

Abstract
Aging in the immune system results in tendency to proinflammatory responses. Intradermal DNA immunization showed Th2 polarized noninflammatory immune responses. We tested here 18-month-old mice which were immunized with Aβ42 peptide, DNA Aβ42 trimer, or 2 different prime boost protocols identical to previous experiments. High Aβ42 antibody levels were found in aged mice which had received peptide immunizations (900 μg/mL plasma), and in mice which had received peptide prime and DNA boost immunizations (500 μg/mL), compared with antibodies in DNA Aβ42 immunized mice with 50 μg/mL. Although we found T-cell proliferation and inflammatory cytokines in mice which had received peptide or prime boost immunization, these were not found in DNA-immunized mice. The results are concordant with proinflammatory responses because of immunosenescence and contrain-dicate the use of Aβ42 peptide immunizations or prime boost immunization protocols for the use in elderly Alzheimer's disease patients. DNA Aβ42 immunization only on the other hand does lead to effective levels of antibodies without inflammatory cytokine or T-cell responses in the aged animal model tested.
Keywords: Alzheimer disease, Aging, Aβ42 immunotherapy, Inflammation, Immunosenescence, Inflammaging
1. Introduction
Alzheimer's disease is the most common form of dementia found in the aging population worldwide. There is no cure for this disease, and treatment options are only symptomatic. Immuno-therapy provides the biggest hope and potential for future treatment options. Clinical trials with passive immunizations are ongoing after a major setback from a first clinical trial in 2001 (AN1792), in which patients received Aβ42 peptide immunizations with Qs21 as adjuvant and 6% of the immunized patients developed meningoencephalitis because of an inflammatory Th1 immune response (Fox et al., 2005; Gilman et al., 2005; Orgogozo et al., 2003). Major efforts are now set on passive immunizations with preformed antibodies and active immunizations with epitope vaccines using only the B-cell epitope for antibody specificity, avoiding the Aβ42 T-cell epitope and thus avoiding a T-cell response (Carillo et al., 2013; Lambracht-Washington and Rosenberg, 2013a; Mangialasche et al., 2010). With focus on a positive outcome from the current clinical trials for Alzheimer's disease (AD) prevention (Bateman et al., 2012; Garber, 2012; Morris et al., 2012), it is likely for the future that safe active immunization will be in demand. Compared with passive immunotherapy active immunization is much less cost intense and can be easily applied to large populations.
Vaccination in the elderly individuals, however, is not as successful as in younger individuals. Aging of the immune system or immunosenescence is at least in part responsible for the decreased immune response and low antibody titers. Immunosenescence can be characterized by several features and both, the innate and adaptive immune system undergo major changes with the aging process. There is a weakened ability to respond to new antigens and a higher bias toward inflammatory immune responses, and these changes received the descriptive term of inflammaging (Franceschi and Campisi, 2014; Franceschi et al., 2007; Giunta et al., 2008). The T-cell receptor repertoire in naïve and memory T-cells changes with age leading to a less diversified and more monoclonal immune response with main involvement of memory cells. Thus, memory B and T-cell responses are unsustained, whereas the naïve B and Tcell pools are much smaller in an aged immune system.
Aging leads to changes in regard to particular CD4 T-cell responses: T helper 1 (Th1) responses with production of the inflammatory cytokine IFNγ are increased, whereas T helper 2 (Th2) responses with production of IL-4, IL-5, and IL-13 are diminished. Immunosenescence leads also to a higher propensity to develop autoimmune responses (Akbar and Henson, 2011; Chen et al., 2009; Gruver et al., 2007; Sakata-Kaneko et al., 2000; Solana et al., 2012; Uciechowski et al., 2008; Vasudev et al., 2014; Vukmanovic-Stejic et al., 2011; Weksler, 2000). Vaccination in the elderly individuals is challenging and in the active Aβ42 peptide immunization clinical trial, AN1792, only a small percentage of patients (23.4%) showed positive antibody titers (Fox et al., 2005; Gilman et al., 2005; Holmes et al., 2008).
We have previously published the absence or downregulation of antigen specific T cells in a DNA Aβ42 immunization mouse model making it safe for possible use in AD patients as the risk for inflammatory autoimmune reactions is low (Lambracht-Washington et al., 2009, 2011). In a DNA and peptide prime-boost immunization approach, we found that antibody levels were increased via the heterologous boost immunizations and that both peptide-boosted immunizations as well as DNA-boosted immunizations worked very well in this regard (Lambracht-Washington et al., 2013). Considering the fact that age is an important factor for the development of AD and that the patient population affected by AD is in the elderly individuals, we repeated the prime-boost experiments in groups of aged mice (18–22 months old) and compared these with the antibody and cellular immune responses in identical immunized adult mice (8–10 months old). The aim of this study is to show how effective and noninflammatory DNA Aβ42 immunization is compared with proinflammatory peptide immunizations in a senescent immune system.
2. Methods
2.1. Animals and immunizations
Mice had been purchased from Jackson Laboratories (Bar Harbor, Maine) and were aged in the institute animal facility. 12- to 18-month-old B6SJLF1 or B6C3F1 mice were immunized with Aβ42 peptide, DNA Aβ42 trimer, or 2 different prime boost protocols identical to previous experiments (Lambracht-Washington et al., 2009, 2013). In brief, intradermal DNA immunizations with plasmid DNA encoding Aβ42 trimer were performed into mouse ear skin using the Helios gene gun (Bio-Rad, Hercules, CA). DNA-coated gold particles were injected onto both sides of the ears consisting of a total of 4 μg DNA per immunization. For Aβ1–42 peptide immunizations, the Aβ peptide (rPeptide Bogart, GA) was dissolved directly in PBS at 4 mg/mL and incubated overnight at 37 °C allowing fibril formations (Walsh et al., 1997). Mice received 100 μg Aβ peptide with QuilA (20 μg per mouse per injection, Sigma) as adjuvant mixed just before the intraperitoneal (i.p) immunizations. In the prime-boost protocol, mice received 3 immunization with DNA Aβ42 trimer followed by 3 immunizations with Aβ42 peptide (DNA prime and peptide boost), or they received 3 Aβ42 peptide immunizations first followed by 3 immunizations with DNA Aβ42 trimer (peptide prime/DNA boost). Groups of 4 mice in 3 groups received either 6 DNA immunizations, 6 peptide immunizations, or 3 DNA prime and 3 peptide-boost immunizations, 3 peptide prime and 3 DNA-boost immunizations in biweekly intervals, respectively. For the direct comparison of adult and aged mice, 16 adult mice were included in the experimental setup which received the respective DNA or peptide immunizations in parallel. In additional experiments, we had compared DNA immunizations in adult and aged Balb/c mice (4× immunized), as well as DNA and peptide immunizations in aged B6SJL mice (8× immunized). Animal use for this study was approved by the UT Southwestern Medical Center Animal Research Committee.
2.2. Plasma and splenocyte collections
Ten days following the final immunization, the mice were euthanized. Blood was collected by cardiac puncture. Spleens were aseptically removed and processed for tissue culture as previously described (Lambracht-Washington et al., 2009).
2.3. Analysis of cell proliferation by carboxyfluorescein succinimidyl ester dilution
This method has been described previously (Lambracht-Washington et al., 2011). Cell were labeled with carboxyfluorescein succinimidyl ester (CFSE, Molecular Probes, Grand Island, NY) and cultured for 6 days in medium or with Aβ42 peptide. After staining of the cells with fluorochrome labeled antibodies to detect CD4 and CD8 T-cell populations (BD Biosciences, San Jose, CA), the fluorescence of the cells was measured using an Accuri C6 Flow Cytometer (Ann Arbor, MI). Data were analyzed with CFlow Plus, and proliferation was quantitated with FCS Express Version 3.
2.4. Enzyme-linked immunosorbent assay and enzyme-linked immunospot (ELISPOT) assays
Enzyme-linked immunosorbent assays (ELISAs) for antibody levels and titers in mouse plasma and cytokine concentrations from cell culture supernatants, and enzyme-linked immunospot (ELISPOT) assays to determine frequencies of cytokine secreting cells were performed according to standard procedures and as previously described (Lambracht-Washington et al., 2009, 2013).
2.5. Intracellular forkhead box P3 staining
Mouse splenocytes were analyzed for expression of the transcription factor forkhead box P3 (Foxp3) directly ex vivo. For intracellular staining, a Foxp3 buffer set (eBioscience, San Diego, CA and Tonbo Biosciences, San Diego, CA) was used in combination with CD4-FITC (clone GK1.5), CD25-PE (clone PC61), and Foxp3-Alexa645 (clone FJK-16s) (eBioscience) antibodies as recommended by the manufacturer.
2.6. Statistics
For statistics (unpaired t test with 2-tailed p values, Mann-Whitney t test, column statistics, 1-way analysis of variance), we used GraphPad Prism version 6 for Windows (San Diego, CA, www.graphpad.com). p-values of ≤0.05 were considered significant.
3. Results
3.1. DNA Aβ42 immunization results in similar levels of anti-Aβ42 antibodies in adult and aged mice
To analyze the effectiveness of DNA Aβ42 immunization in aged animals, we compared directly the antibody levels and titers from 10-month-old adult mice and 18- to 20-month-old aged mice, which had received 6 DNA immunizations or 6 peptide immunizations. Although there was a significant difference for the comparison of the amount of antibody in DNA- and peptide-immunized mice (p = 0.0002 and <0.0001), the direct comparison of adult and aged mice showed no significant differences for the antibody levels found after DNA or peptide immunizations (Fig. 1). Peptide-immunized mice had antibody levels (±standard deviation) of 907.4 (±130.1) μg/mL plasma in the aged mice and 844.4 (±51.02) μg/mL in the adult mice; DNA-immunized mice had antibody levels of 53.9 (±85.74) μg/mL in the aged mice and 29.94 (±22.43) μg/mL in the adult mice. For prime-boost immunizations, we tested 4 groups of aged mice and combined the data in Fig. 1. High Aβ42 antibody levels were found in 18-month-old mice which had received the prime-boost immunizations. Both boost immunizations, DNA and peptide, were effective in increasing the antibody levels. After 3 DNA prime immunizations, aged mice had antibody levels of 19.31 (±16.41) μg/mL which increased to 623.2 (±313.9) μg anti-Aβ42 IgG antibodies per mL plasma in the DNA prime and peptide-boost groups. Three times peptide-immunized aged mice had 116.4 (±13.31) μg/mL anti-Aβ antibodies which increased to 487.2 (±266.0) μg/mL in the peptide prime and DNA-boost groups (p-value for the comparison of the 2 prime-boost groups 0.0885). To compare the effectiveness of the prime-boost regimen between aged and the adult mice which we had published before, we compared the antibody levels in an ELISA with a 1:1000 plasma dilution. For the peptide prime and DNA-boosted groups in the adult mice, we found 518.5 ± 78.6 μg anti-Aβ IgG per mL plasma and for the DNA prime and peptide-boosted groups, we found 429.6 ± 63.8 μg anti-Aβ IgG per mL plasma (data not shown).
Fig. 1.
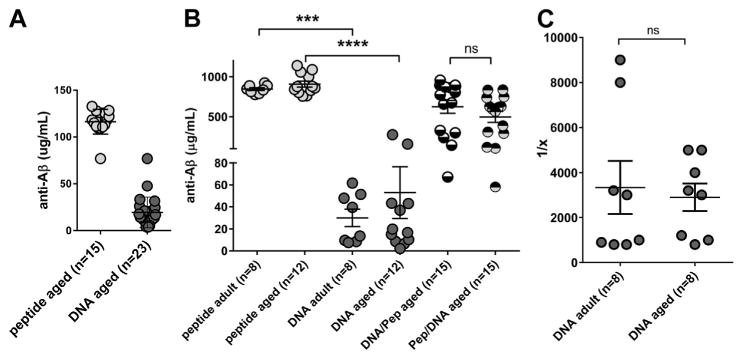
Analyses of the humoral immune responses in adult and aged mice. Antibody levels in the aged mice were analyzed after 3 prime immunizations, DNA Aβ42 and Aβ42 peptide, plasma dilutions 1:200 (A) and after 6 immunizations: peptide and DNA in adult and aged mice (n = 8 or n = 12), and DNA primed and peptide boosted and peptide primed and DNA boosted (n = 15 each group, plasma dilutions 1:1000, (B). Data shown are from the combined analysis of all blood samples in this study. The y axis shows the amount of IgG antibody in μg/mL plasma, the x axis shows the differently immunized mouse groups. In C, anti-Aβ42 IgG antibody titers were compared for DNA-immunized adult and aged mice. Analyses were performed in triplicates from 8 mice in each group; shown are mean and SEM. **** (p<0.0001), *** (p<0.0005), ns (p>0.05). Abbreviation: SEM, standard error of the mean.
As isotype switching is an important process during the adaptive immune response and is influenced by the cytokine milieu present during the cellular interactions, the anti-Aβ antibody isotypes were analyzed and compared between the adult and aged mouse groups. The antibody isotype profile was IgG1 (Th2) in the DNA Aβ42-immunized mice, aged and adult, as shown with the higher IgG1-to-IgG2a ratio (>2), and mixed IgG1/IgG2a (Th1/Th2) in the peptide-immunized mice and in both prime/boost immunization groups highly consistent with our previous reports (IgG1-to-IgG2a ratios ≤1, Table 1). To achieve good comparisons for the antibody levels in adult and aged DNA immunized mice, titer analyses were performed as well. Both groups had anti-Aβ1-42 antibody titers of 1:3000 (titer ± SEM: adult mice, 1:3338 ± 1182; aged mice, 1:2900 ± 613; Fig. 1C). Similar results were obtained when we determined the titers for binding to 2 other Aβ peptides, Aβ3-42 and Aβ1-40, for plasma from DNA immunized mice (data not shown).
Table 1. IgG1-to-IgG2a ratio calculated for the different groups.
| IgG1-to-IgG2a ratio, Th2/Th1 polarization | ||||
|---|---|---|---|---|
|
| ||||
| Aged | Adult | |||
| 6× DNA Aβ1-42 | 2.20, 2.84, 4.25 | Th2 | 5.07, 5.64, 2.31 | Th2 |
| 6× Aβ42 peptide | 1.05, 0.96, 0.83 | Mixed | 1.03, 1.02, 1.01 | Mixed |
| 3× DNA/3× peptide | 1.3, 0.97, 1.02 | Mixed | 1.16 | Mixed |
| 3× peptide/3× DNA | 1.04, 1.11, 0.72 | Mixed | 1.01 | Mixed |
| 4× DNA Aβ1-42 | 2.82 | Th2 | 2.4 | Th2 |
These ratios are indicative of Th1 (<1), Th2 (>2), and mixed T cell (around 1) responses.
Results shown were from the different groups of aged and adult mouse strain comparisons (B6SJLF1, B6C3F1, and BALB/c) analyzed in this study.
3.2. Analyses of a regulatory T-cell immune response in aged DNA Aβ42-immunized mice
In the comparison of 15-month-old B6SJLF1 mice which had received 8 DNA Aβ42 immunizations or 8 Aβ42 peptide immunizations, we found a significant increase in the percentage of CD4+CD25+Foxp3+ T cells (Tregs) for the DNA-immunized mice (p = 0.0016, unpaired t test). Although there was no significant difference in the percentages of T-effector cells (CD4+CD25+Foxp3−) and Tregs in the DNA-immunized mice, the Aβ42 peptide-immunized mice has significantly increased numbers of Teffs in the comparison with Treg numbers in the same mice (p < 0.0001, unpaired t test), and in the comparison with Teff numbers in the parallel analyzed DNA immunized mice (p = 0.0016, unpaired t test, Fig. 2A).
Fig. 2.
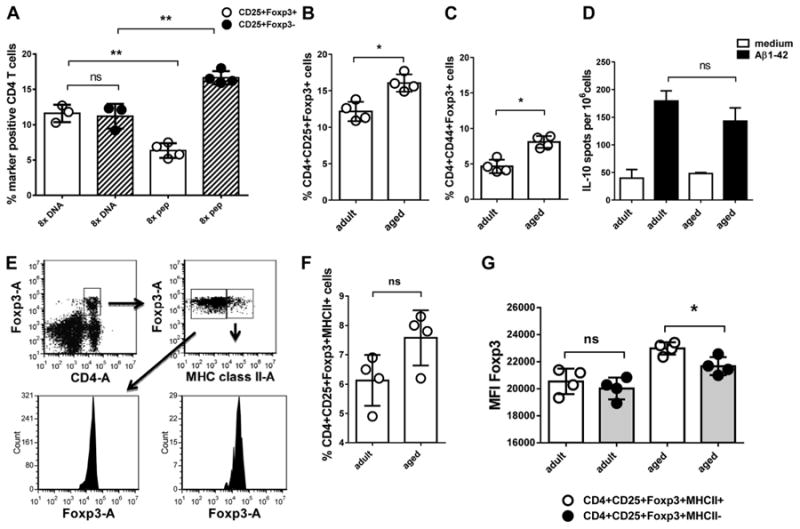
Analyses of the regulatory immune response in adult and aged mice. This figure shows the analyses of CD4+CD25+Foxp3+ cells by flow cytometry and IL-10 secreting cells measured by ELISPOT from 2 different mouse strains. In A, percentages of CD4+CD25+Foxp3+ cells were compared in 15-month-old DNA Aβ42 and Aβ42 peptide immunized B6SJLF1 mice which had received 8 DNA Aβ42 or Aβ42 peptide immunizations, respectively. In B–G, DNA Aβ42-immunized adult and aged Balb/c-Foxp3-EGFP mice were compared, which had received 4four DNA immunizations (n = 4/group). B shows a comparison of the percentages of CD4+CD25+Foxp3+ T cells. In C, the memory T cell marker CD44+ is included in this comparison. In D, IL-10 secretion from adult and aged splenocytes restimulated with Aβ42 peptide was measured by ELISPOT. In E, the gating strategy for the analyses of MHC class II positive Tregs is illustrated (indicated by arrows and boxed cell populations) and shown in F. In G, expression densities (MFI) for Foxp3 were compared on MHC class II negative and positive Tregs for which the gating is also shown in E. ** (p<0.005), * (p<0.05), ns (p>0.05). Abbreviation: MFI, mean fluorescence intensities.
We confirmed these findings in another mouse strain, Balb/c-Foxp3-EGFP, in which all cells expressing the fork head transcription factor Foxp3 are genetically labeled and show enhanced green fluorescence so that we do not have to use the intracellular staining protocol and which allows a direct comparison of cell numbers. In regard to the expected higher numbers of memory T cells (CD4+CD44+) in the aged mice, we analyzed a group of adult (8-month-old) and aged mice (2-month-old) which had received 4 DNA Aβ42 immunizations for expression of CD44 and MHC class II on CD4+CD25+Foxp3+ T cells. The mean percentage of CD4+CD25+Foxp3+ cells was significantly increased in the aged DNA-immunized mice in the comparison with adult DNA-immunized mice (0.0286, Mann-Whitney, Fig. 2B). Although we found no differences in the mean fluorescence intensities between adult and aged mice for CD44 as a memory T-cell marker on CD4+, CD4+Foxp3+, and CD8+ T cells, we found significant differences in the percentage of CD4+CD44+Foxp3+ cells with higher numbers in the aged mice (p = 0.0286, Mann-Whitney, Fig. 2C). In regard to IL-10 secretion on Aβ42 peptide restim-ulation in culture, we found no significant difference between the 2 age groups in an ELISPOT assay: 179 ± 37.5 cytokine secreting cell spots per 106 cells in the adult mice and 143 ± 47.15 spots in the aged mice (p = 0.2000, Mann-Whitney, Fig. 2D). Very low numbers of cytokine secreting cells were found by ELISPOT for the parallel analyzed cytokines IL-4, IL-17, and IFNγ in these 2 mouse groups (data not shown). When we compared the percentages of CD4+Foxp3+ MHC class II+ cells between the adult and aged mice, we observed a slight increase of MHC class II positive Tregs (CD4+CD25+Foxp3+) in the aged mice which was not significant (p = 0.0636, Fig. 2F). However, when we compared the expression densities (mean fluorescence intensities) for Foxp3 on MHC class II positive and MHC class II negative Tregs, we found no difference in the adult mice although again the aged mice showed slightly increased Foxp3 protein expression levels on MHC class II positive Tregs (p-value 0.0175, unpaired t test, Fig. 2G). In humans, functional differences had been described for MHC class II positive and MHC class II negative Tregs (Baecher-Allan et al., 2006).
3.3. Immunized-aged mice had increased levels of inflammatory cytokines, IFNγ, and IL-17
In addition to the antibody isotype data which provide information about the type of immune response elicited (Th1 or Th2), the analysis of cytokine secretion in response to Aβ42 peptide restimulation in vitro provides further insights. ELIPOT analyses with splenocytes from the immunized-aged mice showed confluent cell layers secreting IL-17 or IFNγ after Aβ42 peptide restimulation as well as antigen unspecific T-cell stimulation with an anti-CD3 antibody in peptide-immunized aged mice and the 2 aged prime-boost groups (>1000 spots/106 cells). In aged DNA-immunized mice, we found 10 ± 8 spots for IFNγ-secreting cells and 2 ± 1 spots for IL-17-secreting cells and also confluent but not as dense layers of IFNγ- and IL-17-secreting cells after antigen unspecific anti-CD3 antibody stimulation (>800 spots/106 cells, Fig. 3A). The number of IL-4 secreting cells following Aβ1-42 peptide restimulation in culture was 117.3 ± 20.43 spots per 106 cells in the 6 times peptide-immunized mice, 125 ± 23.64 and 230.2 ± 30.92 spots in the 2 prime-boost groups, and 16.67 ± 7.09 spots in the 6 times DNA-immunized mice. Similar levels of IL-4-producing cells were found for all 4 groups in the wells stimulated with an anti-CD3 antibody: 117.3 ± 20.43, 77 ± 8.66, 78.33 ± 4.16, and 76.0 ± 14.18 spots per 106 cells (1-way analysis of variance for comparison of the groups, p = 0.0812). Similar results were found for the three aged mouse groups analyzed with this assay.
Fig. 3.
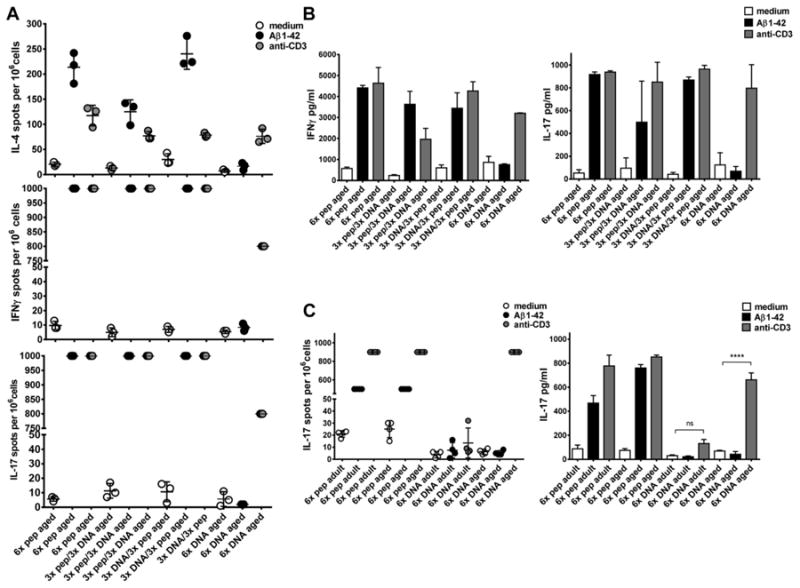
Analyses of the cytokine production in cells from adult and aged mice on restimulation. The secretion of cytokines was measured by ELISPOT, enzyme-linked immunospot and ELISA. In A, ELISPOT assays were compared for IL-4, IFNγ, and IL-17 secreting cells in aged B6SJL mice which had received 6-peptide immunizations, the prime-boost immunizations, or 6 DNA immunizations as indicated on the x axis. The cells had been cultured in medium, Aβ42 peptide, or anti-CD3 antibody containing medium. In B, the cytokine levels for IFNγ and IL-17 were shown for these mice as measured from 72 hours cell culture supernatants. In C, IL-17 secretion was compared between adult and aged DNA Aβ42 and Aβ42 peptide immunized mice in a 48-hour ELISPOT assay (left graph) or as measured from 48 hours cell culture supernatants (right graph). Shown are mean values and SEM. Results are representative for 2 or 3 identical performed experiments. **** (p<0.0001), ns (p>0.05). Abbreviations: ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean.
To analyze the amount of secreted IFNγ and IL-17 cytokine ELISA were performed with supernatants from the restimulated cultures after 72 hours. High levels of IFNγ and IL-17 were found in mice which had been immunized with Aβ42 peptide and in the mice which had received the prime/boost immunizations. IFNγ of 4409 ± 122.4 pg/mL and 917.2 ± 11.5 pg/mL IL-17A were measured for the peptide immunized mice, and 3628 ± 619.1 or 3442 ± 739.7 pg/mL IFNγ and 497.7 ± 208.6 or 868.6 ± 13.1 pg/mL IL-17A in the 2 prime-boost groups, compared with in Aβ42 restimulated cultures from these mice compared with 753.7 ± 37.3 pg/mL IFNγ or 68.5 ± 29.8 pg/mL IL-17A in the DNA-immunized mice (Fig. 3B).
The comparison of adult and aged mice confirmed the predisposition to inflammatory immune responses in aged mice. This was particularly obvious for the IL-17 cytokine responses. Although adult mice which had received DNA immunizations did not secrete IL-17 (not in the ELISPOT assays, not in the cytokine ELISA), cells from the aged mice secreted high levels of IL-17 on the antigen-independent stimulation with an anti-CD3 antibody (661.9 ± 32.8 pg/mL, p-value in comparison with medium control <0.0001 unpaired t test, Fig. 3C). Similar results were obtained for both of the mouse strains, B6SJL/F1 and B6C3/F1 in the comparison of adult and aged Aβ42-immunized mice.
3.4. Aged Aβ42 peptide-immunized mice showed high proliferation levels after Aβ peptide restimulation in vitro
Proliferation was measured with a CFSE dilution assay. No CD4+ T cell and no CD8+ T cell proliferation were found in the aged mice which had received DNA Aβ42 immunizations (Fig. 4A and B). High levels of T cell proliferation was found for Aβ1-42 peptide-immunized mice and both groups of prime/boost immunized mice (Fig. 4A). In 6 times peptide-immunized mice, we found mean CD4 T cell proliferations (±SEM) of 59.37% (±9.70); and in the 2 prime/boost immunized mouse groups, we found 42.67% (±12.91) and 46.04% (±17.33), respectively. These values are significant in the comparisons with proliferation in the medium controls: p-values were 0.0001 (6× peptide), 0.0175 (3× DNA/3× pep) and 0.0243 (3× pep/3× DNA). In 6 times DNA-immunized mice, the proliferation was not increased compared with the medium controls, 9.56% (±3.63) and 11.63% (±1.26), p-value of 0.408. The CD8T cell proliferation were 30.16% (±8.12) for 6 times Aβ42 peptide-immunized mice (p-value of 0.0089 compared with medium control), 32.37% (±6.7) in 3× DNA/3× peptide immunized mice (p = 0.0476) and 17.91% (±4.66) in 3× peptide/3× DNA-immunized mice (p = 0.0107). No CD8 T-cell proliferations was found for 6 times DNA-immunized mice with 8.3% (±1.63) proliferating cells in the medium controls and 11.63% (±4.21) in the Aβ42-peptide cultures (p = 0.2367, Fig. 4A).
Fig. 4.
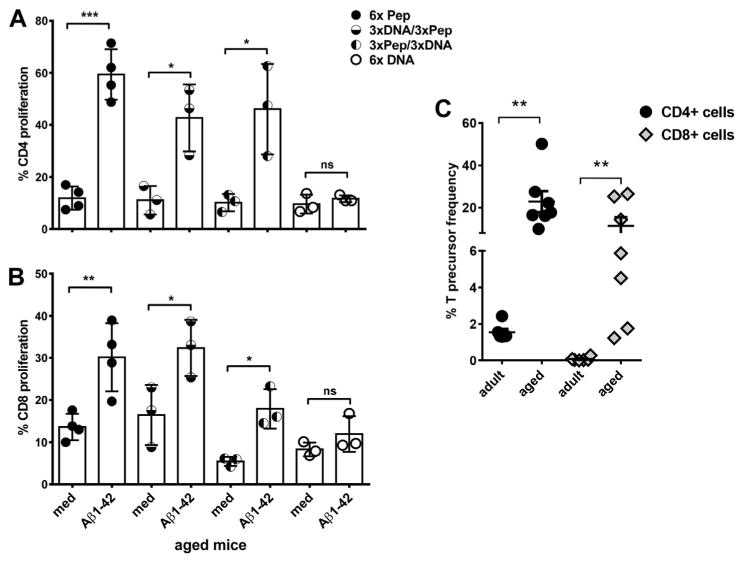
Analyses of T cell proliferation in adult and aged mice with a CFSE proliferation assay. Mouse splenocytes were labeled with CFSE and cultured for 5 days with medium or Aβ42 peptide. After staining with antibodies for CD4 and CD8, CFSE dilution was measured following gating on the respective T cell populations by flow cytometry. Proliferation was measured from triplicate wells for each of the mice analyzed. One circle represents the proliferation found for 1 individual mouse. In A, CD4 proliferation is compared for the differently immunized aged mouse groups (mean values and SEM). In B, CD8 proliferation is shown (mean values and SEM). Using the FCS3 express software, CD4 and CD8 precursor frequencies were calculated based on numbers and daughter generations of divided cells for adult and aged Aβ42 peptide immunized mice (C). Results shown are representative for 3 similar performed experiments. *** (p<0.0005), ** (p<0.005),* (p<0.05), ns (p>0.05). Abbreviations: CFSE, carboxyfluorescein succinimidyl ester; SEM, standard error of the mean.
Consistent with the predicted higher memory T-cell pool in aged mice, we found higher percentages of Aβ42 specific CD4 and CD8 T-cell precursor frequencies in Aβ42 peptide-immunized mice in the comparison of aged and adult mice which was calculated from the CFSE proliferation assays. For CD4+ T cells, we found precursor frequencies of 1.55% ± 0.43% in adult mice and 22.89% ± 13.21% in the aged mice; and for CD8+ T cells, we found precursor frequencies of 0.07% ± 0.01% in adult mice and 11.37% ± 10.84% in the aged mice. Despite a high standard deviation in the aged mice, the data are highly significant in the comparisons of CD4 and CD8 precursor frequencies in adult and aged mice (p = 00,012 for CD4 and CD8, Mann-Whitney test, Fig. 4B).
4. Discussion
We and others had shown previously that DNA-immunization results in a Th2 polarized noninflammatory immune response, and we could show this here also for aged mice. These findings are significant as the patient population for potential Aβ42 immunotherapy is the elderly individuals with the aforementioned changes in the immune system because of immunosenescence (Chen et al., 2009; Solana et al., 2012; Vukmanovic-Stejic et al., 2011). Aging in the immune system results in tendency to proinflammatory responses, and this fact needs major consideration before using an autoantigen, such as Aβ42, to trigger an immune response and production of therapeutic antibodies for removal of excess Aβ42 from brain. This fact had likely also led to the negative side effects and termination of the clinical trial AN1792, in which AD patients had received Aβ42 peptide immunizations together with a Th1 immune response boosting adjuvant and which had led to autoimmune inflammation (Fox et al., 2005; Gilman et al., 2005; Holmes et al., 2008).
Vaccines are generally given via intramuscular or subcutaneous injection of the respective antigen. The ease of access makes the skin to a preferred location and therefore a number of routes to perform these immunizations; the subcutaneous, the trans-cutaneous, and the epidermal routes are available for antigen injection and have been exploited (Nicolas and Guy, 2008). In recent years, the intradermal vaccination route has been proven to be highly effective in elderly humans for a seasonal flu vaccine (Camilloni et al., 2014; Holland et al., 2008). DNA immunizations can be also applied via intramuscular and intradermal injections and the immune response is highly dependent on the respective site of antigen administration. Whereas the intramuscular route results mainly in Th1 responses which can result in inflammation, the intradermal location of antigen injection results in a polarized Th2 response which is under most circumstances noninflammatory and for which we report our results here (Pertmer et al., 1996). Although DNA immunizations had been shown to result in good immune responses in animal models, much less immunogenicity had been reported in humans. However, promising results were achieved when physical methods such as ballistic devices, gene gun or Biojector, or electroporation devices had been used (Drape et al., 2006; Grimes-Serrano et al., 2008; Ledgerwood et al., 2012; Roy et al., 2000; Smith et al., 2010).
The transition of therapeutic antibodies into the brain is hindered by the blood-brain barrier which is generally not permeable for large molecules like antibodies. Therefore, a critique point for AD immunotherapy is that the antibody amount needed for a successful treatment is too large and unrealistic. However, it is actually not known how much antibody is needed for a successful therapy. Mechanisms of antibody action include inhibition of aggregation, activation of microglia, and a peripheral sink mechanism which will all lead to decrease of aggregated Aβ in brain. Anti-Aβ antibodies were detected in CSF of patients which had received active Aβ immunization in AN1792 and were assumed to affect Aβ levels in brain (Hock et al., 2002, 2003; Orgogozo et al., 2003). In a number of blood-brain barrier permeability studies, it was found that permeability increases with age in healthy humans and was further increased in patients with AD in the comparison with healthy control subjects (Farrall and Wardlaw, 2009; Popescu et al., 2009). Results from mice receiving passive Aβ immunization had shown that anti-Aβ antibodies enter the brain and are found in regions with high amyloid concentrations; the ability to enter the brain changes with age and might depend also on features specific to the antibodies (Banks et al., 2007; Bard et al., 2000). The antibody amounts found in aged DNA-immunized mice (mean 53 μg/mL plasma) were higher than the antibody levels, we had found earlier in transgenic APP mice (2.9 μg/mL plasma) and inwhich we found a substantial reduction in plaque load (50%) and total Aβ42 levels (41%) in brain from the immunized mice compared with control mice (Lambracht-Washington and Rosenberg, 2013b; Qu et al., 2006, 2007).
Our results showed the DNA Aβ42 immunization into skin leads to comparable antibody levels in adult and aged mice and based on studies to the optimal sites of immunization of elderly individuals with a flu vaccine, it is very likely that the skin as immunization site in aged AD patients will be the preferred route of application and that sufficient antibody responses will be achieved. In regard to a diminished or increased Th2 T-cell responses which both had been described because of aging (Rink et al., 1998; Sakata-Kaneko et al., 2000; Uciechowski et al., 2008), we did not find differences in the antibody isotype profile nor in the IL-4 cytokine responses between adult and aged DNA Aβ42-immunized mice. Thus, the Th2 immune profile following DNA immunization is not changed because of aging, making it unlikely that this type of immunotherapy would cause inflammation in AD patients.
In regard to changes in the regulatory T cell response, we found high numbers of CD4+CD25+Foxp3+ cells in the aged mice analyzed. In the comparison of Aβ42 peptide and DNA Aβ42-immunized mice, the difference in Tregs as measured in lymphocytes from spleen cells was highly significant. In Aβ42 peptide-immunized mice, we found significant higher numbers of T-effector cells. Actually, a shift to a more regulatory immune response because of aging has been described before and is in general considered to be responsible for the lack of immune surveillance and development of tumors in the elderly individuals (Fulop et al., 2010, 2011; Garg et al., 2014; Schmitt et al., 2013). For DNA Aβ42 immunizations, however, we have described this as a characteristic positive feature as the development of a regulatory immune response minimizes the risk of inflammatory auto-immunity against brain Aβ42 (article submitted). Thus, an increased regulatory T-cell response in the elderly individuals increases the safety of this immunotherapy approach.
In regard to increased inflammatory immune responses, we did find high levels of IFNγ and IL-17 secretion in the aged mice but with significant differences between DNA- and peptide-immunized mice and in the antigen specific (Aβ42) and antigen unspecific (anti-CD3) responses. Peptide-immunized aged mice and the 2 prime boost-immunized mouse groups responded with high IFNγ and high IL-17 secretion on antigen specific as well as antigen un-specific stimulation. Although the adult DNA-immunized mice did not show IL-17 secretion on antigen-specific and antigen-unspecific stimulation, T cells from the aged DNA-immunized mice did not secrete IL-17 on Aβ42 peptide restimulation but secreted high levels in response to the antigen independent signal delivered with an anti-CD3 monoclonal antibody. IL-17 is produced only byasubset of T cells, and naïve T cells do not secrete IL-17 on antigen-independent stimulation, not even in aged mice (data not shown). This showed that the aged DNA-immunized mice had a lower threshold for an inflammatory response but not to signals from the DNA Aβ42 immunization. Comparable results had been described in an allograft rejection model in mice, in which the authors showed for aged mice increased production of IL-17 but not IFNγ during alloactivation because of elevated numbers of memory CD4+ T cells (Tesar et al., 2009).
Different from a previous study inwhich we examined the effect of prime-boost immunizations on the humoral- and cellular-immune responses in adult mice and in which we could show that a DNA Aβ42-boost immunization following Aβ1-42 peptide prime did lead to the downregulation of T-cell proliferation, as well as cytokine secretion (Lambracht-Washington et al., 2013), we found here that thisis not the case inanaged mouse model. CD4 and CD8 T cells proliferated very well in peptide immunized mice as well as in the 2 prime-boost immunization groups. A DNA boost following a peptide prime did not influence the cellular immune response.
Our results are in concordance that immunosenescence in general leads to proinflammatory immune responses and contra-indicate the use of Aβ42-peptide immunizations as well as a pep-tide/DNA prime-boost immunization protocols for possible use in elderly AD patients. However, DNA Aβ42 immunization in the aged mouse leads to effective levels of antibodies to remove excess amyloid from brain (Lambracht-Washington and Rosenberg, 2013b; Qu et al., 2006, 2007, 2010) without inflammatory cytokine or proliferative cell responses. We conclude from these data that the aged patient with AD may deliver a safe and effective clinical response in removal of accumulated amyloid following active DNA Aβ42 immunotherapy.
Acknowledgments
This study was funded by grants from NIH/NIA to the Alzheimer's Disease Center (P30AG12300-19), the Rudman Partnership, Darryl Royal Foundation and McCune Foundation to Roger N. Rosenberg and an RO3 grant to Doris Lambracht-Washington (R03AG042697-02).NIH
Footnotes
Disclosure statement: The authors have nothing to disclose.
References
- Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11:289–295. doi: 10.1038/nri2959. [DOI] [PubMed] [Google Scholar]
- Baecher-Allan CM, Wolf E, Hafler DA. MHC class II expression identifies functionally distinct human regulatory T cells. J Immunol. 2006;176:4622–4631. doi: 10.4049/jimmunol.176.8.4622. [DOI] [PubMed] [Google Scholar]
- Banks WA, Farr SA, Morley JE, Wolf KM, Geylis V, Steinitz M. Anti-amyloid beta protein antibody passage across the blood–brain barrier in the SAMP8 mouse model of Alzheimer's disease: an age-related selective uptake with reversal of learning impairment. Exp Neurol. 2007;206:248–256. doi: 10.1016/j.expneurol.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Dominantly inherited Alzheimer network. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni B, Basileo M, Di Martino A, Donatelli I, Iorio AM. Antibody responses to intradermal or intramuscular MF59-adjuvanted influenza vaccines as evaluated in elderly institutionalized volunteers during a season of partial mismatching between vaccine and circulating A(H3N2) strains. Immun Ageing. 2014;11:10. doi: 10.1186/1742-4933-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo MC, Brashear HR, Logovinsky V, et al. Can we prevent Alzheimer's disease? Secondary “prevention” trials in Alzheimer's disease. Alzheimers Dement. 2013;9:123–131. doi: 10.1016/j.jalz.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Chen WH, Kozlovsky BF, Effros RB, Grubeck-Loebenstein B, Edelman R, Sztein MB. Vaccination in the elderly: an immunological perspective. Trends Immunol. 2009;30:351–359. doi: 10.1016/j.it.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drape RJ, Macklin MD, Barr LJ, Jones S, Haynes JR, Dean HJ. Epidermal DNA vaccine for influenza is immunogenic in humans. Vaccine. 2006;24:4475–4481. doi: 10.1016/j.vaccine.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease–systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–352. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Fulop T, Kotb R, Fortin CF, Pawelec G, de Angelis F, Larbi A. Potential role of immunosenescence in cancer development. Ann N Y Acad Sci. 2010;1197:158–165. doi: 10.1111/j.1749-6632.2009.05370.x. [DOI] [PubMed] [Google Scholar]
- Fulop T, Larbi A, Kotb R, de Angelis F, Pawelec G. Aging, immunity, and cancer. Discov Med. 2011;11:537–550. [PubMed] [Google Scholar]
- Garber K. Genentech's Alzheimer's antibody trial to study disease prevention. Nat Biotechnol. 2012;30:731–732. doi: 10.1038/nbt0812-731. [DOI] [PubMed] [Google Scholar]
- Garg SK, Delaney C, Toubai T, Ghosh A, Reddy P, Banerjee R, Yung R. Aging is associated with increased regulatory T-cell function. Aging Cell. 2014;13:441–448. doi: 10.1111/acel.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, Tan J. Inflammaging as a prodrome to Alzheimer's disease. J Neuroinflammation. 2008;5:51. doi: 10.1186/1742-2094-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes-Serrano Jill M, Lu Shan, Wang Shixia. Current progress of DNA vaccine studies in humans. Expert Rev Vaccines. 2008;7:175–191. doi: 10.1586/14760584.7.2.175. [DOI] [PubMed] [Google Scholar]
- Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J Pathol. 2007;211:144–156. doi: 10.1002/path.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, Davey G, Moritz E, Nitsch RM. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–1275. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Holland D, Booy R, De Looze F, Eizenberg P, McDonald J, Karrasch J, McKeirnan M, Salem H, Mills G, Reid J, Weber F, Saville M. Intradermal influenza vaccine administered using a new microinjection system produces superior immunogenicity in elderly adults: a randomized controlled trial. J Infect Dis. 2008;198:650–658. doi: 10.1086/590434. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Lambracht-Washington D, Qu BX, Fu M, Eagar TN, Stüve O, Rosenberg RN. DNA beta-amyloid (1-42) trimer immunization for Alzheimer disease in a wild-type mouse model. JAMA. 2009;302:1796–1802. doi: 10.1001/jama.2009.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambracht-Washington D, Qu BX, Fu M, Anderson LD, Jr, Stüve O, Eagar TN, Rosenberg RN. DNA immunization against amyloid beta 42 has high potential as safe therapy for Alzheimer's disease as it diminishes antigen specific Th1 and Th17 cell proliferation. Cell Mol Neurobiol. 2011;31:867–874. doi: 10.1007/s10571-011-9680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambracht-Washington D, Qu BX, Fu M, Anderson LD, Jr, Stüve O, Eagar TN, Rosenberg RN. A peptide prime-DNA boost immunization protocol provides significant benefits as a new generation Abeta42 DNA vaccine for Alzheimer disease. J Neuroimmunol. 2013;254:63–68. doi: 10.1016/j.jneuroim.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambracht-Washington D, Rosenberg RN. Advances in the development of vaccines for Alzheimer's disease. Discov Med. 2013a;15:319–326. [PMC free article] [PubMed] [Google Scholar]
- Lambracht-Washington D, Rosenberg RN. Anti-amyloid beta to tau - based immunization: developments in immunotherapy for Alzheimer disease. Immunotargets Ther. 2013b;2:105–114. doi: 10.2147/ITT.S31428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledgerwood JE, Hu Z, Gordon IJ, Yamshchikov G, Enama ME, Plummer S, Bailer R, Pearce MB, Tumpey TM, Koup RA, Mascola JR, Nabel GJ, Graham BS. VRC 304 and VRC 305 study Teams. Influenza virus h5 DNA vaccination is immunogenic by intramuscular and intradermal routes in humans. Clin Vaccin Immunol. 2012;19:1792–1797. doi: 10.1128/CVI.05663-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–707. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- Morris JC, Aisen PS, Bateman RJ, Benzinger TL, Cairns NJ, Fagan AM, Ghetti B, Goate AM, Holtzman DM, Klunk WE, McDade E, Marcus DS, Martins RN, Masters CL, Mayeux R, Oliver A, Quaid K, Ringman JM, Rossor MN, Salloway S, et al. Developing an international network for Alzheimer research: the dominantly inherited Alzheimer network. Clin Inves-tig (Lond) 2012;2:975–984. doi: 10.4155/cli.12.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas JF, Guy B. Intradermal, epidermal and transcutaneous vaccination: from immunology to clinical practice. Expert Rev Vaccines. 2008;7:1201–1214. doi: 10.1586/14760584.7.8.1201. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2002;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Pertmer TM, Roberts TR, Haynes JR. Influenza virus nucleoprotein-specific immunoglobulin G subclass and cytokine responses elicited by DNA vaccination are dependent on the route of vector DNA delivery. J Virol. 1996;70:6119–6125. doi: 10.1128/jvi.70.9.6119-6125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu BO, Toescu EC, Popescu LM, Bajenaru O, Muresanu DF, Schultzberg M, Bogdanovic N. Blood-brain barrier alterations in ageing and dementia. J Neurol Sci. 2009;283:99–106. doi: 10.1016/j.jns.2009.02.321. [DOI] [PubMed] [Google Scholar]
- Qu B, Boyer PJ, Johnston SA, Hynan LS, Rosenberg RN. Aβ42 gene vaccination reduces brain amyloid plaque burden in transgenic mice. J Neurol Sci. 2006;244:151–158. doi: 10.1016/j.jns.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu BX, Xiang Q, Li L, Johnston SA, Hynan LS, Rosenberg RN. Aβ42 gene vaccine prevents Aβ42 deposition in brain of double transgenic mice. J Neurol Sci. 2007;260:204–213. doi: 10.1016/j.jns.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu BX, Lambracht-Washington D, Fu M, Eagar TN, Stüve O, Rosenberg RN. Analysis of three plasmid systems for use in DNA A beta 42 immunization as therapy for Alzheimer's disease. Vaccine. 2010;28:5280–5287. doi: 10.1016/j.vaccine.2010.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink L, Cakman I, Kirchner H. Altered cytokine production in the elderly. Mech Ageing Dev. 1998;102:199–209. doi: 10.1016/s0047-6374(97)00153-x. [DOI] [PubMed] [Google Scholar]
- Roy MJ, Wu MS, Barr LJ, Fuller JT, Tussey LG, Speller S, Culp J, Burkholder JK, Swain WF, Dixon RM, Widera G, Vessey R, King A, Ogg G, Gallimore A, Haynes JR, Heydenburg Fuller D. Induction of antigen-specific CD8+ T cells, T helper cells, and protective levels of antibody in humans by particle-mediated administration of a hepatitis B virus DNA vaccine. Vaccine. 2000;19:764–778. doi: 10.1016/s0264-410x(00)00302-9. [DOI] [PubMed] [Google Scholar]
- Sakata-Kaneko S, Wakatsuki Y, Matsunaga Y, Usui T, Kita T. Altered Th1/Th2 commitment in human CD4+ T cells with ageing. Clin Exp Immunol. 2000;120:267–273. doi: 10.1046/j.1365-2249.2000.01224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt V, Rink L, Uciechowski P. The Th17/Treg balance is disturbed during aging. Exp Gerontol. 2013;48:1379–1386. doi: 10.1016/j.exger.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Smith LR, Wloch MK, Ye M, Reyes LR, Boutsaboualoy S, Dunne CE, Chaplin JA, Rusalov D, Rolland AP, Fisher CL, Al-Ibrahim MS, Kabongo ML, Steigbigel R, Belshe RB, Kitt ER, Chu AH, Moss RB. Phase 1 clinical trials of the safety and immunogenicity of adjuvanted plasmid DNA vaccines encoding influenza A virus H5 hemagglutinin. Vaccine. 2010;28:2565–2572. doi: 10.1016/j.vaccine.2010.01.029. [DOI] [PubMed] [Google Scholar]
- Solana R, Tarazona R, Gayoso I, Lesur O, Dupuis G, Fulop T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol. 2012;24:331–341. doi: 10.1016/j.smim.2012.04.008. [DOI] [PubMed] [Google Scholar]
- Tesar BM, Du W, Shirali AC, Walker WE, Shen H, Goldstein DR. Aging augments IL-17 T-cell alloimmune responses. Am J Transpl. 2009;9:54–63. doi: 10.1111/j.1600-6143.2008.02458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uciechowski P, Kahmann L, Plümäkers B, Malavolta M, Mocchegiani E, Dedoussis G, Herbein G, Jajte J, Fulop T, Rink L. TH1 and TH2 cell polarization increases with aging and is modulated by zinc supplementation. Exp Gerontol. 2008;43:493–498. doi: 10.1016/j.exger.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Vasudev A, Ying CT, Ayyadhury S, Puan KJ, Andiappan AK, Nyunt MS, Binte Shadan N, Mustafa S, Low I, Rotzschke O, Fulop T, Ng TP, Larbi A. γ/δ T cell subsets in human aging using the classical α/β T cell model. J Leukoc Biol. 2014;96:647–655. doi: 10.1189/jlb.5A1213-650RR. [DOI] [PubMed] [Google Scholar]
- Vukmanovic-Stejic M, Rustin MH, Nikolich-Zugich J, Akbar AN. Immune responses in the skin in old age. Curr Opin Immunol. 2011;23:525–531. doi: 10.1016/j.coi.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22367. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- Weksler ME. Changes in the B-cell repertoire with age. Vaccine. 2000;18:1624–1628. doi: 10.1016/s0264-410x(99)00497-1. [DOI] [PubMed] [Google Scholar]