

Abstract
c-Met is a receptor tyrosine kinase that upon binding of its ligand, hepatocyte growth factor (HGF), activates downstream pathways with diverse cellular functions that are important in organ development and cancer progression. Anomalous c-Met signalling has been described in a variety of cancer types, and the receptor is regarded as a novel therapeutic target. In breast cancer there is a need to develop new treatments, particularly for the aggressive subtypes such as triple-negative and basal-like cancer, which currently lack targeted therapy. Over the last two decades, much has been learnt about the functional role of c-Met signalling in different models of breast development and cancer. This work has been complemented by clinical studies, establishing the prognostic significance of c-Met in tissue samples of breast cancer. While the clinical trials of anti-c-Met therapy in advanced breast cancer progress, there is a need to review the existing evidence so that the potential of these treatments can be better appreciated. The aim of this article is to examine the role of HGF/c-Met signalling in in vitro and in vivo models of breast cancer, to describe the mechanisms of aberrant c-Met signalling in human tissues, and to give a brief overview of the anti-c-Met therapies currently being evaluated in breast cancer patients. We will show that the HGF/c-Met pathway is associated with breast cancer progression and suggest that there is a firm basis for continued development of anti-c-Met treatment, particularly for patients with basal-like and triple-negative breast cancer.
Introduction
The receptor tyrosine kinase (RTK) c-Met was originally identified as the product of a transforming gene generated from a chemically transformed osteosarcoma cell line [1]. In 1991, c-Met was discovered to be the receptor for hepatocyte growth factor (HGF), a protein that had previously been shown to promote hepatocyte growth in culture [2,3]. Mutations in the MET gene were subsequently described in hereditary and sporadic papillary renal cell carcinomas [4]. Since then, dysregulation of c-Met signalling has been identified in a variety of malignant and premalignant lesions, including those arising in the breast, lung, stomach, pharynx, colorectum and cervix [5-10]. Accordingly, the utility of targeting c-Met in different cancer types is now being evaluated in clinical trials [11].
New therapeutic targets are needed in breast cancer, particularly in patients with triple-negative (TN) breast cancer and the related basal-like (BL) subgroup of breast cancer. Although distinct, BL tumours can be considered an aggressive subgroup of TN cancers, and both are characterised by a lack of oestrogen receptor and c-erbB2 (Her2) expression, limiting systemic treatment options [12,13]. Since their discovery, the literature regarding c-Met and HGF in the breast has grown rapidly, and there is now a need to consolidate the findings from these studies to better understand the relevance of anti-c-Met therapy in breast cancer.
The aim of this review is to explore the roles of HGF/c-Met signalling in breast development, different in vitro and in vivo models of breast cancer, and the various mechanisms of aberrant c-Met signalling identified in breast cancer tissue. We will also outline the anti-c-Met compounds currently being investigated as possible breast cancer treatments.
Structure and function
c-Met is first produced as a 170 kDa precursor that then undergoes proteolytic cleavage, generating a 50 kDa α-subunit and a 145 kDa β-subunit [3,14]. The extracellular α-subunit is attached to the transmembrane β-subunit by a disulphide bond (reviewed in [15]). A Sema domain, a PSI domain (so-called because it is present in plexins, semaphorins and integrins) and four IPT domains (immunoglobulin-like fold shared by plexins and transcription factors) make up the extracellular portion of c-Met. The intracellular aspect contains three further domains: the juxtamembrane region, which is important in downgrading kinase activity following Ser 975 phosphorylation; the catalytic domain that harbours the Y1234 and Y1235 residues; and the multifunctional carboxy-terminal docking site [15].
The only known mammalian agonistic ligand for c-Met is HGF (also known as scatter factor) [16]. As is the case with c-Met, HGF is secreted first as a precursor, which must then be activated by proteases, resulting in the formation of a mature heterodimer composed of an α-chain and a β-chain [17].
When HGF binds to c-Met, the receptor undergoes autophosphorylation of the Y1234 and Y1235 residues in the kinase domain [14]. Subsequently, tyrosine residues in the docking site (Y1349 and Y1356) become phosphorylated, permitting binding of adaptor molecules including growth factor receptor-bound protein 2, growth factor receptor-bound protein 2-associated binder 1 and Shc [14,15]. These molecules facilitate downstream signalling through several pathways, such as the Rac1/Cdc42 pathway, the phosphoinositide 3-kinase/Akt pathway, signal transducer and activator of transcription 3 and the Erk/mitogen-activated protein kinase cascade [15,18]. Together, these pathways regulate cellular proliferation, motility, migration, invasion and tubulogenesis [18].
The only other ligand known to bind c-Met in mammals is decorin, a leucine-rich proteoglycan [16]. Decorin has been shown to antagonise c-Met signalling by promoting intracellular degradation of the receptor, resulting in suppression of c-Met-mediated cell migration and growth [16].
In common with other RTKs, c-Met is regulated by the ubiquitin ligase, Cbl [19,20]. Following c-Met activation, phosphorylation of the Y1003 residue in the juxtamembrane region recruits Cbl to c-Met, permitting polyubiquitination and degradation of the receptor [19,20]. Although c-Met internalisation is part of the process of signal attenuation, trafficking of the receptor within endosomes, under the control of protein kinase C, results in sustained signalling and is necessary for HGF-mediated migration [21-23] (Figure 1).
Figure 1.
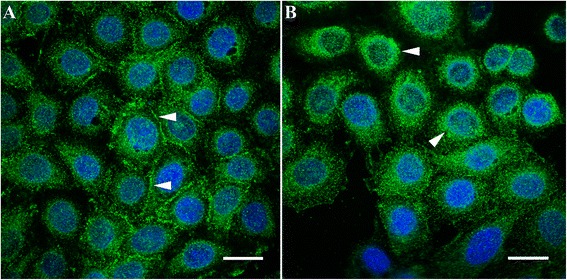
Trafficking of c-Met in MDA-MB-468 cells. (A) In resting cells there is prominent membrane expression of the receptor (white arrowheads). (B) Following hepatocyte growth factor stimulation there is internalisation of the receptor and a predominantly perinuclear, granular pattern of staining (white arrowheads), consistent with the presence of c-Met within endosomes. Immunofluorescence (green, c-Met; blue, nuclei); ×63 objective under oil immersion. Scale bars represent 20 μm.
Hepatocyte growth factor/c-Met signalling in breast development
The relationship between HGF and c-Met during development is complex and dynamic. In a study of mouse embryos, Andermarcher and colleagues described a shift in expression of HGF and c-Met from gastrulation to early organogenesis [24]. It was noted that while HGF and c-Met were coexpressed in endodermal and mesodermal cells during gastrulation, the onset of organogenesis coincided with HGF being localised to the mesenchyme and c-Met to the epithelium and endothelium [24]. The authors suggested that this change from an autocrine to a paracrine relationship may reflect the different roles of the pathway at different stages of development [24].
The HGF/c-Met pathway may also have distinct roles in the different compartments of the mammary epithelium. Exposure of luminal epithelial and myoepithelial cells to HGF evokes contrasting effects in the two cell types [25]. The growth rate of luminal cells increased fivefold to ninefold with HGF compared with controls, and no morphological changes were seen. In contrast, HGF had no effect on myoepithelial cell growth but did induce extensive branch formation [25]. RT-PCR analysis showed c-Met expression was higher in luminal cells than in myoepithelial cells, which may explain these differences. The authors of this study considered the developmental relevance of this arrangement, and hypothesised that the myoepithelial cells lay down the ductal framework that the luminal cells proliferate and migrate along, thus populating and extending the ductal system [25].
Other studies utilising primary murine mammary epithelial cells in in vivo models have further emphasised the importance of the HGF/c-Met pathway in mammary development [26,27]. Overexpression of HGF in primary murine mammary epithelial cells and subsequent transplantation of these cells into the cleared mammary fat pads of mice resulted in a marked increase in ductal branches/bifurcations, along with an increase in the size and number of ductal end buds [26,27]. Importantly, immunohistochemistry revealed an increase in basal/myoepithelial marker expression (smooth muscle actin, cytokeratin 14 and p63) and a reduction in luminal marker expression (cytokeratin 18 and oestrogen receptor) compared with control mice [27]. This finding led the authors to suggest that c-Met signalling directs progenitor cells towards a basal phenotype over luminal differentiation [27], which is reflected in the pattern of c-Met staining seen in different breast cancer subtypes (as discussed later).
Aberrant c-Met signalling in breast cancer
A broad range of mechanisms may result in aberrant c-Met signalling, including activating gene mutations, gene amplification, protein overexpression, increased ligand-dependent paracrine stimulation and the acquisition of autocrine signalling [28] (Table 1). In breast cancer, the majority of studies have looked at the significance of protein overexpression of c-Met and the relationship between levels of the receptor and prognostic factors/survival.
Table 1.
Mechanisms of aberrant c-Met signalling in invasive breast cancer
| Mechanism | Frequency/prognostic significance in breast cancer | Reference |
|---|---|---|
| Gene mutation | MET mutations are uncommon; HGF promoter region mutations occur in 15 to 51% of breast cancers | [30,31] |
| Gene amplification | MET amplification is uncommon, occurring in 0 to 8% of breast cancers; MET copy number is positively correlated with TN tumours | [32,33] |
| Patients with trastuzumab-treated Her2-positive metastatic breast cancer show MET amplification in 27.7% of cases and HGF amplification in 39.3% of cases; patients with MET-amplified Her2-positive tumours have a shorter time to progression | [34] | |
| Autocrine signalling | HGF and MET mRNA detected in tumour cells in all breast cancers analysed, with strongest positivity at the advancing edge of the tumour | [35] |
| On IHC, autocrine pattern of staining seen in 46.6% of tumours | [37] | |
| Paracrine signalling | On IHC, paracrine pattern seen in 59.1% of tumours; paracrine signalling is associated with a worse outcome when c-Met staining is more intense at the tumour front | [68] |
| C-Met activity (phosphorylation) | Using RPPA, 47.9% of tumours showed high phospho-c-Met expression; inconsistent relationship with molecular subtype; high phospho-c-Met associated with an increased risk of tumour recurrence | [43,44] |
Frequency and prognostic significance of the different mechanisms of aberrant c-Met signalling in invasive breast cancer, identified in studies using human tissue samples. HGF, hepatocyte growth factor, IHC, immunohistochemistry, RPPA, reverse-phase protein arrays; TN, triple negative.
Gene mutation
Following the discovery of mutations in the tyrosine kinase domain of MET in hereditary and sporadic papillary renal cell carcinomas [4], MET mutations have also been found in up to 30% of cancers of unknown primary origin [29]. These mutations include those in the SEMA domain and the juxtamembrane domain and an activating mutation in the tyrosine kinase domain [29]. However, few studies have assessed the frequency of MET mutations in primary breast cancer. In a small study comprised of 11 patients with breast cancer (including six patients that showed loss of heterozygosity in the region of the MET gene), no mutations in the tyrosine kinase domain of the MET were identified [30], suggesting that this is not a common event in breast cancer. In contrast, a mutation in the HGF promoter region referred to as the deoxyadenosine tract element appears to be a frequent event, having been identified in 15% of European breast cancer patients and over 50% of African Americans with breast cancer [31]. A truncation mutation in the deoxyadenosine tract element activates the HGF promoter in breast cancer cells, leading to the formation of an HGF/c-Met autocrine loop [31].
Gene amplification
Amplification of the MET gene (located on chromosome 7), like mutation, is unusual in invasive breast cancer: in a study of 155 patients, Carracedo and colleagues did not identify MET amplification at all (although 22% of tumours showed low-grade polysomy) [32]. Elsewhere, in a much larger study, Gonzalez-Angulo and colleagues found increased copy numbers of MET in a minority of cases (82 out of 971 tumours studied) [33]. Although a high copy number of MET was not an independent predictor of recurrence-free survival, these workers did note lower recurrence-free survival rates in the MET-amplified group on univariate analysis [33]. Moreover, there was a positive correlation between MET copy number and TN status [33].
Amplification of MET may also be important in other molecular subtypes of breast cancer: in a study of 130 Her2-positive breast cancers, both MET and HGF amplification were associated with trastuzumab failure and patients with MET amplified tumours had a shorter time to progression [34].
Autocrine/paracrine signalling and c-Met activation
Several lines of evidence suggest that HGF-dependent c-Met signalling (both paracrine and autocrine) is an important mediator of breast cancer progression [35-38]. HGF and c-Met are frequently coexpressed in invasive breast cancers: c-Met in epithelial cells and HGF in epithelial cells (autocrine pattern) and/or stromal cells (paracrine pattern) [35-37]. HGF/c-Met coexpression is often strongest at the infiltrative margins of tumours [35,37]. Moreover, when tumours demonstrate this strong coexpression at the advancing edge, there is a significant correlation with high tumour grade, an increased proliferation index and reduced survival, compared with cancers that are negative for coexpression [37]. Expression of matriptase (an activator of HGF) is positively correlated with both c-Met and HGF in invasive breast cancer, and high levels of c-Met and matriptase are associated with reduced 30-year survival at univariate analysis [38].
While analysis of HGF/c-Met coexpression provides useful insight into the role of ligand-dependent c-Met activation, measuring c-Met phosphorylation would also take into account ligand-independent c-Met activation, theoretically giving a more global readout of c-Met signalling. Unfortunately, the detection of c-Met phosphorylation in human formalin-fixed, paraffin-embedded samples is complicated by the poor stability of phospho-epitopes in general [39,40] and by the limited sensitivity and specificity of phospho-specific antibodies [41,42]. It is therefore perhaps not surprising that few studies have investigated the prognostic significance of c-Met phosphorylation in invasive breast cancer. Two studies that have managed to identify phospho-c-Met in breast cancer did so using reverse-phase protein analysis, with contrasting results [43,44]. In a study of 107 primary breast cancer patients, Hochgräfe and colleagues found higher phospho-c-Met expression (pY1234/5) in TN tumours [43], whereas Raghav and colleagues found no difference in phospho-c-Met expression (pY1235) between different molecular subtypes but did find higher recurrence rates in patients whose tumours showed high levels of pY1235 [44].
Protein overexpression
c-Met protein overexpression, as assessed by immunohistochemistry (IHC)/immunofluorescence, is now generally accepted to be a poor prognostic factor in invasive breast cancer [6,38,45-51] (Table 2). Exactly what constitutes overexpression is less clear, and several different scoring methods and cutoff points were utilised in these studies, resulting in a variable proportion of cases being classified as c-Met-positive (15 to 63%) [6,38,45-49,51]. Of course, the characteristics of the study population and the choice of antibody for the IHC assay are additional variables that may influence the proportion of c-Met-positive cases. Two particular issues related to c-Met IHC that deserve further comment are the reproducibility of staining from commercially available antibodies and the domain of the receptor targeted by the antibody.
Table 2.
Relationship between c-Met expression and prognostic factors
| Prognostic parameter | Relationship | Reference |
|---|---|---|
| Age at presentation | No established relationship | [6,46,49,50] |
| Tumour size | Most studies have found no relationship | [36,46,49,53] |
| We found inverse correlation between c-Met expression and tumour size | [50] | |
| Lymph node status | Most studies show no relationship | [6,49,53] |
| We found higher c-Met expression in node-negative tumours | [50] | |
| Tumour grade | Mixed; some studies show no association | [6,36,48] |
| Some studies show increased c-Met expression in high-grade tumours | [46,49] | |
| One study showed increased c-Met in low-grade tumours | [53] | |
| Histological subtype | Increased c-Met in tubular carcinoma, decreased in lobular carcinoma | [50] |
| Molecular subtype | Increased c-Met in basal-like breast cancer | [50,51,55,82] |
| Survival | Increased c-Met associated with reduced survival | [6,38,45-51] |
In an analysis of six different commercial c-Met antibodies (five of which recognised the protein at western blot), Pozner-Moulis and colleagues found a low correlation between c-Met expression on different sections of the same tissue microarrays stained with the same antibody [52]. Moreover, when different lots of the same monoclonal antibody were applied to the same tumour, marked differences were seen in the staining pattern. These findings suggest that many c-Met antibodies may not be providing a reproducible evaluation of c-Met expression [52]. Several studies have also commented on the importance of selecting c-Met antibodies that target the intracellular domain, since expression of this part of the receptor appears to have more prognostic relevance than those directed against the extracellular region [38,47,52]. Thus, there is now a need to develop standardised guidelines for the methodology (such as the use of validated anti-c-Met antibodies) and interpretation of c-Met IHC.
Most studies have found no association between c-Met expression and established prognostic factors, such as age at presentation, tumour size and lymph node status [6,36,46,49,53], perhaps explaining why c-Met expression retains prognostic power after correcting for these factors on multivariate analysis [38,45-48,50]. Interestingly, a large recent study utilising breast cancers from 924 patients did find a positive correlation between c-Met expression and both increasing tumour size and nodal involvement; c-Met-overexpressing tumours were associated with worse survival, but not on multivariate analysis [51]. With regard to tumour grade there is no consensus, with some studies finding no association [6,36,48], other studies finding increased c-Met expression in high-grade tumours [46,49] and one study identifying more frequent immunoreactivity in grade 1 tumours compared with grade 3 cancers (75% versus 43.8%, respectively) [53].
In our own analysis we identified significantly different levels of c-Met expression in two special histological subtypes of breast cancer: levels were lower in the E-Cadherin-negative invasive lobular carcinomas and higher in tubular carcinomas [50] (Figure 2), a well-differentiated tumour subtype characterised by angulated tubules [54]. These observations are reminiscent of findings from the aforementioned studies on mammary development, where HGF stimulated tubule formation in murine mammary epithelial cells [26,27]. We also demonstrated, for the first time, that c-Met protein expression was independently associated with BL breast cancer (Figure 3), a finding supported by the results from the most recent IHC analyses [50,51]. Together these findings indicate that patients with BL cancer should be included in clinical trials of anti-c-Met therapy.
Figure 2.
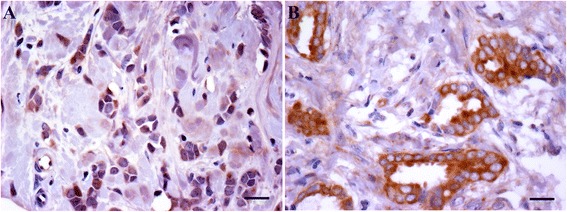
c-Met expression varies between histological subtypes of breast cancer. (A) Invasive lobular carcinoma characterised by discohesive tumour cells with low c-Met expression. (B) Tubular carcinoma with cohesive tumour cells arranged in angulated tubules with strong expression of c-Met. Immunohistochemistry, ×40 objective. Scale bars represent 20 μm.
Figure 3.
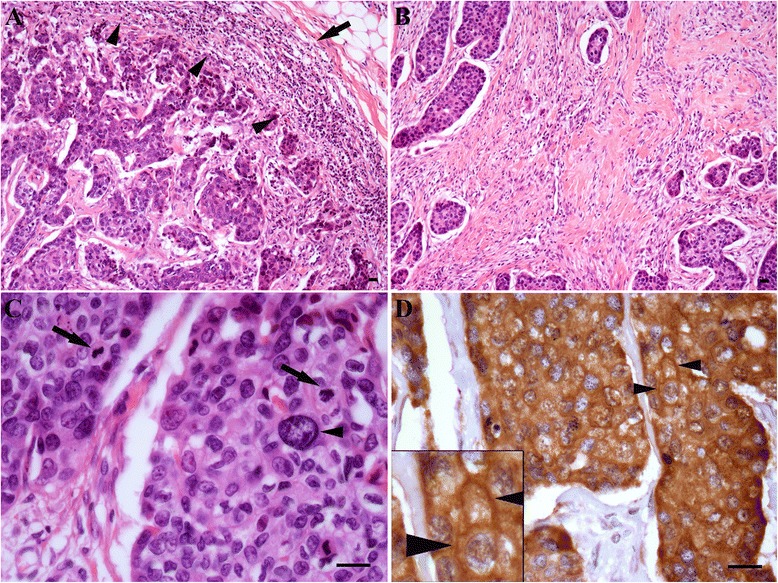
c-Met expression in basal-like breast cancer. Characteristic features of basal-like breast cancer (images are from the same tumour). (A) Circumscribed tumour front (arrowheads) and associated chronic inflammatory cell infiltrate (arrow). (B) Tumour fibrosis. (C) High-grade cytology, with nuclear enlargement and pleomorphism (arrowheads), along with prominent mitotic figures (arrows). Haematoxylin and eosin; (A) and (B) × 10 objective, (C) × 40 objective. Scale bars represent 20 μm. (D) High cytoplasmic and membranous (arrowheads) expression of c-Met. Immunohistochemistry, ×40 objective. Scale bar represents 20 μm. Inset image is at 200% magnification.
Hepatocyte growth factor/c-Met signalling in breast cancer cells
A variety of breast cancer cell lines (BCLs) have been used to study the role of HGF and c-Met in a range of cellular processes implicated in the progression of breast cancer. Although these BCLs include those representative of the luminal, Her2-overexpressing and BL subtypes, MET overexpression at the RNA level and c-Met protein overexpression are more often seen in the BL BCLs [55,56].
Tubulogenesis
The extent of tubule formation is a key component of the grading system in invasive breast cancer [57]. A lack of tubular differentiation is a feature of high-grade tumours, which are associated with a poorer outlook than their low-grade counterparts [57]. Tubule formation has been observed in T47D and MCF7 cells in response to HGF treatment and c-Met expression has been identified at confocal microscopy in T47D cells that bordered luminal structures [58]. However, the relationship seems complex. When similar experiments were performed on colon carcinoma cell lines, low doses of HGF (1 to 10 ng/ml) stimulated tubule formation, but higher doses (up to 100 ng/ml) appeared to abrogate this phenomenon [58].
Migration and invasion
The promigratory and proinvasive effects of HGF have been shown in several BCLs, including MCF7, MCF10.DCIS and MDA-MB-231 [59-62]. Administration of HGF, either in the form of recombinant HGF, conditioned media from HGF-secreting fibroblasts or by way of co-culture with HGF-secreting fibroblasts, has been shown to significantly increase migration and invasion in wound closure and transwell invasion assays [59-62]. In addition, adipose-derived mesenchymal cells isolated from lipoaspirates express variable levels of HGF and co-culture of these cells with MDA-MB-231 cells resulted in increased migration [63]. These studies are supported by work demonstrating that NK4, a variant form of HGF that competitively inhibits HGF binding [64], reduces HGF-mediated c-Met phosphorylation and inhibits HGF-induced scattering and invasion of MCF7 and MDA-MB-231 cells [59].
Numerous studies have sought to uncover the mechanisms through which HGF/c-Met signalling contributes to the migratory and invasive phenotype in breast cancer, particularly focusing on pathways associated with epithelial adhesion [65-67]. E-Cadherin is a key component of adherens junctions (specialised intraepithelial junctions) [68,69], and is regarded by some as a tumour suppressor important in the prevention of cell migration, invasion and metastasis [69]. In MCF7 cells, E-Cadherin and c-Met are co-localised at the cell membrane in regions of cell–cell contact [65,70]. Following treatment with HGF, lysates from these cells show a reduction in E-Cadherin expression, and immunofluorescent studies demonstrate asymmetric accumulation of c-Met and E-Cadherin in the cytosol, ultimately leading to complete internalisation of both proteins after 2 hours [61,70].
In addition to favouring epithelial dissociation via E-Cadherin downregulation/internalisation, there is some evidence that HGF/c-Met signalling contributes to breast cancer progression by promoting cancer cell adhesion to components of the extracellular matrix [66]. HGF treatment increased adhesion of MtLn3 rat mammary adenocarcinoma cells to laminin, type 1 collagen and fibronectin, compared with control cells [66]. Furthermore, treatment with HGF was associated with lamellipodia formation, focal adhesion kinase phosphorylation and focal adhesion kinase expression at focal contacts, suggesting that c-Met and focal adhesion kinase cooperate to promote cancer cell/substrate adhesion [66].
Proteolytic pathway regulation is another mechanism influenced by HGF/c-Met signalling in the in vitro setting [60]. Conditioned media from HGF-secreting fibroblasts and recombinant HGF treatment resulted in increased secretion of both urokinase-type plasminogen activator and its receptor (urokinase-type plasminogen activator receptor) by different ductal carcinoma in situ cell lines (MCF10.DCIS cells and SUM102 cells). Increased collagen IV degradation was also demonstrated, along with increased numbers of invasive outgrowths in three-dimensional cultures of ductal carcinoma in situ cells when in the presence of HGF [60]. Together, these findings implicate HGF-secreting fibroblasts in the progression of ductal carcinoma in situ to invasive cancer [60].
Cell survival
HGF/c-Met signalling has been associated with both pro-apoptotic and anti-apoptotic effects [71]. Using the murine hepatocellular carcinoma cell line Hepa1-6, Wang and colleagues established that c-Met and the death receptor FAS formed a complex; they proposed a model in which c-Met sequesters FAS, thus preventing ligand-independent activation (due to clustering) and FAS ligand/FAS binding, resulting in cell survival [71]. In this model, high levels of HGF (or FAS ligand) would cause dissociation of the c-Met/FAS complex, leaving the cells vulnerable to FAS-mediated apoptosis. Such a model would explain the paradoxical effects of HGF/c-Met on cell survival [71].
A similar mechanism may also exist in breast cancer cells, where treatment of preneoplastic MCF-10AT breast epithelial cells with anti-FAS (an activator of FAS signalling) induced c-Met/FAS complex dissociation and apoptosis [72]. It has also been shown that HGF protects MDA-MB-453 breast cancer cells from adriamycin-induced apoptosis [73]. Preincubation of these cells with HGF blocked adriamycin-mediated FAS ligand upregulation and inhibited the reduction in levels of the anti-apoptotic protein Bcl-XL [73,74].
Cross-talk with other receptor tyrosine kinases
It is widely appreciated that c-Met can cross-talk with a variety of other cell surface receptors (reviewed in [75]). In breast cancer, cross-talk between c-Met and members of the c-erbB family in particular has received considerable interest. HGF has been shown to trans-activate epidermal growth factor receptor (EGFR) in PyVmT mouse mammary carcinoma cells [76]. Moreover, the EGFR inhibitor gefitinib blocked HGF-mediated proliferation in PyVmT cells, migration in PyVmT cells and NMuMG cells, and invasion in PyVmT cells, NMuMG cells and MDA-MB-231 cells [76]. The authors went on to show that gefitinib effects c-Met activation in an EGFR-dependent process (as opposed to directly targeting c-Met) by finding no effect on c-Met activation when EGFR-null/c-Met expressing haematopoietic 32D cells were treated with the inhibitor [76].
Similarly, another member of the c-erbB family – Her2 – has been noted to cross-talk with c-Met in HCC1954 breast cancer cells, which overexpress c-Met and Her2 [77]. In HCC1954 cells, knockdown of MET resulted in increased Her2 phosphorylation and, conversely, knockdown of Her2 was associated with an increase in c-Met activity [77].
The relationship between c-Met and other RTKs has important implications for the development of resistance to anti-RTK therapies already in clinical use – now a significant problem in breast cancer treatment [77]. Indeed, in the EGFR tyrosine kinase inhibitor-resistant cell line SUM229, c-Met is phosphorylated and thought to stimulate EGFR phosphorylation in the presence of EGFR inhibitors in a Src-mediated process [78]. Likewise, treatment of the Her2-overexpressing BT-474 and SKBR3 cells with trastuzumab upregulated c-Met protein expression in just 48 hours [79]. HGF-mediated c-Met phosphorylation in these cells opposed trastuzumab-mediated growth inhibition by abrogating p27 induction [79]. c-Met therefore plays an important role in breast cancer cell function and signalling by virtue of its ability to interact with other RTKs.
In vivo models of HGF/c-Met-mediated tumour formation
The in vivo effects of aberrant HGF/c-Met signalling have been explored in different mouse models [80-83]. Mice harbouring the whey acidic protein WAP-HGF transgenic construct show elevated HGF expression in mammary epithelium, compared with wild-type mice, and go on to develop mammary tumours characterised by a high Ki67 proliferation index, a reduced progesterone receptor immunoreactivity and areas of squamous differentiation (a feature of BL breast cancers) [80,84].
Squamous metaplasia was also detected in a high proportion (65%) of mammary tumours that developed in mice with mutationally activated MET [82]. The majority of these tumours also expressed the basal cytokeratin, cytokeratin 5 [82]. Elsewhere, Ponzo and colleagues studied transgenic mice that express oncogenic MET in the mammary epithelium, under the control of the murine mammary tumour virus promoter [81]. About one-half of the tumours that developed in these mice showed variable histological patterns which included BL features [85] such as squamous/spindle cell differentiation, high nuclear grade, necrosis and lymphocytic infiltration [81]. These tumours also expressed cytokeratin 5/6 and cytokeratin 14 (another basal cytokeratin) on IHC [81]. Interestingly, in a subsequent study these workers found that loss of TP53, in addition to oncogenic MET expression, was associated with the formation of tumours with a claudin-low profile, a recently described subgroup of TN tumours that is distinct from the BL subtype [83,86].
Anti-c-Met therapy in invasive breast cancer
There are various strategies for antagonising HGF/c-Met signalling: antibodies can be directed against c-Met; HGF itself can be targeted with antibodies; and the catalytic function of c-Met can be opposed with tyrosine kinase inhibitors, which account for the majority of anti-c-Met compounds under investigation [87]. Four therapies currently in phase II clinical trials for the treatment of advanced TN breast cancer are tivantinib (also known as ARQ197) [ClinicalTrials.gov:NCT 01575522], cabozantinib (alternatively known as XL184) [ClinicalTrials.gov:NCT 01738438], MetMab (onartuzumab) [ClinicalTrials.gov:NCT 01186991] and foretinib (XL880) [ClinicalTrials.gov:NCT 01147484] [11] (Table 3).
Table 3.
Anti-c-Met therapies currently under investigation in clinical trials for breast cancer [11]
| Compound | Target/mechanism of action | ClinicalTrials.gov identifier |
|---|---|---|
| Tivantinib (ARQ197) | c-Met/non-ATP kinase inhibitor | NCT 01575522 |
| Cabozantinib (XL184) | c-Met and VEGFR, along with RET, KIT, AXL/kinase inhibitor | NCT 01738438 |
| Foretinib (XL880) | c-Met and VEGFR, along with KIT, Flt-3, PDGFR, Tie-2/kinase inhibitor | NCT 01147484 |
| MetMab (onartuzumab) | c-Met/anti-c-Met antibody | NCT 01186991 |
PDGFR, platelet-derived growth factor receptor; VEGFR, vascular endothelial growth factor receptor.
As well as breast cancer, the anti-c-Met monoclonal antibody MetMab is also being trialled in lung cancer and colon cancer [88]. Unlike many other anti-c-Met antibodies, MetMab is monovalent – therefore it does not promote dimerisation when it binds to c-Met, thus avoiding the agonistic effects associated with similar therapies [88].
Tivantinib belongs to the c-Met tyrosine kinase inhibitor class of c-Met antagonists, and is a non-ATP competitive inhibitor of the receptor [89,90]. A phase I trial of tivantinib in 51 patients with solid tumours (including two patients with breast cancer) found the inhibitor to be well tolerated, with fatigue, nausea and vomiting being the most common adverse effects [90]. Furthermore, when pretreatment and on-treatment tumour biopsies were compared, there was a reduction in total and phosphorylated c-Met expression in the on-treatment samples, suggesting that tivantinib inhibited intra-tumoural c-Met signalling [90].
Cabozantinib is another small molecule inhibitor of c-Met, which targets a range of tyrosine kinases, including RET, KIT and AXL, but particularly c-Met and VEGFR2 [91]. Cabozantinib not only inhibits c-Met phosphorylation in vivo, but also promotes tumour hypoxia and cell death and inhibits the growth of MDA-MB-231 tumours in a dose-dependent manner [91]. It has been suggested that the ability of cabozantinib to inhibit both c-Met and vascular endothelial growth factor receptor 2 may actually counter the c-Met-dependent resistance noted when only the vascular endothelial growth factor pathway is targeted [91].
Foretinib is a small molecule kinase inhibitor that principally targets c-Met and vascular endothelial growth factor receptor [92]. Foretinib inhibits HGF-induced c-Met phosphorylation, inhibits tumour cell growth in hypoxic and normoxic conditions, and has been shown to reduce tumour cell burden in an in vivo model of lung cancer [92]. A phase I study in patients with a wide variety of solid organ cancers (including one breast cancer patient) found the inhibitor to be safe and noted a partial response in 7.5% of patients and stable disease in a further 55% of patients [93].
Conclusion
Much progress has been made in our understanding of c-Met/HGF signalling in recent years, and there is now convincing in vitro and in vivo evidence that this is an important pathway in mammary development and cancer progression. Clinical studies have confirmed the prognostic significance of c-Met expression in breast cancer and highlight the potential of c-Met inhibitors as a novel form of targeted therapy. The possible role of c-Met signalling in promoting BL breast cancer is noteworthy, and merits further investigation in the experimental and clinical trial settings. The outcomes of ongoing and future clinical trials of anti-c-Met therapy will be eagerly anticipated, but issues such as receptor cross-talk and resistance may need to be addressed if treatment efficacy is to be maximised. It is also important to stratify patients appropriately, and the development of standardised prognostic/predictive assays will be crucial in identifying those subgroups of patients most likely to benefit from anti-c-Met therapy.
Acknowledgements
The authors apologise that they have been unable to cite all relevant studies, due to space constraints. CMH-Y is funded by a Cancer Research UK Clinical Research Fellowship. JLJ is funded by the Breast Cancer Campaign Tissue Bank.
Abbreviations
- BCL
Breast cancer cell line
- BL
Basal-like
- EGFR
Epidermal growth factor receptor
- HGF
Hepatocyte growth factor
- IHC
Immunohistochemistry
- RTK
Receptor tyrosine kinase
- TN
Triple negative
Footnotes
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Colan M Ho-Yen, Email: Colan.HoYen@stgeorges.nhs.uk.
J Louise Jones, Email: l.j.jones@qmul.ac.uk.
Stephanie Kermorgant, Email: s.kermorgant@qmul.ac.uk.
References
- 1.Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122:1450–9. doi: 10.1016/0006-291X(84)91253-1. [DOI] [PubMed] [Google Scholar]
- 3.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–4. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 5.Walker F, Kermorgant S, Daraï E, Madelenat P, Cremieux AC, Hénin D, et al. Hepatocyte growth factor and c-Met in cervical intraepithelial neoplasia: overexpression of proteins associated with oncogenic human papillomavirus and human immunodeficiency virus. Clin Cancer Res. 2003;9:273–84. [PubMed] [Google Scholar]
- 6.Lengyel E, Prechtel D, Resau JH, Gauger K, Welk A, Lindemann K, et al. C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer. 2005;113:678–82. doi: 10.1002/ijc.20598. [DOI] [PubMed] [Google Scholar]
- 7.Kim CH, Kim J, Kahng H, Choi EC. Change of E-cadherin by hepatocyte growth factor and effects on the prognosis of hypopharyngeal carcinoma. Ann Surg Oncol. 2007;14:1565–74. doi: 10.1245/s10434-006-9320-5. [DOI] [PubMed] [Google Scholar]
- 8.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 9.De Oliveira AT, Matos D, Logullo AF, DA Silva SR, Neto RA, Filho AL, et al. MET Is highly expressed in advanced stages of colorectal cancer and indicates worse prognosis and mortality. Anticancer Res. 2009;29:4807–11. [PubMed] [Google Scholar]
- 10.Li Y, Chen CQ, He YL, Cai SR, Yang DJ, He WL, et al. Abnormal expression of E-cadherin in tumor cells is associated with poor prognosis of gastric carcinoma. J Surg Oncol. 2012;106:304–10. doi: 10.1002/jso.23008. [DOI] [PubMed] [Google Scholar]
- 11.ClinicalTrials.gov. US National Institutes of Health, Bethesda, MD. http://www.clinicaltrials.gov. Accessed Jun 2014.
- 12.Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14:1368–76. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 13.Ho-Yen C, Bowen RL, Jones JL. Characterization of basal-like breast cancer: an update. Diagn Histopathol. 2012;18:104–11. doi: 10.1016/j.mpdhp.2011.12.002. [DOI] [Google Scholar]
- 14.Hanna JA, Bordeaux J, Rimm DL, Agarwal S. The function, proteolytic processing, and histopathology of Met in cancer. Adv Cancer Res. 2009;103:1–23. doi: 10.1016/S0065-230X(09)03001-2. [DOI] [PubMed] [Google Scholar]
- 15.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–48. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 16.Goldoni S, Humphries A, Nyström A, Sattar S, Owens RT, McQuillan DJ, et al. Decorin is a novel antagonistic ligand of the Met receptor. J Cell Biol. 2009;185:743–54. doi: 10.1083/jcb.200901129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 18.Gherardi E, Birchmeier W, Birchmeier C, Vande WG. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 19.Abella JV, Peschard P, Naujokas MA, Lin T, Saucier C, Urbé S, et al. Met/hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol Cell Biol. 2005;25:9632–45. doi: 10.1128/MCB.25.21.9632-9645.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peschard P, Park M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene. 2007;26:1276–85. doi: 10.1038/sj.onc.1210201. [DOI] [PubMed] [Google Scholar]
- 21.Kermorgant S, Zicha D, Parker PJ. PKC controls HGF-dependent c-Met traffic, signalling and cell migration. EMBO J. 2004;23:3721–34. doi: 10.1038/sj.emboj.7600396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kermorgant S, Parker PJ. Receptor trafficking controls weak signal delivery: a strategy used by c-Met for STAT3 nuclear accumulation. J Cell Biol. 2008;182:855–63. doi: 10.1083/jcb.200806076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrow-McGee R, Kermorgant S. Met endosomal signalling: in the right place, at the right time. Int J Biochem Cell Biol. 2014;49:69–74. doi: 10.1016/j.biocel.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Andermarcher E, Surani MA, Gherardi E. Co-expression of the HGF/SF and c-met genes during early mouse embryogenesis precedes reciprocal expression in adjacent tissues during organogenesis. Dev Genet. 1996;18:254–66. doi: 10.1002/(SICI)1520-6408(1996)18:3<254::AID-DVG6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 25.Niranjan B, Buluwela L, Yant J, Perusinghe N, Atherton A, Phippard D, et al. HGF/SF: a potent cytokine for mammary growth, morphogenesis and development. Development. 1995;121:2897–908. doi: 10.1242/dev.121.9.2897. [DOI] [PubMed] [Google Scholar]
- 26.Yant J, Buluwela L, Niranjan B, Gusterson B, Kamalati T. In vivo effects of hepatocyte growth factor/scatter factor on mouse mammary gland development. Exp Cell Res. 1998;241:476–81. doi: 10.1006/excr.1998.4028. [DOI] [PubMed] [Google Scholar]
- 27.Gastaldi S, Sassi F, Accornero P, Torti D, Galimi F, Migliardi G, et al. Met signaling regulates growth, repopulating potential and basal cell-fate commitment of mammary luminal progenitors: implications for basal-like breast cancer. Oncogene. 2013;32:1428–40. doi: 10.1038/onc.2012.154. [DOI] [PubMed] [Google Scholar]
- 28.Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3:S21–35. doi: 10.1177/1758834011422557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stella GM, Benvenuti S, Gramaglia D, Scarpa A, Tomezzoli A, Cassoni P, et al. MET mutations in cancers of unknown primary origin (CUPs) Hum Mutat. 2011;32:44–50. doi: 10.1002/humu.21374. [DOI] [PubMed] [Google Scholar]
- 30.Bièche I, Champème MH, Lidereau R. Infrequent mutations of the MET gene in sporadic breast tumours. Int J Cancer. 1999;82:908–10. doi: 10.1002/(SICI)1097-0215(19990909)82:6<908::AID-IJC22>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 31.Ma J, DeFrances MC, Zou C, Johnson C, Ferrell R, Zarnegar R. Somatic mutation and functional polymorphism of a novel regulatory element in the HGF gene promoter causes its aberrant expression in human breast cancer. J Clin Invest. 2009;119:478–91. doi: 10.1172/JCI36640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carracedo A, Egervari K, Salido M, Rojo F, Corominas JM, Arumi M, et al. FISH and immunohistochemical status of the hepatocyte growth factor receptor (c-Met) in 184 invasive breast tumors. Breast Cancer Res. 2009;11:402. doi: 10.1186/bcr2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez-Angulo AM, Chen H, Karuturi MS, Chavez-MacGregor M, Tsavachidis S, Meric-Bernstam F, et al. Frequency of mesenchymal–epithelial transition factor gene (MET) and the catalytic subunit of phosphoinositide-3-kinase (PIK3CA) copy number elevation and correlation with outcome in patients with early stage breast cancer. Cancer. 2013;119:7–15. doi: 10.1002/cncr.27608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minuti G, Cappuzzo F, Duchnowska R, Jassem J, Fabi A, O’Brien T, et al. Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. Br J Cancer. 2012;107:793–9. doi: 10.1038/bjc.2012.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tuck AB, Park M, Sterns EE, Boag A, Elliott BE. Coexpression of hepatocyte growth factor and receptor (Met) in human breast carcinoma. Am J Pathol. 1996;148:225–32. [PMC free article] [PubMed] [Google Scholar]
- 36.Jin L, Fuchs A, Schnitt SJ, Yao Y, Joseph A, Lamszus K, et al. Expression of scatter factor and c-met receptor in benign and malignant breast tissue. Cancer. 1997;79:749–60. doi: 10.1002/(SICI)1097-0142(19970215)79:4<749::AID-CNCR12>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 37.Edakuni G, Sasatomi E, Satoh T, Tokunaga O, Miyazaki K. Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol Int. 2001;51:172–8. doi: 10.1046/j.1440-1827.2001.01182.x. [DOI] [PubMed] [Google Scholar]
- 38.Kang JY, Dolled-Filhart M, Ocal IT, Singh B, Lin CY, Dickson RB, et al. Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res. 2003;63:1101–5. [PubMed] [Google Scholar]
- 39.Baker AF, Dragovich T, Ihle NT, Williams R, Fenoglio-Preiser C, Powis G. Stability of phosphoprotein as a biological marker of tumor signaling. Clin Cancer Res. 2005;11:4338–40. doi: 10.1158/1078-0432.CCR-05-0422. [DOI] [PubMed] [Google Scholar]
- 40.Dua R, Zhang J, Parry G, Penuel E. Detection of hepatocyte growth factor (HGF) ligand-c-MET receptor activation in formalin-fixed paraffin embedded specimens by a novel proximity assay. PLoS One. 2011;6:e15932. doi: 10.1371/journal.pone.0015932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarvius M, Paulsson J, Weibrecht I, Leuchowius KJ, Andersson AC, Wählby C, et al. In situ detection of phosphorylated platelet-derived growth factor receptor beta using a generalized proximity ligation method. Mol Cell Proteomics. 2007;6:1500–9. doi: 10.1074/mcp.M700166-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Blokzijl A, Friedman M, Pontén F, Landegren U. Profiling protein expression and interactions: proximity ligation as a tool for personalized medicine. J Intern Med. 2010;268:232–45. doi: 10.1111/j.1365-2796.2010.02256.x. [DOI] [PubMed] [Google Scholar]
- 43.Hochgräfe F, Zhang L, O’Toole SA, Browne BC, Pinese M, Porta Cubas A, et al. Tyrosine phosphorylation profiling reveals the signaling network characteristics of Basal breast cancer cells. Cancer Res. 2010;70:9391–401. doi: 10.1158/0008-5472.CAN-10-0911. [DOI] [PubMed] [Google Scholar]
- 44.Raghav KP, Wang W, Liu S, Chavez-MacGregor M, Meng X, Hortobagyi GN, et al. cMET and phospho-cMET protein levels in breast cancers and survival outcomes. Clin Cancer Res. 2012;18:2269–77. doi: 10.1158/1078-0432.CCR-11-2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghoussoub RA, Dillon DA, D’Aquila T, Rimm EB, Fearon ER, Rimm DL. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;82:1513–20. doi: 10.1002/(SICI)1097-0142(19980415)82:8<1513::AID-CNCR13>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 46.Camp RL, Rimm EB, Rimm DL. Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma. Cancer. 1999;86:2259–65. doi: 10.1002/(SICI)1097-0142(19991201)86:11<2259::AID-CNCR13>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 47.Tolgay Ocal I, Dolled-Filhart M, D’Aquila TG, Camp RL, Rimm DL. Tissue microarray-based studies of patients with lymph node negative breast carcinoma show that met expression is associated with worse outcome but is not correlated with epidermal growth factor family receptors. Cancer. 2003;97:1841–8. doi: 10.1002/cncr.11335. [DOI] [PubMed] [Google Scholar]
- 48.Chen HH, Su WC, Lin PW, Guo HR, Lee WY. Hypoxia-inducible factor-1alpha correlates with MET and metastasis in node-negative breast cancer. Breast Cancer Res Treat. 2007;103:167–75. doi: 10.1007/s10549-006-9360-3. [DOI] [PubMed] [Google Scholar]
- 49.Zagouri F, Bago-Horvath Z, Rössler F, Brandstetter A, Bartsch R, Papadimitriou CA, et al. High MET expression is an adverse prognostic factor in patients with triple-negative breast cancer. Br J Cancer. 2013;108:1100–5. doi: 10.1038/bjc.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho-Yen CM, Green AR, Rakha EA, Brentnall AR, Ellis IO, Kermorgant S, et al. C-Met in invasive breast cancer: is there a relationship with the basal-like subtype? Cancer. 2014;120:163–71. doi: 10.1002/cncr.28386. [DOI] [PubMed] [Google Scholar]
- 51.Kim YJ, Choi JS, Seo J, Song JY, Lee SE, Kwon MJ, et al. MET is a potential target for use in combination therapy with EGFR inhibition in triple-negative/basal-like breast cancer. Int J Cancer. 2014;134:2424–36. doi: 10.1002/ijc.28566. [DOI] [PubMed] [Google Scholar]
- 52.Pozner-Moulis S, Cregger M, Camp RL, Rimm DL. Antibody validation by quantitative analysis of protein expression using expression of Met in breast cancer as a model. Lab Invest. 2007;87:251–60. doi: 10.1038/labinvest.3700515. [DOI] [PubMed] [Google Scholar]
- 53.Nakopoulou L, Gakiopoulou H, Keramopoulos A, Giannopoulou I, Athanassiadou P, Mavrommatis J, et al. c-met tyrosine kinase receptor expression is associated with abnormal beta-catenin expression and favourable prognostic factors in invasive breast carcinoma. Histopathology. 2000;36:313–25. doi: 10.1046/j.1365-2559.2000.00847.x. [DOI] [PubMed] [Google Scholar]
- 54.Harris G, Pinder SE, O’Malley FP. Invasive carcinoma: special types. In: O’Malley FP, Pinder SE, editors. Breast pathology. Philadelphia: Elsevier; 2006. pp. 201–23. [Google Scholar]
- 55.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adélaïde J, Cervera N, et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25:2273–84. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 56.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991;19:403–10. doi: 10.1111/j.1365-2559.1991.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 58.Tsarfaty I, Resau JH, Rulong S, Keydar I, Faletto DL, Vande Woude GF. The met proto-oncogene receptor and lumen formation. Science. 1992;257:1258–61. doi: 10.1126/science.1387731. [DOI] [PubMed] [Google Scholar]
- 59.Hiscox S, Parr C, Nakamura T, Matsumoto K, Mansel RE, Jiang WG. Inhibition of HGF/SF-induced breast cancer cell motility and invasion by the HGF/SF variant, NK4. Breast Cancer Res Treat. 2000;59:245–54. doi: 10.1023/A:1006348317841. [DOI] [PubMed] [Google Scholar]
- 60.Jedeszko C, Victor BC, Podgorski I, Sloane BF. Fibroblast hepatocyte growth factor promotes invasion of human mammary ductal carcinoma in situ. Cancer Res. 2009;69:9148–55. doi: 10.1158/0008-5472.CAN-09-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hung CM, Kuo DH, Chou CH, Su YC, Ho CT, Way TD. Osthole suppresses hepatocyte growth factor (HGF)-induced epithelial–mesenchymal transition via repression of the c-Met/Akt/mTOR pathway in human breast cancer cells. J Agric Food Chem. 2011;59:9683–90. doi: 10.1021/jf2021489. [DOI] [PubMed] [Google Scholar]
- 62.Ayoub NM, Akl MR, Sylvester PW. Combined γ-tocotrienol and Met inhibitor treatment suppresses mammary cancer cell proliferation, epithelial-to-mesenchymal transition and migration. Cell Prolif. 2013;46:538–53. doi: 10.1111/cpr.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eterno V, Zambelli A, Pavesi L, Villani L, Zanini V, Petrolo G, et al. Adipose-derived mesenchymal stem cells (ASCs) may favour breast cancer recurrence via HGF/c-Met signaling. Oncotarget. 2014;5:613–33. doi: 10.18632/oncotarget.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Date K, Matsumoto K, Shimura H, Tanaka M, Nakamura T. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett. 1997;420:1–6. doi: 10.1016/S0014-5793(97)01475-0. [DOI] [PubMed] [Google Scholar]
- 65.Hiscox S, Jiang WG. Association of the HGF/SF receptor, c-met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem Biophys Res Commun. 1999;261:406–11. doi: 10.1006/bbrc.1999.1002. [DOI] [PubMed] [Google Scholar]
- 66.Beviglia L, Kramer RH. HGF induces FAK activation and integrin-mediated adhesion in MTLn3 breast carcinoma cells. Int J Cancer. 1999;83:640–9. doi: 10.1002/(SICI)1097-0215(19991126)83:5<640::AID-IJC13>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 67.Reshetnikova G. Met receptor subcellular localization depends on E-cadherin function. ScientificWorldJournal. 2007;7:2009–11. doi: 10.1100/tsw.2007.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 69.Rodriguez FJ, Lewis-Tuffin LJ, Anastasiadis PZ. E-cadherin’s dark side: possible role in tumor progression. Biochim Biophys Acta. 1826;2012:23–31. doi: 10.1016/j.bbcan.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matteucci E, Ridolfi E, Desiderio MA. Hepatocyte growth factor differently influences Met-E-cadherin phosphorylation and downstream signaling pathway in two models of breast cells. Cell Mol Life Sci. 2006;63:2016–26. doi: 10.1007/s00018-006-6137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, DeFrances MC, Dai Y, Pediaditakis P, Johnson C, Bell A, et al. A mechanism of cell survival: sequestration of Fas by the HGF receptor Met. Mol Cell. 2002;9:411–21. doi: 10.1016/S1097-2765(02)00439-2. [DOI] [PubMed] [Google Scholar]
- 72.Shen K, Novak RF. Fas-signaling and effects on receptor tyrosine kinase signal transduction in human breast epithelial cells. Biochem Biophys Res Commun. 1997;230:89–93. doi: 10.1006/bbrc.1996.5896. [DOI] [PubMed] [Google Scholar]
- 73.Fan S, Wang JA, Yuan RQ, Rockwell S, Andres J, Zlatapolskiy A, et al. Scatter factor protects epithelial and carcinoma cells against apoptosis induced by DNA-damaging agents. Oncogene. 1998;17:131–41. doi: 10.1038/sj.onc.1201943. [DOI] [PubMed] [Google Scholar]
- 74.Gao M, Fan S, Goldberg ID, Laterra J, Kitsis RN, Rosen EM. Hepatocyte growth factor/scatter factor blocks the mitochondrial pathway of apoptosis signaling in breast cancer cells. J Biol Chem. 2001;276:47257–65. doi: 10.1074/jbc.M106791200. [DOI] [PubMed] [Google Scholar]
- 75.Lai AZ, Abella JV, Park M. Crosstalk in Met receptor oncogenesis. Trends Cell Biol. 2009;19:542–51. doi: 10.1016/j.tcb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 76.Bonine-Summers AR, Aakre ME, Brown KA, Arteaga CL, Pietenpol JA, Moses HL, et al. Epidermal growth factor receptor plays a significant role in hepatocyte growth factor mediated biological responses in mammary epithelial cells. Cancer Biol Ther. 2007;6:561–70. doi: 10.4161/cbt.6.4.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paulson AK, Linklater ES, Berghuis BD, App CA, Oostendorp LD, Paulson JE, et al. MET and ERBB2 are coexpressed in ERBB2+ breast cancer and contribute to innate resistance. Mol Cancer Res. 2013;11:1112–21. doi: 10.1158/1541-7786.MCR-13-0042. [DOI] [PubMed] [Google Scholar]
- 78.Mueller KL, Hunter LA, Ethier SP, Boerner JL. Met and c-Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res. 2008;68:3314–22. doi: 10.1158/0008-5472.CAN-08-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shattuck DL, Miller JK, Carraway KL, 3rd, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008;68:1471–7. doi: 10.1158/0008-5472.CAN-07-5962. [DOI] [PubMed] [Google Scholar]
- 80.Gallego MI, Bierie B, Hennighausen L. Targeted expression of HGF/SF in mouse mammary epithelium leads to metastatic adenosquamous carcinomas through the activation of multiple signal transduction pathways. Oncogene. 2003;22:8498–508. doi: 10.1038/sj.onc.1207063. [DOI] [PubMed] [Google Scholar]
- 81.Ponzo MG, Lesurf R, Petkiewicz S, O’Malley FP, Pinnaduwage D, Andrulis IL, et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc Natl Acad Sci U S A. 2009;106:12903–8. doi: 10.1073/pnas.0810402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Graveel CR, DeGroot JD, Su Y, Koeman J, Dykema K, Leung S, et al. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc Natl Acad Sci U S A. 2009;106:12909–14. doi: 10.1073/pnas.0810403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Knight JF, Lesurf R, Zhao H, Pinnaduwage D, Davis RR, Saleh SM, et al. Met synergizes with p53 loss to induce mammary tumors that possess features of claudin-low breast cancer. Proc Natl Acad Sci U S A. 2013;110:E1301–10. doi: 10.1073/pnas.1210353110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fulford LG, Easton DF, Reis-Filho JS, Sofronis A, Gillett CE, Lakhani SR, et al. Specific morphological features predictive for the basal phenotype in grade 3 invasive ductal carcinoma of breast. Histopathology. 2006;49:22–34. doi: 10.1111/j.1365-2559.2006.02453.x. [DOI] [PubMed] [Google Scholar]
- 85.Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT, et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19:264–71. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 86.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gentile A, Trusolino L, Comoglio PM. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008;27:85–94. doi: 10.1007/s10555-007-9107-6. [DOI] [PubMed] [Google Scholar]
- 88.Yano S, Nakagawa T. The current state of molecularly targeted drugs targeting HGF/Met. Jpn J Clin Oncol. 2014;44:9–12. doi: 10.1093/jjco/hyt188. [DOI] [PubMed] [Google Scholar]
- 89.Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signalling pathway in cancer. Eur J Cancer. 2010;46:1260–70. doi: 10.1016/j.ejca.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yap TA, Olmos D, Brunetto AT, Tunariu N, Barriuso J, Riisnaes R, et al. Phase I trial of a selective c-MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol. 2011;29:1271–9. doi: 10.1200/JCO.2010.31.0367. [DOI] [PubMed] [Google Scholar]
- 91.Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10:2298–308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 92.Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69:8009–16. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- 93.Eder JP, Shapiro GI, Appleman LJ, Zhu AX, Miles D, Keer H, et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res. 2010;16:3507–16. doi: 10.1158/1078-0432.CCR-10-0574. [DOI] [PubMed] [Google Scholar]