR <0.001) (5). Cluster analysis of these 1,201 genes into ten clusters genes are shown in Figure 7. Clusters 2 and 8 are shown with box plots for those genes in the two patient cohorts. These statistical analyses showed that the only differences that could be found between the genomics of the two cohorts were quantitative and not qualitative. The genomic patterns of the complicated patients showed higher deviations from normal gene expression and these changes quantitatively persisted longer than those same genes in the uncomplicated patients. Qualitatively, no genes were recruited, were omitted, or were regulated in a different direction in the genomic signature of the WBCs when complications developed (5).
Figure 7.

Differences in gene expression patterns between patients with a complicated and uncomplicated clinical recovery. Heat map of 1,201 genes whose expression was at least twofold different at any time point when compared with controls (CTRL) for patients with a complicated (Comp) or uncomplicated recovery (Uncomp). (A) Cluster analysis of the two cohorts. The brackets to the right of the cluster indicate cluster 2 and 8 shown in B and C, respectively. (B) One cluster of genes whose expression was up-regulated in patients with a complicated recovery. (C) One cluster of genes whose expression was down-regulated in patients with a complicated recovery. © 2011 Xiao et al.5 Originally published in Journal of Experimental Medicine.2011 Nov;208(13):2581-2590. doi: 10.1084/jem.20111354.
Of these 1,201 genes that were significantly different by two-fold at least at one time point, there were only 63 genes that were different between those two cohorts at the two-fold level at all time points (FDR <0.001) (FIGURE 8, left panel) (35). More than two thirds of these genes were directly related to protective immunity. In fact, adaptive immunity pathways showed most notably the suppression of genes involved in antigen presentation (e.g. HLA-DQ, HLA-DR) and with interferon and interferon-inducible genes (e.g. IFIT 1,2,3,5). These HLA Class II and other interferon responsive genes that are downstream of the interferons are greatly suppressed in complicated patients relative to those genes in the uncomplicated patients. This means that there are two distinct genomic patterns or signatures that can be closely associated with very different patient outcomes (35).
Figure 8.

Differences in gene expression patterns of the 63 genes. (A) Pearson correlation between the microarray and nanoString™ expression level of the 63 genes found to be differentially regulated between uncomplicated and complicated patient cohorts. Values represent fold change over control expression values. Source: Cuenca et al.35 © 2013 Lippincott Williams & Wilkins. Reproduced with permission from Wolters Kluwer Health. (B) Variance normalized mean expression for all the genes up-regulated and down-regulated differentially between the two cohorts, and fitted curve. The differences in expression between the two cohorts could be primarily explained by the magnitude of the response and the duration.
The Two Major Objectives of the Glue Grant
Among others, there are two objectives that continue to motivate the Glue Grant investigators even today: the development of diagnostics to predict patient outcome and the identification of drug targets that might alter patient outcome. We did not anticipate that more than half of the few genes that were different between the two patient cohorts would be downstream of interferon gamma (IFN gamma). This discovery alone begs to be considered for a therapeutic intervention. To explain this rationale, we have identified a genomic signature that is closely associated with an adverse patient outcome. Many of the genes in this signature are downstream of the interferons (alpha, beta, and gamma). We believe that exogenous IFN gamma might alter this genomic pattern or signature toward the uncomplicated signature and as a result, this improved genomic signature might be clinically relevant and result in improved patient outcomes (i.e. a statistically significant reduction in TTR to <14 days). If this is true, this means that the patient would be discharged many days before s/he might have otherwise been predicted to have been discharged.
In summary, there were 5,136 genes changed at a two-fold level in the circulating WBCs of patients compared to normal volunteers. These changes were long-lasting and in the complicated patients, more than 50% of these genes remained abnormal even at 28 days post-injury. Of the patients discharged during those 28 days, the genomics were still grossly abnormal at the time of hospital discharge (5). In burn injuries, there were genomic changes present even after one year. Eight of ten innate immunity pathways were up-regulated and nine of ten adaptive immunity pathways were down-regulated after these serious injuries. There were two distinct patient cohorts, those who had an uncomplicated recovery and those with many complications. The genomic response in both these patient cohorts was identical qualitatively but different quantitatively. There were no new genes responding differently in the complicated patients relative to the uncomplicated patients. In the two patient cohorts, there were quantitative differences at the two-fold level at all time points in only 63 genes.
To put these findings on a gene level in terms of innate and adaptive immunity, there could be disagreement whether a gene belongs to one or another. Using categorizations from our publication in Nature (27), the data for these genes categorized by innate versus adaptive immunity are shown along with centroids in FIGURE 9. Nearly immediately after injury, there is a very significant up-regulation in the innate immunity genes and simultaneously, a very significant, almost mirror image, down-regulation in the adaptive immunity genes. These transcriptional changes in innate and adaptive immunity likely occur within hours of injury, while the phenotypic changes ultimately seen in our patients do not appear for days.
Figure 9.
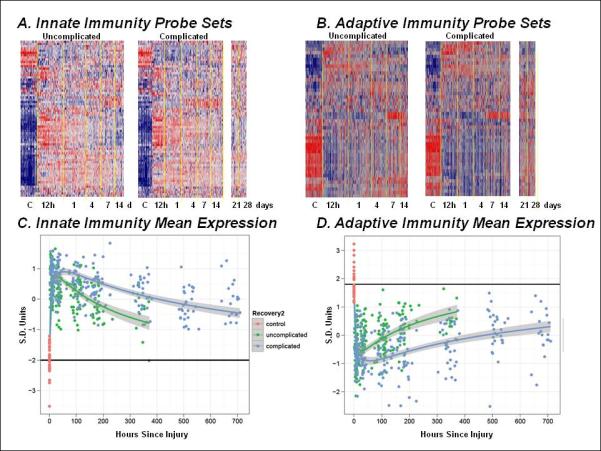
Expression patterns of selected genes involved in innate and adaptive immunity 152 genes directly associated with either innate or adaptive immune processes were selected, and their expression patterns evaluated in the uncomplicated and complicated patient cohorts. Panel A and B represent cluster analyses of the probe sets involved in innate and adaptive immunity, respectively, while Panels C and D represent a summary of the variance normalized mean gene expression for the individual probe sets. The primary differences in the patterns of gene expression are in the duration of the response, rather than in the magnitude or the timing of the changes.
A New Paradigm
These findings suggest a more relevant paradigm to genomically describe our patients after serious injury (FIGURE 10) (5). In terms of transcription, there is an immediate genomic response that triggers up-regulation of the innate immune response and a down-regulation of the adaptive immune response. These transcriptional changes are statistically more dramatic and longer lasting in the complicated than in the uncomplicated patient. It is likely that these genomic changes translate into the phenotypic changes of SIRS and CARS commonly seen on our critical care patients. There are features that differ from our traditional understanding about these syndromes: the transcriptional responses leading to SIRS and CARS are simultaneous, the CARS response is not a compensatory response, and the transcriptional responses are continuous in nature and do not wax and wane. This new insight is important because these immune responses have been triggered transcriptionally in the early hours post-injury. If an intervention intended to alter components of the innate immune or the adaptive immune response is contemplated, then it likely must be planned very early post-injury.
Figure 10.

A genomic storm: refining the immune, inflammatory paradigm in trauma (A) The current paradigm explains complications of severe injury as a result of excessive proinflammatory responses (SIRS) followed temporally by compensatory antiinflammatory responses (CARS) and suppression of adaptive immunity. A second-hit phenomenon results from sequential insults, which leads to more severe, recurrent SIRS and organ dysfunction. (B) The proposed new paradigm involves simultaneous and rapid induction of innate (both pro- and antiinflammatory genes) and suppression of adaptive immunity genes. Complicated recoveries are delayed, resulting in a prolonged, dysregulated immune–inflammatory state. © 2011 Xiao et al.5 Originally published in Journal of Experimental Medicine.2011 Nov;208(13):2581-2590. doi: 10.1084/jem.20111354.
Modulating Post-injury Immunity
These transcriptional changes are consistent with dramatically up-regulated innate inflammatory responses that are seen in our ICU patients. However, regarding the suppressed adaptive response, it is not entirely obvious that boosting this response is necessarily a good strategy. Forty percent of the new peptides and proteins that appear in plasma after these injuries are likely self-antigens (6-12,57). Processing these self-antigens and developing a specific response could trigger autoimmunity and death of the host.
On the other hand, maintenance of the critical components of the innate response including for example Fc-mediated phagocytosis, opsonization, and anti-bacterial peptides are essential to resolve nosocomial infections if they develop. Although adaptive immune responses develop over many days and weeks, they are not necessarily the reason that nosocomial infections resolve and the patients survive.
The Genomics of Murine Injury Versus Human Injury on a Molecular Basis
Now having described the human genomic response to injury, a reasonable question to ask is how well do the genomics of murine injury models mimic the human response on a molecular basis? Referring to Figure 5 (upper left nine squares), there are the Pearson Correlations (R2) between human burns, trauma, and endotoxemia (5). The remainder of the figure compares mouse burns, trauma, and endotoxemia to each other and to the human conditions (60). Looking at this figure, the genomic responses between the various mouse conditions (burns, trauma, and endotoxemia) are not reproducible among the three injuries nor do the mouse responses for any of the three injuries reproduce the genomics of the human conditions.
To understand this figure, it is important to recognize that zero correlation means randomness. For example, mouse trauma reproduces human trauma at a 5% correlation – near randomness. Further studies show that when the murine trauma model includes more extensive injury, the correlations improve to as much as 0.1 or 10% explanation of the variance (61). It should be recognized that the mouse in no way is treated exactly as we treat humans with an intensive care unit, for example, and therefore, the relative degree of injury is relatively less. However, the fact remains that there is only 10% or less reproducibility of the phenomenon on a molecular basis, which creates a philosophical decision about whether 10% reproducibility is “good enough” to mimic human injury sufficiently to design therapies for humans (62).
The Murine Versus Human Genomics on a Pathway Basis
On a pathway basis, each of the three human conditions reproduces the responses of the other two conditions to a very high degree. In contrast however, the mouse models neither mimic other murine conditions nor do they mimic the human conditions to any significant degree on a pathway basis (60).
As an example, FC-mediated phagocytosis in monocytes and neutrophils, which is an essential innate immune response to injury, is poorly mimicked at the pathway level between the three mouse models and it is poorly correlated with those comparable pathways in the human conditions (60). This is a basic component of the innate immune response that is necessary to resolve nosocomial infections (pneumonias and wound infections) in our patients. These failures are also true for the down-regulated pathways. This lack of correlation at the molecular level of pathways is very troublesome and threatens the development of human therapeutics by necessarily requiring an efficacious effect to be demonstrated in a murine model.
Genomic Comparisons for Other Human Diseases – Sepsis, ARDS, and Infections
A reasonable question to ask is whether the poor correlation is only true for injuries like burns and trauma or endotoxemia. Using the data available in public databases including GEO, we can ask how well does the human genomic response to injury and endotoxemia mimic genomic responses for other human conditions including sepsis, ARDS, and infections. The answer is that there is an excellent correlation among the human responses to these other three conditions, which is nearly identical to those genomic responses seen in burns and trauma. In stark contrast, we see a familiar pattern emerging – the murine models of sepsis, ARDS, and infections not only poorly reproduce the genomic responses in other murine models, but also the genomic responses in these other murine models fail to mimic human genomic responses in the comparable human conditions (60).
There are multiple other very important differences that merit recognition and discussion in the genomic responses seen in our mice models as compared to the comparable human conditions (60). In terms of the genomic response, the number of genes that are changed significantly is very different; the murine response is far more attenuated relative to the response in humans. It is possible that humans became far more sensitive to the stress of injury as a result of evolution in our genomics with a survival benefit achieved by developing sensitivity in our immune response. Unlike the evolution of the mouse in which after reproduction, the lifespan is brief, today, patients, who would have died in distant times, now fill our hospital beds and present us with the current day clinical challenges commonly seen in our modern day ICUs.
This difference in immune sensitivity is easily seen by examining the massive difference in the human versus the mouse response to endotoxin, which is highly tolerant to this immune stimulant (60). For example, the lethal doses of endotoxin is 5-25 mg/kg for most mice strains while a dose that is a million-fold less (30 ng/kg) has been reported to cause shock in humans (63). This sensitivity is not only to bacterial products including endotoxin but also to the microbe as well. There is a comparable major resistance to bacterial loads that is better tolerated by mice relative to that of humans by a million-fold.
If one restricts the comparisons between mice and humans to only those genes that change in both species and change in the same direction, then the correlations can be made to appear better (62,64). In Takao and Miyakawa 2014, the authors used p<0.05 and fold-change > 1.2 (introducing significant uncertainties) to compare human burns and mouse infection models and identified 13,586 and 3,116 changed genes respectively. Only 1,992 are common to both species and of these, only 1,608 genes change in the same direction. Therefore, out of the 13,586 genes that changed in the burn injury, only 12% (1,608) are changed in the same direction for both species. This leaves close to 90% of the genes that change significantly in human disease not modeled in mice. If one only considers a highly selected subset of genes that happen to behave similarly to develop a model, it becomes philosophical as to whether the results of the model should be trusted. Even with these reduced p-value and fold-change requirements, the correlation R2 improved to only 0.25 or 25% of the variation explained.
Another possible reason for the differences in the correlations between the human conditions and the murine models is that the injuries in the murine models are less severe than those seen in humans. In more recent studies, the injuries in the murine model were made to be more severe but even with these changes the R2 correlation increases to only 0.15 - 0.2 (61). These findings should not be surprising when one considers a very recent study, which evaluates the transcriptional landscapes of tissues comparing mice to humans (66). In their studies, “Overall, our results indicate that there is considerable RNA expression diversity between humans and mice, well beyond what was described previously, likely reflecting the fundamental physiological differences between these two organisms”.
The Same Response Times but Different Recovery Times for Genes
Another important difference involves the genomic response times in the human conditions versus murine models (60,65). The response time (time when the gene fold change reaches half of the maximum change) is very comparable in humans and mice; the vast majority of genes in both humans and mice have responded to near maximal levels within 12 hours post-injury. However, the massive differences are seen when comparing recovery times (time when the gene fold change returns back to half after it reached the maximum). The recovery times are markedly longer in humans compared to those in mice. For example, HLA-DR, a very commonly-used example of HLA Class II receptors, is suppressed for many months after human burn injuries while in trauma, a similar depression occurs, but over a shorter time period. In human endotoxemia, there is no consistent effect upon HLA-DR expression. In each of the murine model systems, no consistent changes are seen at all in any of the models.
So How Should Murine Models be Used
Despite the many differences seen at the molecular level, studies in mice are very useful to provide valuable information about how molecules interact. Testing and understanding molecular interactions in animal models are essential but before any statements about their relevance in human disease, confirmation that these same molecules are active in the human condition should be required. Often a similar appearing phenotype can be created in an animal model, but the molecular details to create the phenotype may or may not be identical to a comparable phenotype in the human condition. In fact, the likelihood might be a near random one. It's not the mouse's fault that drugs developed to work in mice fail in humans. It is more likely that we failed to understand the human diseases at a detailed molecular level. To demonstrate the point that our understanding about molecules in human diseases are limited, there were two publications in highly cited journals within weeks of each other about Eritoran, which is a synthetic lipid A antagonist that blocks the binding of endotoxin to the MD2-TLR4 complex. It was thought that activation of this complex is very important in our response to inflammation and infections, but when tested in critically ill patients, the drug had no beneficial effects (67) while in mice, it protected from lethal influenza infections (68).
To be fair, animal models have also been very helpful to develop symptomatic therapies and to slow pathological processes for diseases, for example, in two rodent arthritis models of rheumatoid arthritis - rat adjuvant-induced arthritis and mouse collagen-induced arthritis (69). The rat model reproduces the pain symptoms quite well and much has been learned about non-steroidal anti-inflammatory drugs. The mouse collagen model has been particularly helpful to understand lymphocytic and monocytic as well as cytokine roles in the development of rheumatoid arthritis. Anti-TNF antibody therapy has become a gold standard in the treatment of rheumatoid arthritis and much has come from work with the murine model. In addition to our lack of understanding about the molecular details of human diseases, there are many other confounding features that are poorly mimicked in animal models (69). Animal models tend to be acute while the comparable human conditions are often chronic and these models most often do not enlighten about the cause(s) of human disease.
What Have We Learned from the Glue Grant Program?
The Glue Grant Program has made substantial contributions to improve our understanding about how humans respond to potentially lethal injuries. The computational analysis of these complex data is ongoing today and we continue to work to make biological sense of the human genomic and proteomic responses. There are so many insights yet to come from the data sets generated by the Glue Grant Program. Soon to be published data from studies of the separated cells (neutrophils, T-cells, and monocytes) and data identifying splice variants of many of the responsive genes and control regions of the genome will provide the field with even greater information. With these new insights, we have the promise for a better modeling of organ failure, prediction of patient outcome, and possible new therapeutics to improve the patient outcome. Our genomic data from peripheral tissues (muscle, fat, and skin) in burn injured patients will provide us with a vastly improved understanding of the tissue-specific metabolic responses to severe injury.
If the Glue Grant has taught us anything, we now know that assuming a molecular mechanism found in a murine model is automatically relevant in humans is folly. Over the past 30 years to better understand human disease, our field has come to depend upon research heavily dominated by studies in mice. Recent data from the Glue Grant Program and other groups have shown that this reliance on murine data should come with limited expectations. This is not a failure of mice and their models but more our limited understanding of human disease. I believe that our genomics and proteomics data point us only in one direction – we are only going to better understand human diseases by studying diseases in humans.
There are multiple technologies that are already in place that can begin to provide us with a much better understanding of the molecular details of human diseases in humans. Quantitative imaging including dynamic MRI and positron emission tomography (PET) can now provide mechanistic information non-invasively in patients. Furthermore, the new genomic and proteomic technologies in use by our program and others now require only very small amounts of tissues (possibly from as little as that obtained by skinny needles) to derive genome-wide sequence and expression (21,36) and high throughput proteomic (57) information in patients.
The compelling need to improve our understanding of the human response to injury remains high because despite many billions of dollars and over 100 clinical trials, there has not been a single successful drug or biological agent found for sepsis therapeutics (70,71). We still fail to understand the basic problem inherent in the inflammatory response in sepsis. The only currently effective treatments remain the judicious usage of fluids and antibiotics along with other (surgical with incisions or interventional) physical treatments.
Efficacy will never come from murine studies unless the detailed molecular mechanism or pathways modeled in the mouse are found to be the same as those in the human disease. Our biggest challenge in studying the inflammatory response to potentially lethal injuries is to temper those harmful components of the innate immune response and to retain the essential components of that response for survival and successful recovery after the injury. In my view, this can only be accomplished by studying human inflammation in humans. And with that said, I wish to conclude. Thank you very much.
Acknowledgement
The magnitude of the clinical, genomic, and proteomic data reported here required the efforts of many individuals at participating institutions. In particular, I wish to acknowledge the supportive research environment created and sustained by the participants in the Glue Grant Program:
Lily Altstein, Ph.D., Henry V. Baker, Ph.D., Ulysses G.J. Balis, M.D., Paul E. Bankey, M.D., Ph.D., Timothy R. Billiar, M.D., Bernard H. Brownstein, Ph.D., Steven E. Calvano, Ph.D., David G. Camp II, Ph.D., J. Perren Cobb, M.D., Mitchell J. Cohen, M.D., Alex G. Cuenca. M.D., Joseph Cuschieri, M.D., Ronald W. Davis, Ph.D., Asit K. De, Ph.D., Philip A. Efron, M.D., Celeste C. Finnerty, Ph.D., Richard L. Gamelli, M.D., Nicole S. Gibran, M.D., Hong Gao, Ph.D., Brian G. Harbrecht, M.D., Douglas L. Hayden, M.A., Laura Hennessy, R.N., David N. Herndon, M.D., Shari E. Honari, R.N., Jureta Horton, Ph.D. (deceased), Marc G. Jeschke, M.D., Ph.D., Jeffrey L. Johnson, M.D., Matthew B. Klein, M.D., M. Cecelia Lopez, B.S., Stephen F. Lowry, M.D. (deceased), Ronald V. Maier, M.D., Philip H. Mason, Ph.D., Grace P. McDonald-Smith, M.Ed., Bruce A. McKinley, Ph.D., Carol L. Miller-Graziano, Ph.D., Michael N. Mindrinos, Ph.D., Joseph P. Minei, M.D., Lyle L. Moldawer, Ph.D., Ernest E. Moore, M.D., Frederick A. Moore, M.D., Avery B. Nathens, M.D., Ph.D., M.P.H., Grant E. O'Keefe, M.D., M.P.H., Laurence G. Rahme, Ph.D., Daniel G. Remick, M.D., David A. Schoenfeld, Ph.D., Junhee Seok, Ph.D., Michael B. Shapiro, M.D., Richard D. Smith, Ph.D., Jason Sperry, M.D., John D. Storey, Ph.D., Robert Tibshirani, Ph.D., Ronald G. Tompkins, M.D., Sc.D., Mehmet Toner, Ph.D., H. Shaw Warren, M.D., Michael A. West, M.D., PhD., Bram Wispelwey, M.D., Wing H. Wong, Ph.D., Wenzhong Xiao, Ph.D., Weihong Xu, Ph.D.
Source of Funding: Supported by U54GM62119 entitled Inflammation and the Host Response to Injury, a Large Scale Collaborative Research Program (“Glue Grant”) funded by the National Institute of General Medical Sciences. ClinicalTrials.gov numbers: NCT00257231 and This study was supported by the U.S. National Institutes of Health, National Institute of General Medical Sciences, Large Scale Collaborative Project, U54GM062119. ClinicalTrials.gov numbers: NCT00257231 and NCT00257244
I want to thank President William Cioffi for the privilege and honor to present this 40th Fitts Oratory. It is my pleasure to share with you, the membership of the Association, my perspective of the findings by a National Institute of General Medical Sciences-supported Large-scale Collaborative Program entitled “Inflammation and the Host Response to Injury”, which is otherwise known as “The Glue Grant”.
Footnotes
Classifications: organ dysfunction, nosocomial infection, inflammation, mortality, shock, trauma, intensivist, evidence-based medicine
The author has no conflicting financial interests.
REFERENCES
- 1.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. the Human Genome Project Investigators The sequence of the human genome. Science. 2001 Feb;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 2.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995 Oct;270:467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 3.The International HapMap Consortium The international hapmap project. Nature. 2003 Dec;246:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 4.Bone RC. A piece of my mind. Maumee: my Walden pond. JAMA. 1996 Dec;276:1931. [PubMed] [Google Scholar]
- 5.Xiao WZ, Mindrinos MN, Seok JH, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, et al. the Inflammation and Host Response to Injury Large-Scale collaborative Research Program A genomic storm in critically injured humans. J Exp Med. 2011 Nov;208(13):2581–90. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen Y, Jacobs JM, Camp DG, 2nd, Fang R, Moore RJ, Smith RD, Xiao W, Davis RW, Tompkins RG. Ultra-high efficiency strong cation exchange LC/RPLC/MS/MS for high dynamic range characterization of the human plasma proteome. Anal Chem. 2004 Feb;76(4):1134–44. doi: 10.1021/ac034869m. [DOI] [PubMed] [Google Scholar]
- 7.Qian WJ, Jacobs JM, Camp DG, 2nd, Monroe ME, Moore RJ, Gritsenko MA, Calvano SE, Lowry SF, Xiao W, Moldawer LL, et al. Comparative proteome analyses of human plasma following in vivo lipopolysaccharide administration using multidimensional separations coupled with tandem mass spectrometry. Proteomics. 2005 Feb;5(2):572–84. doi: 10.1002/pmic.200400942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qian WJ, Monroe ME, Liu T, Jacobs JM, Anderson GA, Shen Y, Moore RJ, Anderson DJ, Zhang R, Calvano SE, et al. Inflammation and the Host Response to Injury Large Scale Collaborative Research Program Quantitative proteome analysis of human plasma following in vivo lipopolysaccharide administration using 16O/18O labeling and the accurate mass and time tag approach. Mol Cell Proteomics. 2005 May;4(5):700–9. doi: 10.1074/mcp.M500045-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu T, Qian WJ, Gritsenko MA, Xiao W, Moldawer LL, Kaushal A, Monroe ME, Varnum SM, Moore RJ, Purvine SO, et al. Inflammation and the Host Response to Injury Large Scale Collaborative Research Program High dynamic range characterization of the trauma patient plasma proteome. Mol Cell Proteomics. 2006 Oct;5(10):1899–913. doi: 10.1074/mcp.M600068-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qian WJ, Liu T, Petyuk VA, Gritsenko MA, Petritis BO, Polpitiya AD, Kaushal A, Xiao W, Finnerty CC, Jeschke MG, et al. Inflammation and the Host Response to Injury Large Scale Collaborative Research Program Large-scale multiplexed quantitative discovery proteomics enabled by the use of an (18)O-labeled “universal” reference sample. J Proteome Res. 2009 Jan;8(1):290–9. doi: 10.1021/pr800467r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian WJ, Petritis BO, Kaushal A, Finnerty CC, Jeschke MG, Monroe ME, Moore RJ, Schepmoes AA, Xiao W, Moldawer LL, et al. Inflammation and the Host Response to Injury Large Scale Collaborative Research Program Plasma proteome response to severe burn injury revealed by 18O-labeled “universal” reference-based quantitative proteomics. J Proteome Res. 2010 Sep;9(9):477989. doi: 10.1021/pr1005026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou JY, Krovvidi RK, Gao Y, Gao H, Petritis BO, De A, Miller-Graziano C, Bankey PE, Petyuk VA, Nicora CD, et al. Inflammation and the Host Response to Injury Large Scale Collaborative Research Program Trauma-associated human neutrophil alterations revealed by comparative proteomics profiling. Proteomics Clin Appl. 2013 Aug;7(7-8):571–83. doi: 10.1002/prca.201200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De AK, Miller-Graziano CL, Calvano SE, Laudanski K, Lowry SF, Moldawer LL, Remick DG, Jr, Rajicic N, Schoenfeld D, Tompkins RG. Selective activation of peripheral blood T cell subsets by endotoxin infusion in healthy human subjects corresponds to differential chemokine activation. J Immunol. 2005 Nov;175(9):6155–62. doi: 10.4049/jimmunol.175.9.6155. [DOI] [PubMed] [Google Scholar]
- 14.Laudanski K, Miller-Graziano C, Xiao W, Mindrinos MN, Richards DR, De A, Moldawer LL, Maier RV, Bankey P, Baker HV, et al. Cell-specific expression and pathway analyses reveal alterations in trauma-related human T cell and monocyte pathways. Proc Natl Acad Sci U S A. 2006 Oct;103(42):15564–9. doi: 10.1073/pnas.0607028103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sethu P, Anahtar M, Moldawer LL, Tompkins RG, Toner M. Continuous flow microfluidic device for rapid erythrocyte lysis. Anal Chem. 2004 Nov;76(21):6247–53. doi: 10.1021/ac049429p. [DOI] [PubMed] [Google Scholar]
- 16.Murthy SK, Sin A, Tompkins RG, Toner M. Effect of flow and surface conditions on human lymphocyte isolation using microfluidic chambers. Langmuir. 2004 Dec;20(26):11649–55. doi: 10.1021/la048047b. [DOI] [PubMed] [Google Scholar]
- 17.Revzin A, Sekine K, Sin A, Tompkins RG, Toner M. Development of a microfabricated cytometry platform for characterization and sorting of individual leukocytes. Lab Chip. 2005 Jan;5(1):30–7. doi: 10.1039/b405557h. [DOI] [PubMed] [Google Scholar]
- 18.Sin A, Murthy SK, Revzin A, Tompkins RG, Toner M. Enrichment using antibody-coated microfluidic chambers in shear flow: model mixtures of human lymphocytes. Biotechnol Bioeng. 2005 Sep;91(7):816–26. doi: 10.1002/bit.20556. [DOI] [PubMed] [Google Scholar]
- 19.Sethu P, Moldawer LL, Mindrinos MN, Scumpia PO, Tannahill CL, Wilhelmy J, Efron PA, Brownstein BH, Tompkins RG, Toner M. Microfluidic isolation of leukocytes from whole blood for phenotype and gene expression analysis. Anal Chem. 2006 Aug;78(15):5453–61. doi: 10.1021/ac060140c. [DOI] [PubMed] [Google Scholar]
- 20.Russom A, Sethu P, Irimia D, Mindrinos MN, Calvano SE, Garcia I, Finnerty C, Tannahill C, Abouhamze A, Wilhelmy J, et al. Inflammation and Host Response to Injury Large Scale Collaborative Research Program Microfluidic leukocyte isolation for gene expression analysis in critically ill hospitalized patients. Clin Chem. 2008 May;54(5):891–900. doi: 10.1373/clinchem.2007.099150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irimia D, Mindrinos M, Russom A, Xiao W, Wilhelmy J, Wang S, Heath JD, Kurn N, Tompkins RG, Davis RW, et al. Genome-wide transcriptome analysis of 150 cell samples. Integr Biol (Camb) 2009 Jan;1(1):99–107. doi: 10.1039/b814329c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kotz KT, Xiao W, Miller-Graziano C, Qian WJ, Russom A, Warner EA, Moldawer LL, De A, Bankey PE, Petritis BO, et al. Inflammation and the Host Response to Injury Collaborative Research Program Clinical microfluidics for neutrophil genomics and proteomics. Nat Med. 2010 Sep;16(9):1042–7. doi: 10.1038/nm.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenbach AE, Koria P, Goverman J, Kotz KT, Gupta A, Yu M, Fagan SP, Irimia D, Tompkins RG. Microfluidics for T-lymphocyte cell separation and inflammation monitoring in burn patients. Clin Transl Sci. 2011 Feb;4(1):63–8. doi: 10.1111/j.1752-8062.2010.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warner EA, Kotz KT, Ungaro RF, Abouhamze AS, Lopez MC, Cuenca AG, Kelly-Scumpia KM, Moreno C, O'Malley KA, Lanz JD, et al. Microfluidics-based capture of human neutrophils for expression analysis in blood and bronchoalveolar lavage. Lab Invest. 2011 Dec;91(12):1787–95. doi: 10.1038/labinvest.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feezor RJ, Baker HV, Mindrinos M, Hayden D, Tannahill CL, Brownstein BH, Fay A, MacMillan S, Laramie J, Xiao W, et al. Inflammation and Host Response to Injury, Large-Scale Collaborative Research Program Whole blood and leukocyte RNA isolation for gene expression analyses. Physiol Genomics. 2004 Nov;19(3):247–54. doi: 10.1152/physiolgenomics.00020.2004. [DOI] [PubMed] [Google Scholar]
- 26.Cobb JP, Mindrinos MN, Miller-Graziano C, Calvano SE, Baker HV, Xiao W, Laudanski K, Brownstein BH, Elson CM, Hayden DL, et al. Inflammation and Host Response to Injury Large-Scale Collaborative Research Program Application of genome-wide expression analysis to human health and disease. Proc Natl Acad Sci U S A. 2005 Mar;102(13):4801–6. doi: 10.1073/pnas.0409768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, et al. Inflammation and Host Response to Injury Large Scale Collaborative Research Program A network-based analysis of systemic inflammation in humans. Nature. 2005 Oct;437(7061):1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 28.Storey JD, Xiao W, Leek JT, Tompkins RG, Davis RW. Significance analysis of time course microarray experiments. Proc Natl Acad Sci U S A. 2005 Sep;102(36):12837–42. doi: 10.1073/pnas.0504609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayden D, Lazar P, Schoenfeld D, Inflammation and the Host Response to Injury Investigators Assessing statistical significance in microarray experiments using the distance between microarrays. PLoS One. 2009 Jun 16;4(6):e5838. doi: 10.1371/journal.pone.0005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warren HS, Elson CM, Hayden DL, Schoenfeld DA, Cobb JP, Maier RV, Moldawer LL, Moore EE, Harbrecht BG, Pelak K, et al. Inflammation and Host Response to Injury Large Scale Collaborative Research Program A genomic score prognostic of outcome in trauma patients. Mol Med. 2009 Jul-Aug;15(7-8):220–7. doi: 10.2119/molmed.2009.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajicic N, Finkelstein DM, Schoenfeld DA, Inflammation and Host Response to Injury Research Program Investigators Analysis of the relationship between longitudinal gene expressions and ordered categorical event data. Stat Med. 2009 Sep;28(22):2817–32. doi: 10.1002/sim.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou B, Xu W, Herndon D, Tompkins R, Davis R, Xiao W, Wong WH, Toner M, Warren HS, Schoenfeld DA, et al. Inflammation and Host Response to Injury Program Analysis of factorial time-course microarrays with application to a clinical study of burn injury. Proc Natl Acad Sci U S A. 2010 Jun;107(22):9923–8. doi: 10.1073/pnas.1002757107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajicic N, Cuschieri J, Finkelstein DM, Miller-Graziano CL, Hayden D, Moldawer LL, Moore E, O'Keefe G, Pelik K, Warren HS, et al. Inflammation and the Host Response to Injury Large Scale Collaborative Research Program Identification and interpretation of longitudinal gene expression changes in trauma. PLoS One. 2010 Dec 20;5(12):e14380. doi: 10.1371/journal.pone.0014380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desai KH, Tan CS, Leek JT, Maier RV, Tompkins RG, Storey JD, Inflammation and the Host Response to Injury Large-Scale Collaborative Research Program Dissecting inflammatory complications in critically injured patients by within-patient gene expression changes: a longitudinal clinical genomics study. PLoS Med. 2011 Sep;8(9):e1001093. doi: 10.1371/journal.pmed.1001093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuenca AG, Gentile LF, Lopez MC, Ungaro R, Liu H, Xiao W, Seok J, Mindrinos MN, Ang D, Baslanti TO, et al. the Inflammation and Host Response to Injury Collaborative Research Program Development of a genomic metric that can be rapidly used to predict clinical outcome in severely injured trauma patients. Crit Care Med. 2013 May;41(5):1175–85. doi: 10.1097/CCM.0b013e318277131c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu W, Seok J, Mindrinos MN, Schweitzer AC, Jiang H, Wilhelmy J, Clark TA, Kapur K, Xing Y, Faham M, et al. Inflammation and Host Response to Injury Large-Scale Collaborative Research Program Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci U S A. 2011 Mar;108(9):3707–12. doi: 10.1073/pnas.1019753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leek JT, Monsen E, Dabney AR, Storey JD. EDGE: extraction and analysis of differential gene expression. Bioinformatics. 2006 Feb 15;22(4):507–8. doi: 10.1093/bioinformatics/btk005. [DOI] [PubMed] [Google Scholar]
- 38.Storey JD, Dai JY, Leek JT. The optimal discovery procedure for large-scale significance testing, with applications to comparative microarray experiments. Biostatistics. 2007 Apr;8(2):414–32. doi: 10.1093/biostatistics/kxl019. [DOI] [PubMed] [Google Scholar]
- 39.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007 Sep;3(9):1724–35. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seok J, Xu W, Gao H, Davis RW, Xiao W. JETTA: junction and exon toolkits for transcriptome analysis. Bioinformatics. 2012 May;28(9):1274–5. doi: 10.1093/bioinformatics/bts134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seok J, Xu W, Davis RW, Xiao W. RASA: robust alternative splicing analysis for human transcriptome arrays. Bioinformatics. 2014 doi: 10.1038/srep11917. Accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryu SY, Qian WJ, Camp DG, Smith RD, Tompkins RG, Davis RW, Xiao W. Detecting differential protein expression in large-scale population proteomics. Bioinformatics. 2014 Oct;30(19):2741–6. doi: 10.1093/bioinformatics/btu341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minei JP, Cuschieri J, Sperry J, Moore EE, West MA, Harbrecht BG, O'Keefe GE, Cohen MJ, Moldawer LL, Tompkins RG, et al. Inflammation and the Host Response to Injury Collaborative Research Program The changing pattern and implications of multiple organ failure after blunt injury with hemorrhagic shock. Crit Care Med. 2012 Apr;40(4):1129–35. doi: 10.1097/CCM.0b013e3182376e9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuschieri J, Johnson JL, Sperry J, West MA, Moore EE, Minei JP, Bankey PE, Nathens AB, Cuenca AG, Efron PA, et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program Benchmarking outcomes in the critically injured trauma patient and the effect of implementing standard operating procedures. Ann Surg. 2012 May;255(5):993–9. doi: 10.1097/SLA.0b013e31824f1ebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nathens AB, McMurray MK, Cuschieri J, Durr EA, Moore EE, Bankey PE, Freeman B, Harbrecht BG, Johnson JL, Minei JP, et al. The practice of venous thromboembolism prophylaxis in the major trauma patient. J Trauma. 2007 Mar;62(3):557–62. doi: 10.1097/TA.0b013e318031b5f5. [DOI] [PubMed] [Google Scholar]
- 46.Nathens AB, Johnson JL, Minei JP, Moore EE, Shapiro M, Bankey P, Freeman B, Harbrecht BG, Lowry SF, McKinley B, et al. Inflammation and the Host Response to Injury Investigators Inflammation and the host response to injury, a large-scale collaborative project: patient-oriented research core – standard operating procedures for clinical care. I. Guidelines for mechanical ventilation of the trauma patient. J Trauma. 2005 Sep;59(3):764–769. [PubMed] [Google Scholar]
- 47.Minei JP, Nathens AB, West M, Harbrecht BG, Moore EE, Shapiro MB, Bankey PE, Johnson JL, Freeman B, McKinley BA, et al. Inflammation and the Host Response to Injury Investigators Inflammation and the host response to injury, a large-scale collaborative project: patient-oriented research core – standard operating procedures for clinical care. II. Guidelines for prevention, diagnosis, and treatment of ventilator-associated pneumonia (VAP) in the trauma patient. J Trauma. 2006 May;60(5):1106–13. doi: 10.1097/01.ta.0000220424.34835.f1. [DOI] [PubMed] [Google Scholar]
- 48.Moore FA, McKinley BA, Moore EE, Nathans AB, West M, Shapiro MB, Bankey P, Freeman B, Harbrecht BG, Johnson JL, et al. Inflammation and the Host Response to Injury Collaborative Research Program Inflammation and the host response to injury, a large-scale collaborative project: patient-oriented research core – standard operating procedures for clinical care. III. Guidelines for shock resuscitation. J Trauma. 2006 Jul;61(1):82–9. doi: 10.1097/01.ta.0000225933.08478.65. [DOI] [PubMed] [Google Scholar]
- 49.West MA, Shapiro MB, Nathens AB, Johnson JL, Moore EE, Minei JP, Bankey PE, Freeman B, Harbrecht BG, McKinley BA, et al. Inflammation and the Host Response to Injury Collaborative Research Program Inflammation and the host response to injury, a large-scale collaborative project: patient-oriented research core – standard operating procedures for clinical care. IV. Guidelines for transfusion in the trauma patient. J Trauma. 2006 Aug;61(2):436–9. doi: 10.1097/01.ta.0000232517.83039.c4. [DOI] [PubMed] [Google Scholar]
- 50.Shapiro MB, West MA, Nathens AB, Harbrecht BG, Moore FA, Bankey PE, Freeman B, Johnson JL, McKinley BA, Minei JP, et al. Inflammation and the Host Response to Injury Collaborative Research Program Inflammation and the host response to injury, a large-scale collaborative project: patient-oriented research core – standard operating procedures for clinical care. V. Guidelines for sedation and analgesia during mechanical ventilation general overview. J Trauma. 2007 Oct;63:945–50. doi: 10.1097/TA.0b013e318142d21b. [DOI] [PubMed] [Google Scholar]
- 51.Harbrecht BG, Minei JP, Shapiro MB, Nathens AB, Moore EE, West MA, Bankey PE, Cuschieri J, Johnson JL, Maier RV, Inflammation and the Host Response to Injury Collaborative Research Program Inflammation and the host response to injury, a large-scale collaborative project: patient-oriented research core – standard operating procedures for clinical care. VI. Blood glucose control in the critically ill trauma patient. J Trauma. 2007 Sep;63(3):703–8. doi: 10.1097/TA.0b013e31811eadea. [DOI] [PubMed] [Google Scholar]
- 52.West MA, Moore EE, Shapiro MB, Nathens AB, Cuschieri J, Johnson JL, Harbrecht BG, Minei JP, Bankey PE, Maier RV, Inflammation and the Host Response to Injury Collaborative Research Program Inflammation and the host response to injury, a large-scale collaborative project: patient-oriented research core--standard operating procedures for clinical care. VII--Guidelines for antibiotic administration in severely injured patients. J Trauma. 2008 Dec;65(6):1511–19. doi: 10.1097/TA.0b013e318184ee35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Keefe GE, Shelton M, Cuschieri J, Moore EE, Lowry SF, Harbrecht BG, Maier RV, Inflammation and the Host Response to Injury Collaborative Research Program Inflammation and the host response to injury, a large-scale collaborative project: patient-oriented research core – standard operating procedures for clinical care. VIII – Nutritional support of the trauma patient. J Trauma. 2008 Dec;65(6):1520–28. doi: 10.1097/TA.0b013e3181904b0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans HL, Cuschieri J, Moore EE, Shapiro MB, Nathens AB, Johnson JL, Harbrecht BG, Minei JP, Bankey PE, Maier RV, et al. Inflammation and Host Response to Injury Investigators Inflammation and the host response to injury, a Large-Scale Collaborative Project: patient-oriented research core standard operating procedures for clinical care. IX. Definitions for complications of clinical care of critically injured patients. J Trauma. 2009 Aug;67(2):384–8. doi: 10.1097/TA.0b013e3181ad66a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuschieri J, Freeman B, O'Keefe G, Harbrecht BG, Bankey P, Johnson JL, Minei JP, Sperry J, West M, Nathens A, et al. Inflammation and the Host Response to Injury Collaborative Research Program Inflammation and the host response to injury a large-scale collaborative project: patient-oriented research core standard operating procedure for clinical care. X. Guidelines for venous thromboembolism prophylaxis in the trauma patient. J Trauma. 2008 Oct;65(4):944–50. doi: 10.1097/TA.0b013e3181826df7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silver GM, Klein MB, Herndon DN, Gamelli RL, Gibran NS, Altstein L, McDonald-Smith GP, Tompkins RG, Hunt JL, Inflammation and the Host Response to Injury, Collaborative Research Program Standard operating procedures for the clinical management of patients enrolled in a prospective study of Inflammation and the Host Response to Injury. J Burn Care Res. 2007 Mar-Apr;28(2):222–30. doi: 10.1097/BCR.0B013E318031AA44. [DOI] [PubMed] [Google Scholar]
- 57.Shen Y, Kim J, Strittmatter EF, Jacobs JM, Camp DG, 2nd, Fang R, Tolié N, Moore RJ, Smith RD. Characterization of the human blood plasma proteome. Proteomics. 2005 Oct;5(15):4034–45. doi: 10.1002/pmic.200401246. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Hunger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses in injury. Nature. 2010;464:104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu J, Zhang X, Elayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013 Feb;110(9):3507–12. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gentile LF, Nacionales DC, Lopez MC, Vanzant E, Cuenca A, Cuenca AG, Ungaro R, Baslanti TO, McKinley BA, Bihorac A, et al. A better understanding of why murine models of trauma do not recapitulate the human syndrome. Crit Care Med. 2014 Jun;42(6):1406–13. doi: 10.1097/CCM.0000000000000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warren HS, Tompkins RG, Moldawer LL, Seok JH, Xu WH, Mindrinos MN, Maier RV, Xiao WH, Davis RW. Mice are not men. Proc Natl Acad Sci USA. 2014 doi: 10.1073/pnas.1414857111. Letter to the Editor www.pnas.org/cgi/doi/10.10734/pnas.1414857111. [DOI] [PMC free article] [PubMed]
- 63.Warren HS, Tompkins RG, Mindrinos MN, Xiao W, Davis RW, et al. Of men, not mice, and inflammation. Proc Natl Acad Sci U S A. 2013 Aug;110(34):E3151. doi: 10.1073/pnas.1308943110. Reply to Cauwels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2014 doi: 10.1073/pnas.1401965111. www.pnas.org/cgi/doi/10.1073/pnas.1401965111. [DOI] [PMC free article] [PubMed]
- 65.Tompkins RG, Warren HS, Mindrinos MN, Xiao W, Davis RW, et al. To study human inflammatory diseases in humans. Proc Natl Acad Sci U S A. 2013 Sep;110(36):E3371. doi: 10.1073/pnas.1307452110. Reply to Osterburg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin S, Lin Y, Nery JR, Urich MA, Breschi A, Davis CA, Dobin A, Zaleski C, Beer MA, Chapman WC, Gingeras TR, Ecker JR, Snyder MP. Comparison of the transcriptional landscapes between human and mouse tissues. Proc Natl Acad Sci U S A. 2014 Nov 20; doi: 10.1073/pnas.1413624111. pii: 201413624. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugemier T, Perrotin D, Tidswell M, et al. ACCESS Study Group Effect of Eritoran, an antagoinist of MD2-TLR4, on mortality in patients with severe sepsis. The ACCESS Randomized Trial. JAMA. 2013 Mar 20;309(11):1154–62. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 68.Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioanni TL, Weiss J, et al. The TLR4 antagonist, Eritoran, protects mice from lethal influenza infection. Nature. 2013 May 23;497(7450):498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Webb DR. Animal models of human disease: inflammation. Biochem Pharmacol. 2014 Jan;87(1):121–30. doi: 10.1016/j.bcp.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 70.Annane D. Improving clinical trials in the critically ill: unique challenge--sepsis. Crit Care Med. 2009 Jan;37(1 Suppl):S117–128. doi: 10.1097/CCM.0b013e318192078b. [DOI] [PubMed] [Google Scholar]
- 71.Deans KJ, Haley M, Natanson C, Eichacker PQ, Minneci PC. Novel therapies for sepsis: a review. J Trauma. 2005 Apr;58(4):867–74. doi: 10.1097/01.ta.0000158244.69179.94. [DOI] [PubMed] [Google Scholar]