

Abstract
Many viruses have evolved fusion-mediating glycoproteins that couple the energy released from irreversible protein refolding to the work of membrane fusion. The viral fusion proteins require a triggering event to undergo a cascade of tightly regulated conformational changes. Different isolates of the paramyxovirus SV5 fusion (F) protein have either a short (20-residue) or long (42-residue) cytoplasmic tail (CT), and a long CT suppresses fusion activity in a sequence-specific manner. Addition of a domain to the F protein CT, which has the propensity to form a three-helix bundle, stabilizes the F protein and increases the energy required for fusion activation. Quantitative cell–cell fusion assays and measurement of ectodomain conformation by monoclonal antibody reactivity indicate that this suppression of fusion by the long CT or addition of a three-helix bundle occurs at a step preceding initial membrane merger. The data suggest that F protein activation involves CT signaling to the ectodomain.
Keywords: membrane fusion, fusion activation, paramyxoviruses, class 1 fusion protein
Enveloped viruses enter cells by fusing the viral envelope with a cellular membrane. To that end, they have evolved fusion-mediating glycoproteins that couple the energy released from irreversible protein refolding to the work of membrane fusion. The proteins fold initially into a metastable form and then undergo a cascade of tightly regulated conformational changes, releasing the fusion proteins to a lower energy state. These viral fusion machines are one-time use machines that do not require ATP as an energy source.
Class I viral fusion proteins of enveloped viruses are trimeric. They are synthesized as a precursor that has to be cleaved to yield a membrane-anchored subunit that, typically at the newly generated N terminus, contains a hydrophobic fusion peptide. The membrane-anchored subunit also contains two heptad repeats, one proximal to the fusion peptide (heptad repeat A) and the other proximal to the transmembrane (TM) domain (heptad repeat B). Most class I fusion proteins have a receptor-binding domain that clamps the fusion-causing domain such that the protein is triggered only after receptor binding when the target membrane is within range. Examples of class I fusion proteins are hemagglutinin (HA) of influenza virus, gp160 of HIV-1, the envelope glycoprotein (Env) of other retroviruses, and fusion (F) protein of paramyxoviruses (reviewed in refs. 1 and 2).
Triggering of fusion is usually thought to involve only changes within the extracellular ectodomain of the fusion proteins, and fusion is triggered either by receptor binding or by the low pH found in endosomal compartments. The fusion peptide inserts into target membranes (3) with formation of a transient pre-hairpin structure (4, 5). Then, protein refolding occurs, with heptad repeat A and heptad repeat B refolding into a six-helix bundle (6HB) structure with the fusion peptide and the TM domain located in close proximity in the same membrane (reviewed in ref. 2). The available data suggest that it is the free energy released on forming the 6HB that brings the two opposing membranes together (5, 6). The 6HB structure is very stable to thermal denaturation and is thought to represent the final form of the class I fusion proteins after fusion has occurred.
The cytoplasmic tail (CT) of many class I viral fusion proteins has usually been thought to have an important role in viral assembly and budding (reviewed in ref. 7). However, especially for retroviruses, several lines of evidence suggest a connection between the Env CT and membrane fusion. For several retro-viruses [e.g., Moloney murine leukemia virus (MoMLV) and Mason–Pfizer monkey virus], the Env CT is cleaved by the viral protease after virus release, and this cleavage activates Env fusion activity (8–10). Truncation of the CT of the HIV-1 gp160 membrane proximal subunit (gp41) increases fusion activity. HIV-1 virions lacking protease activity show impaired fusion activity, and the HIV gp41 CT is uncleaved (11). For MoMLV, it has been proposed that the uncleaved CT forms a trimeric structure (12) that suppresses fusion unless cleavage occurs.
Activation of most paramyxovirus F proteins occurs at neutral pH and is thought to be triggered by steps including (i) binding of the viral attachment protein (HN or H) to its cell surface receptor, (ii) HN interacting with F, and (iii) the HN/F interaction leading to changes in F that mediate membrane fusion (5, 13). Most paramyxovirus F proteins require coexpression of their homotypic HN protein to lower the energy of activation of fusion. However, the SV5 isolate W3A F protein mediates fusion when expressed alone, although to a reduced level than when coexpressed with HN (14, 15). Furthermore, recent mutagenesis studies of SV5 W3A F proteins with changes at residues S443, L447, and I449, a region of F either linked to or part of the 6HB, are destabilized and cause hyperfusion in an HN-independent manner. It has been hypothesized that this region plays two distinct structural and functional roles in the prefusion and 6HB conformation and this region constitutes a regulatory switch segment (16, 17).
The CT of F proteins of the SV5 isolates W3A and WR are 20 residues long (Fig. 1A). These F proteins cause extensive cell–cell fusion whereas SV5 isolates of canine (T1) and porcine (SER) origin, although of overall very similar sequence to the W3A F protein, contain an extended CT of 42 residues (Fig. 1). The T1 and SER F proteins cause reduced or no detectable syncytia formation (18, 19), but truncation of the T1 or SER F protein CT to the 20 residues found in W3A/WR F protein restores cell–cell fusion to W3A/WR-like levels (18, 19).
Fig. 1.

Schematic diagram of the amino acid sequence of the paramyxovirus SV5 fusion (F) protein. (A) Alignment of the amino acid sequence of the F protein from SV5 strains W3A, WR, and SER. Amino acid differences between SV5 isolates are highlighted. The anomalous HN-independent fusion activity of W3A F protein was mapped to residue 22 (16, 22). (B) Amino acid sequences of F protein CT of SV5 isolates W3A, SER, T1, and MEL and W3A F containing a long CT, F551. SER and T1 F encode a CT 22 residues longer, and MELFaCT five residues longer, than the CT of W3A and WR F proteins. F551 encodes the point mutation K529N and has a serine codon (residue 530) substituted for a stop codon, permitting translational read-through to a downstream in-frame stop codon. The amino acid sequence of the extended portion of the F protein CT in F551 is identical to that of canine isolate of SV5 (T1) and almost identical to an SV5 porcine isolate (SER) (18, 19).
We report here data providing a mechanistic link between the paramyxovirus fusion protein ectodomain and its CT in fusion activation. We provide data that suggest that molecular interactions formed by the extended CT of some SV5 F proteins affect a stage in F protein fusion activation, possibly preventing release of heptad repeat B that is required for downstream refolding events. Furthermore, we hypothesize that the CT of paramyxovirus fusion proteins can regulate differentially virus–cell vs. cell–cell fusion.
Materials and Methods
Cells and Viruses. Madin–Darby bovine kidney, CV-1, 293T, Vero, baby hamster kidney-21F (BHK-21F), and BSR T7/5 cells were maintained as described (17, 20).
Recombinant Plasmid Vectors and Oligonucleotide-Directed Mutagenesis. Mutations in F were generated by oligonucleotide-directed mutagenesis by using the SV5 infectious cDNA clone pBH276 (21) as the template for PCR. Amplified sequences were then cloned into either the expression vector pGEM2 or pCAGGS or into the SV5 infectious cDNA clone. Recovered SV5 containing F mutations were obtained as described (21). Mutations were confirmed by nucleotide sequencing using an ABI Prism 3100-Avant genetic analyzer (Applied Biosystems).
Expression of Mutant F Proteins. Expression of WT SV5 strain W3A F protein and mutant F proteins was by either (i) pCAGGS mammalian expression system or (ii) recombinant vaccinia virus-T7 RNA polymerase expression system as described (5).
Syncytium Formation Induced by WT and Mutant F Proteins. Monolayers of BHK-21F cells were transfected with pCAGGS HN and pCAGGS constructs encoding WT W3A and mutant F proteins by using Effectene transfection reagent (Qiagen, Valencia, CA). At 14–20 h posttransfection, cells were stained with HEMA 3 staining system (Fisher Diagnostics) and photographed as described (17).
Metabolic Labeling and Immunoprecipitation of Polypeptides. Cells expressing WT W3A and mutant F proteins were metabolically labeled with 50 μCi/ml (1 Ci = 37 GBq) 35S Pro-mix (Amersham Pharmacia Biotech) in 1 ml of DMEM deficient in cysteine and methionine (DMEM met-cys-) for 15 min. After incubation for varying times (chase), cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (20), and lysates were immunoprecipitated with rabbit sera specific for F1 and F2 and subjected to SDS/PAGE on 15% acrylamide gels as described (20). The stability of F trimers to SDS denaturation was examined by immunoprecipitating lysates with mAb 21-1 (22) and dissolving the proteins in gel-loading buffer containing 2% SDS. Samples were heated to various temperatures for 5 min, and proteins were analyzed on 3.5% acrylamide gels. Quantification of radiolabeled protein bands was done by using a Fuji BioImager 1000 and imagequant 3.3 software (Fuji Medical System, Stamford, CT).
Surface Expression of Mutant F Proteins. Surface expression of WT and mutant F proteins was calculated by determining the amount of the total labeled protein available at the cell surface for biotinylation as described (23).
Flow Cytometry. Quantification of both the number of cells expressing F protein and the level of F expression was performed by flow cytometry, using mAb F1a (24), 6-7, or 21-1 (22) as described (16). Fluorescence intensity of 10,000 cells was measured by using a FACSCalibur flow cytometer (Becton Dickinson).
Reporter Gene Cytoplasmic Content-Mixing Fusion Assays. Cytoplasmic content mixing was assessed by using a luciferase reporter gene assay. Vero cells were used to express the luciferase gene (under the control of the T7 RNA polymerase promoter) and HN and F proteins, and, at 18 h posttransfection, the cells were overlaid with BSR-T7/5 cells, which constitutively express T7 RNA polymerase. The luciferase activity after the fusion of the two cell cultures was measured as described (17).
Lipidic and Aqueous Dye Transfer. Fusion assays between human erythrocytes (RBCs) double-labeled with the aqueous probe 6-carboxyfluorescein and the lipid probe octadecyl rhodamine B chloride (R18) (Molecular Probes) and CV-1 cells expressing F and HN proteins were performed as described (17).
Results
Extension of the CT of the SV5 Fusion Protein Does Not Affect Its Intracellular Cleavage or Cell-Surface Transport. To investigate the mechanism by which the length of the SV5 F protein CT may regulate fusion, the translational stop codon of the F protein of SV5 strain W3A (CT 20 residues) was changed to serine to permit synthesis of a protein that terminated at the subsequent downstream termination codon. The mutant F protein (F551) encodes a 42-aa CT tail (Fig. 1B) that is identical to that of the SV5 natural canine isolate T1 and almost identical to that of the SV5 natural porcine isolate SER. Thus, F551 permits evaluation of the mechanism of CT inhibition of fusion in the context of the well characterized paramyxovirus W3A F protein. We also changed WT W3A F and F551 proteins such that they encoded the destabilizing mutation S443P (5, 16) because SV5 strains MEL, T1, and SER contain naturally a proline at residue 443. Furthermore, SV5 strain MEL has a CT with five additional residues than that of W3A F protein (R. Randall, personal communication) (Fig. 1B), allowing for analysis of a natural isolate with an intermediate length CT.
Analysis of the kinetics of cleavage of the WT W3A and altered F proteins from the precursor Fo to the biologically active subunits F1 and F2 showed very similar kinetics (Fig. 5A, which is published as supporting information on the PNAS web site), suggesting that the mutants were not altered in their rate of intracellular transport to the trans-Golgi network. Furthermore, the relative cell-surface expression level of the F proteins, as determined by cell-surface biotinylation and immunoprecipitation of F, indicated that the mutant F proteins had relative cell-surface densities similar to WT W3A F (Fig. 5 B and C). Thus, the mutants were not debilitated in cell-surface expression levels.
Extension of the CT of the F Protein Decreases Syncytia Formation. To examine the effect of extending the F protein CT (F551) on syncytia formation, BHK-21F cells coexpressing transiently an equivalent surface density of the W3A HN protein and the WT W3A F or mutant F proteins F551, F-S443P, or F551-S443P were photographed at 14 h posttransfection (Fig. 2A). Although WT W3A F caused obvious syncytia formation and F-S443P caused extensive syncytia formation, F551 caused greatly reduced syncytia formation that was barely detectable. In contrast, incorporation of S443P into F-551 (F551-S443P) restored significant syncytium formation. Expression of SV5 MEL F, an F protein that contains naturally proline residues at positions 22 and 443 (analogous to W3A F-S443P) and a CT containing 5 extra residues in addition to the 20 residues found in W3A F protein, showed extensive syncytia formation.
Fig. 2.

Cell–cell fusion of WT W3A and mutant F proteins. (A) Syncytium formation in BHK-21F cells cotransfected with HN and WT or mutant F proteins. (B and C) Cell–cell fusion in Vero cells using the luciferase reporter assay. Vero cells were transfected with plasmids to coexpress WT W3A or mutant F proteins, HN protein, and a luciferase reporter construct. (D) Cell–cell fusion of F mutants at different temperatures. Human erythrocytes (RBCs) were dual labeled with the lipidic dye octadecyl rhodamine (R18) and the aqueous dye 6-carboxyfluorescein and bound on ice to CV-1 cells expressing HN and either WT W3A or mutant F proteins. The temperature was raised to 29°C, 37°C, and 42°C for 10 min to activate fusion (and dye transfer). Shown is the quantification of the number of fusion events observed per microscope field (n = 6–11 fields) at each temperature for the indicated F proteins. (E) mAb reactivity of mutant F proteins. The RMFI for mutant F proteins was determined by flow cytometry and expressed as a percentage of that of WT F protein. mAb F1a reactivity is affected by the length of the CT. mAb 6-7 and 21-1 reactivity is increased by the presence of P22 and P443 but is not affected by the length of the CT.
To quantify fusion, a luciferase reporter assay was used (17) in which the content mixing of two cytoplasms permits production of luciferase after cell–cell fusion mediated by SV5 WT HN and WT W3A F or mutant F proteins. The presence of the longer CT (F-551) led to a 2- to 3-fold decrease in cell–cell fusion as compared with WT W3A F. F551-S443P led to recovery of cell–cell fusion to an extent approaching that found for F-S443P (Figs. 2B). These data confirm data obtained from the syncytium assay and indicate that the extended CT on F551 is capable of reducing the level of cell–cell fusion mediated by WT W3A F, and that F-S443P overcomes much of the inhibitory activity. In the luciferase assay, MEL F induced a level of fusion similar to that observed for F-S443P (Fig. 2B). Parallel data were obtained for fusion activity when the F mutations were incorporated into recombinant SV5 (Fig. 6, which is published as supporting information on the PNAS web site).
A previous mutagenesis study of the CT residues of SER F protein suggested that the fusion inhibition of SER F is sequence specific because changing leucine residues 539 and 548 to alanine increased the level of fusion (25). To investigate further sequence specificity of the F551 CT in inhibiting fusion, we first generated a similar mutant, F551-L359/548A, and we observed that cell–cell fusion was restored to levels approximating those of WT W3A F (Fig. 2C). To extend these experiments, a mutant F551 CT was generated (F551-sc) in which residues 530–551 (the extended portion) were scrambled. F551-sc caused fusion levels analogous to those of WT W3A F. The sequence specificity of the fusion inhibition suggests that the extended portion of the F551 CT forms interactions that are either intramolecular or intermolecular with other viral or cellular proteins.
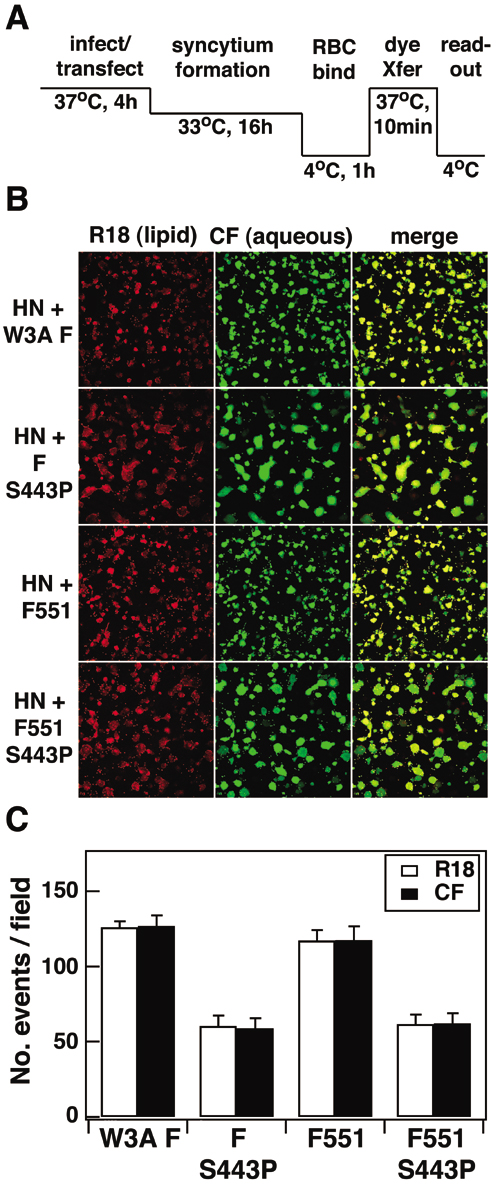
To test the affect of the F mutants on steps late in fusion, we performed a dye transfer fusion assay using dual-labeled (lipidic R18 and aqueous carboxyfluorescein) human RBCs and CV-1 cells expressing W3A HN and WT W3A F or mutant F proteins. To trigger fusion, the temperature was raised from 4°C to 29°C, 37°C, or 42°C for 10 min. Cells were returned to 4°C before examination by confocal microscopy to assess dye transfer. In all cases, lipidic dye transfer was coincident with aqueous dye transfer (content mixing), and in no case was lipidic dye transfer only (hemifusion) observed. As shown in Fig. 2D (for example of raw data, see Fig. 7A, which is published as supporting information on the PNAS web site), the F551 protein-expressing cells showed a reduced extent of fusion as compared with WT W3A F protein at either 29°C or 37°C, but the extent of F551 content mixing increased at 42°C to levels similar to WT W3A F. As expected, the presence of the destabilizing mutant at position 443 restored the fusion inhibition seen for F551 even at low temperatures as observed for F-S443P (Fig. 2D and ref. 16). The sequence-specific effect of the extended portion of the CT was also confirmed because F551-L539/548A led to dye transfer levels very similar to WT W3A F protein (Fig. 2C). Further support that the F mutations do not cause a hemifusion phenotype (lipid mixing only) was shown by other experiments in which the target cells were labeled with lipidic and aqueous fluorescent dyes (Fig. 8, which is published as supporting information on the PNAS web site).
Antibody Reactivity as a Probe for Conformational Differences Among the F Protein Mutants. We used three mAbs specific for F [mAb F1a (24) and mAbs 6-7 and 21-1 (22)] that are conformation specific (16, 22). mAbs 6-7 and 21-1 were raised against an F protein containing a proline residue at positions 22 and 443. These mAbs react very poorly with W3A F containing L22 and S443, react better with W3A F containing L22 and P443, and react well with a W3A F protein containing P22 and P443 (17, 22). HeLa cells transiently expressing equivalent surface densities of the F protein (determined by biotinylation) were incubated with the F-specific mAbs, and the relative mean fluorescence intensity (RMFI) was determined by flow cytometry (Fig. 2E). mAb F1a reacted better with F proteins that encode WT W3A F length CT (20 aa) than F proteins containing an extended CT, as a 2- to 3-fold decrease in antibody reactivity compared with WT W3A F was observed for F551 and F551-S443P. mAbs 6-7 and 21-1 exhibited a 2- to 3-fold increase in reactivity with F protein mutants containing S443P, regardless of the length of the CT (Fig. 2E). The mAb reactivity data suggest that the conformational effects of CT length detected by mAb F1a are distinct from the conformational effects of the S443P mutation detected by mAbs 6-7 and 21-1.
Addition of a Three-Helix Bundle (3HB) Domain to the F Protein CT Suppresses Cell–Cell Fusion. We wished to test the hypothesis that the extended portion of the F551 CT (residues 530–551) reduces (inhibits) fusion because it forms either intra- or intermolecular interactions that stabilize a metastable intermediate in the cascade of conformational changes that occur as the cleaved F protein proceeds from its primary metastable form down an energy gradient to its final very stable postfusion form. Because of the complexity of a CT linked to a TM domain, determination of the structure of a CT is not a simple problem. Therefore, a surrogate F protein was generated that in addition to the WT W3A F protein CT (20 residues) contains 28 C-terminal residues with the propensity to form a three-helix bundle (3HBii) connected by means of a flexible linker. This sequence was predicted to form a trimer based on knowledge of knob-into-hole interactions for helical packing, and this notion was confirmed when 3HBii was shown experimentally to form a 3HB (26). We also generated a mutant (3HBaa) in which the critical isoleucine residues required for 3HB formation were changed to alanine (Fig. 3A). In comparison with WT W3A F, F-3HBii and F-3HBaa when expressed transiently in cells showed a slower SDS/PAGE mobility as expected for an extended CT and the kinetics of cleavage of Fo to F1 and F2 for F-3HBii and F-3HBaa were only slightly slower than those for WT W3A F (Fig. 5A). Cell-surface expression levels (determined by biotinylation) were very similar for WT W3A F, F-3HBii, and F-3HBaa (Fig. 5 B and C). Thus, any differences in fusion activity are not due to differences in cell-surface density of F protein.
Fig. 3.

A cytoplasmic three-helix bundle (3HBii) domain stabilizes the SV5 F protein trimer. (A) Schematic diagram of WT W3A F containing a CT with a flexible linker (SGGSGG) and the 3HBii domain. The amino acid sequence of 3HBii [derived from GCN4-p-II (26)] and a mutant 3HBaa designed to ablate the isoleucine knob-into-hole helical interactions of 3HBii, and thus prevent 3HB formation, is shown. (B) Cell–cell fusion of WT F, F-3HBii, and F-3HBaa (with and without the S443P mutation) was measured by using the luciferase reporter gene activity assay. (C) Quantification of the average number of fusion events from 7–10 microscope fields for each F protein was determined by using the R18/6-carboxyfluorescein dye transfer assay. (D) mAb reactivity (RMFI) of transfected cells having very similar levels of F protein surface expression (as determined by biotinylation) of WT and mutant F proteins as measured by flow cytometry.
Cell–cell fusion activity for F-3HBii and F-3HBaa and derivative mutants was first measured by using the luciferase reporter gene assay. As shown in Fig. 3B, F-3HBii showed less fusion activity than WT W3A F protein. Similarly, incorporation of 3HBii into F-S443P showed a great reduction in its ability to induce hyperfusion. F-3HBaa showed very similar fusion activity to WT W3A F, and incorporation of S443P into F-3HBaa showed hyperfusion activity similar to F-S443P (Fig. 3B).
We also used the dye transfer assay to access the temperature dependence of fusion activation. As shown in Fig. 3C (see also Fig. 7B for an example of raw data), F-3HBii showed essentially no fusion activity at any of the temperatures tested. In contrast, F-3HBaa showed fusion activity that was similar to that of WT W3A F. F-3HBii-S443P did not show hyperfusion activity like that found for F-3HBaa-S443P. In this assay, the level of fusion activity for F-3HBii was lower than in the luciferase assay. We attribute this difference to the time duration over which fusion is measured: 4 h for the reporter gene assay and 10 min for the dye transfer assay. Taken together, the fusion activity data suggest that the addition of the 3HBii domain to the F CT forms a protein structure (presumably a 3HB) that suppresses fusion and the F CT intramolecular interactions reduce the hyperfusion effect of introducing the S443P mutation into the F ectodomain.
F Protein Gains mAb Reactivity upon Heating, and Addition of 3HBii Suppresses This Gain in Epitope Exposure. Flow cytometry was performed on cells expressing at their cell surface equivalent amounts of F protein. mAb F1a detected changes in the length of the CT regardless of effects on fusion (Fig. 3D). mAbs 6-7 and 21-1 reacted with F containing S443P that was independent of the length of the CT. Heating the WT W3A F protein to 50°C for 10 min (22) and returning to 4°C before mAb binding showed an increased reactivity of WT W3A to mAb 21-1, suggesting increased exposure of an epitope recognized by this mAb (Fig. 3D). F-3HBii-S443P, but not F-3HBaa-S443P, showed low mAb 21-1 reactivity, suggesting suppression of the epitope exposed by S443P. The affect of heating F-3HBii with and without S443P on mAb 21-1 reactivity was also examined. As shown in Fig. 3D, heating F-3HBii to 50°C yielded a smaller relative increase in reactivity with mAb 21-1 than for WT W3A. Moreover, heating F-3HBii-S443P to 50°C did not bring about the level of mAb 21-1 reactivity observed for F-S443P. Taken together, the mAb reactivity data suggest that the addition of 3HBii to the F CT suppresses exposure of the epitope recognized by mAb 21-1, which is associated with either heating the F protein or having an F protein containing S443P. Thus, the simplest interpretation of the data is that the addition of 3HBii to the F CT affects a stage in the F activation process before hemifusion.
F551 and F-3HBii Show Increased Trimer Stability in SDS Solution. We observed that, for WT W3A F protein, a fraction of the F trimer failed to dissociate completely into monomers in 2% SDS solution unless the samples were heated to >75°C (Fig. 4A). Although F is immunoprecipitated in these assays, the residual trimers and dimers observed are not due to a spurious effect of an interaction with the mAb, as in 2% SDS the IgG chains dissociate from F at 37–45°C (data not shown). Thus, we reasoned we might be able to use the “residual trimer assay” as a surrogate for examining trimer stability. As shown in Fig. 4, WT W3A F trimer species were lost at 75°C whereas F551 residual trimer species were stable until heating to at least 80°C. In contrast, F551-L539/548A behaved like WT W3A F, with residual trimers dissociating at 75°C. F-3HBii residual trimers were stable until 80–85°C, but F-3HBaa residual trimers were stable only until heating to 75°C. Thus, there is an excellent correlation for the predicted effect of the nature of the CT in forming specific protein–protein interactions and stabilizing the F protein before fusion activation (F551 and F-3HBii) with increased temperature of residual trimer dissociation.
Fig. 4.

SV5 F protein trimer stability. Wt W3A and mutant F proteins were expressed transiently in 293T cells, and proteins were metabolically labeled. F proteins were immunoprecipitated by using mAb 21-1. This mAb recognizes all F proteins used equivalently, as can be observed in Figs. 2 and 3. Before analysis by SDS/PAGE on 3.5% acrylamide gels, samples were heated at the temperatures indicated. (A) WT W3A F protein in SDS solution heated at various temperatures before SDS/PAGE. m, d, and t indicate F species with mobilities expected for F monomer, dimer, and trimer (30). (B–F) Thermal stability of trimer and dimer F species of WT and mutant F proteins. Ramps are in intervals of 5°C. The total amount of F protein in each lane was quantified by using a PhosphorImager (Molecular Dynamics), and each F protein was found to be within 30% of each other. The amount of thermal energy required to dissociate the F trimers into monomers is approximately in the order F-3HBii > F551 > F-3HBaa = F551-L539/548A = WT W3A F.
Discussion
The data presented here have implications for the mechanism of activation of paramyxovirus fusion proteins and the overall mechanism of virus-mediated membrane fusion. The data suggest that the natural CT extension on the F protein of some paramyxovirus SV5 strains inhibits transmission of TM signaling from the CT to the ectodomain, preventing an ectodomain conformational change. The lack of the ectodomain conformational change increases the energy threshold required for fusion activation. F551 shows suppressed fusion as compared with WT W3A F protein, yet F551 containing the mutations L539/548A show fusion levels equivalent to WT W3A F, suggesting it is not the length of the CT per se that inhibits fusion but a property of the protein sequence. This concept is reinforced by the observation that scrambling the sequence of F551, or adding 34 CT residues to WT W3A F (F-3HBaa), still yields levels of fusion comparable with WT W3A F. However, when a sequence with the propensity to form a specific trimeric structure is added to the F CT (F-3HBii), the protein does not cause fusion in the dye-transfer assay even on heating cells to 50°C (data not shown). Thus, by making the CT a presumptive stable structure, fusion activation is suppressed.
The reactivity of the WT W3A F protein with mAbs 6-7 and 21-1 is low in comparison with the reactivities of F-S443P and F551-S443P. However, heating WT W3A F to 50°C (followed by return to 4°C) increases its reactivity to these mAbs. This finding suggests that WT W3A F and F-S443P have conformations that differ from each other and that WT W3A F is trapped energetically in a metastable form that is at a higher energy level than that of F-S443P. Thus, heating to 50°C allows F protein to achieve the lower energy state that reacts better with mAb 21-1. To a large degree, addition of the 3HB to F that contains the S443P mutation (F-3HBii-S443P) prevents the transition to the protein form recognized by mAbs 6-7 and 21-1 and greatly inhibits fusion, even compared with WT W3A F at 42°C. Thus, by forming a presumptive specific CT structure, changes in the F ectodomain at a stage of fusion before hemifusion is prevented. F551 represents a natural extended CT, and, because F551-S443P does show increased fusion and increased mAb reactivity, the data suggest that the stabilizing effect on fusion activity of this CT is less than that caused by F molecules containing an artificial CT sequence (F-3HBii and F-3HBii-S443P). Consistent with the idea that F551 and F-3HBii increase the energy threshold for fusion activation was the finding that F551 and F-3HBii show increased trimer thermostability in SDS solution. The F TM domains must rotate in the plane of the membrane before the 6HB can form, thus providing a possible explanation for the effect of a stable CT structure on inhibiting fusion. Constraining the movements of the TM domains may correspondingly constrain the locations and potential interactions of the adjacent heptad repeat B regions, thereby influencing the energetics of F activation. Alternatively, a stable CT structure might prevent clustering of trimeric pre-hairpin intermediates that are thought to form a fusion prepore complex.
An involvement of the CT in membrane fusion may be general for class I fusion proteins, including some retroviruses [e.g., Moloney murine leukemia virus, Mason–Pfizer monkey virus, and HIV-1 (8–12)]. Truncation of Env (by protease or protein design) leads to differences in conformation-specific mAb reactivity for both murine leukemia virus (27) and HIV-1 (28) Env proteins, suggesting a conformational change on truncation of the Env CT.
For SV5 isolate SER, it has been reported that F protein fusion activation requires both low pH conditions found in endosomes (pH 4.8–6.2) and coexpression of HN (29). Truncation of the SER F protein CT to 20 residues abolishes both the low pH requirement and the requirement for HN coexpression (29). However, the control of low pH activation of the SER F protein is unlikely to reside in the CT because F551 does not show an increase in fusion (syncytia formation) after low pH treatment (our unpublished work). In addition to differing in CT tail length, SER and W3A F proteins differ by six residues in their ectodomains, and thus it seems logical that one or more of the ectodomain residues that differ between SER and W3A controls the low pH-dependent activation.
The paramyxovirus SV5 T1 and SER isolates are viable viruses. They grow to normal infectivity titers for SV5 (18, 19) but fail to cause syncytia. For SER, a low pH fusion activation mechanism precludes syncytia formation. The growth of recovered SV5-F551 in a direct comparison with WT W3A SV5 in MDCK cells shows slightly smaller plaques and has a 1 log lower titer [107 plaque-forming units (pfu)/ml] after 96 h postinfection than WT W3A SV5 (108 pfu/ml) (Fig. 6). The lack of syncytia formation by recovered SV5-F551 but good growth of the virus suggest there may be important mechanistic differences between virus–cell and cell–cell fusion. Syncytia formation causes cell death and limits virus yields in tissue culture; hence, the canine (T1) and porcine (SER) SV5 isolates may have evolved a longer CT to prevent syncytia formation in their animal hosts. In contrast, the W3A and WR isolates were isolated from monkey kidney cells in which syncytia were observed. It is possible that the short CT of W3A and WR are both the result of tissue culture adaptation.
Our experiments have focused on intersubunit interactions that stabilize the F trimer and inhibit fusion. It is also possible that interactions of the CT with other proteins may modulate fusion activity. Thus, interactions with other proteins that stabilize F trimers would inhibit fusion activity whereas those that destabilize trimers would promote fusion activity. This suggestion provides a general mechanism by which the activity of membrane fusion proteins can be regulated by interactions of their cytoplasmic domains.
Supplementary Material
Acknowledgments
We thank Reay Paterson and Andreas Matouschek for helpful discussions and Masato Tsurudome (Mie University, Tsu-Shi, Japan) and Rick Randall (St. Andrews University, St. Andrews, Scotland) for providing the mAbs 6-7, 21-1, and F1a. We thank Frederic Cohen, Richard Compans, Robert Doms, Douglas Lyles, and Rick Randall for helpful comments on the manuscript. This work was supported in part by Research Grant AI-23173 from the National Institute of Allergy and Infectious Diseases. C.J.R. is an Associate and R.A.L. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations: TM, transmembrane; F, paramyxovirus SV5 fusion; 6HB, six-helix bundle; 3HB, three-helix bundle; CT, cytoplasmic tail; Env, envelope glycoprotein; RMFI, relative mean fluorescence intensity; BHK-21F, baby hamster kidney-21F.
References
- 1.Skehel, J. J. & Wiley, D. C. (2000) Annu. Rev. Biochem. 69, 531-569. [DOI] [PubMed] [Google Scholar]
- 2.Eckert, D. M. & Kim, P. S. (2001) Annu. Rev. Biochem. 70, 777-810. [DOI] [PubMed] [Google Scholar]
- 3.Hernandez, L. D., Hoffman, L. R., Wolfsberg, T. G. & White, J. M. (1996) Annu. Rev. Cell Dev. Biol. 12, 627-661. [DOI] [PubMed] [Google Scholar]
- 4.Furuta, R. A., Wild, C. T., Weng, Y. & Weiss, C. D. (1998) Nat. Struct. Biol. 5, 276-279. [DOI] [PubMed] [Google Scholar]
- 5.Russell, C. J., Jardetzky, T. S. & Lamb, R. A. (2001) EMBO J. 20, 4024-4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melikyan, G. B., Markosyan, R. M., Hemmati, H., Delmedico, M. K., Lambert, D. M. & Cohen, F. S. (2000) J. Cell Biol. 151, 413-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitt, A. P. & Lamb, R. A. (2004) Curr. Top. Microbiol. Immunol. 283, 145-196. [DOI] [PubMed] [Google Scholar]
- 8.Green, N., Shinnick, T. M., Witte, O., Ponticelli, A., Sutcliffe, J. G. & Lerner, R. A. (1981) Proc. Natl. Acad. Sci. USA 78, 6023-6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rein, A., Mirro, J., Haynes, J. G., Ernst, S. M. & Nagashima, K. (1994) J. Virol. 68, 1773-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brody, B. A., Rhee, S. S. & Hunter, E. (1994) J. Virol. 68, 4620-4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murakami, T., Ablan, S., Freed, E. O. & Tanaka, Y. (2004) J. Virol. 78, 1026-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor, G. M. & Sanders, D. A. (2003) Virology 312, 295-305. [DOI] [PubMed] [Google Scholar]
- 13.Crennell, S., Takimoto, T., Portner, A. & Taylor, G. (2000) Nat. Struct. Biol. 7, 1068-1074. [DOI] [PubMed] [Google Scholar]
- 14.Paterson, R. G., Hiebert, S. W. & Lamb, R. A. (1985) Proc. Natl. Acad. Sci. USA 82, 7520-7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horvath, C. M., Paterson, R. G., Shaughnessy, M. A., Wood, R. & Lamb, R. A. (1992) J. Virol. 66, 4564-4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paterson, R. G., Russell, C. J. & Lamb, R. A. (2000) Virology 270, 17-30. [DOI] [PubMed] [Google Scholar]
- 17.Russell, C. J., Kantor, K. L., Jardetzky, T. S. & Lamb, R. A. (2003) J. Cell Biol. 163, 363-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito, M., Nishio, M., Komada, H., Ito, Y. & Tsurudome, M. (2000) J. Gen. Virol. 81, 719-727. [DOI] [PubMed] [Google Scholar]
- 19.Tong, S., Li, M., Vincent, A., Compans, R. W., Fritsch, E., Beier, R., Klenk, C., Ohuchi, M. & Klenk, H. D. (2002) Virology 301, 322-333. [DOI] [PubMed] [Google Scholar]
- 20.Paterson, R. G. & Lamb, R. A. (1993) in Molecular Virology: A Practical Approach, eds. Davidson, A. & Elliott, R. M. (IRL Oxford Univ. Press, Oxford), pp. 35-73.
- 21.He, B., Paterson, R. G., Ward, C. D. & Lamb, R. A. (1997) Virology 237, 249-260. [DOI] [PubMed] [Google Scholar]
- 22.Tsurudome, M., Ito, M., Nishio, M., Kawano, M., Komada, H. & Ito, Y. (2001) J. Virol. 75, 8999-9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leser, G. P., Ector, K. J. & Lamb, R. A. (1996) Mol. Biol. Cell 7, 155-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Randall, R. E., Young, D. F., Goswami, K. K. A. & Russell, W. C. (1987) J. Gen. Virol. 68, 2769-2780. [DOI] [PubMed] [Google Scholar]
- 25.Seth, S., Vincent, A. & Compans, R. W. (2003) J. Virol. 77, 167-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harbury, P. B., Zhang, T., Kim, P. S. & Alber, T. (1993) Science 262, 1401-1407. [DOI] [PubMed] [Google Scholar]
- 27.Aguilar, H. C., Anderson, W. F. & Cannon, P. M. (2003) J. Virol. 77, 1281-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edwards, T. G., Wyss, S., Reeves, J. D., Zolla-Pazner, S., Hoxie, J. A., Doms, R. W. & Baribaud, F. (2002) J. Virol. 76, 2683-2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seth, S., Vincent, A. & Compans, R. W. (2003) J. Virol. 77, 6520-6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell, R., Paterson, R. G. & Lamb, R. A. (1994) Virology 199, 160-168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}