

Abstract
Tau is required for the induction of long-term depression (LTD) of synaptic transmission in the hippocampus. Here we probe the role of tau in LTD, finding that an AMPA receptor internalization mechanism is impaired in tau KO mice, and that LTD causes specific phosphorylation at the serine 396 and 404 residues of tau. Surprisingly, we find that phosphorylation at serine 396, specifically, is critical for LTD but has no role in LTP. Finally, we show that tau KO mice exhibit deficits in spatial reversal learning. These findings underscore the physiological role for tau at the synapse and identify a behavioral correlate of its role in LTD.
Keywords: hippocampus, LTD, synaptic plasticity
Introduction
The canonical role of tau in neuronal function is as a microtubule-associated protein that is important for microtubule stabilization and axon elongation (Weingarten et al., 1975; Drubin et al., 1985; Cáceres and Kosik, 1990). Much of our attention when considering tau has been directed toward developing an understanding of neurodegenerative tauopathies (Ballatore et al., 2007), where hyperphosphorylation of tau and its mislocalization to dendritic regions are considered key aspects of tau-related cytopathogenesis (Kowall and Kosik, 1987). Recently, however, tau has been observed in dendritic regions under physiological conditions (Ittner et al., 2010; Mondragón-Rodríguez et al., 2012; Kimura et al., 2014). In concert with this, novel roles for tau in neuronal function have begun to emerge (Morris et al., 2011), and tau is receiving increased attention as an intracellular signaling protein, particularly at the synapse.
It is known that the function of tau is strongly affected by its phosphorylation status, influencing its ability to interact with microtubules and various kinases (Reynolds et al., 2008; Fischer et al., 2009), as well as altering its conformational status (Jeganathan et al., 2008; Fischer et al., 2009), its localization and association with membranes (Hoover et al., 2010; Usardi et al., 2011; Pooler et al., 2012), its cleavage (Mondragón-Rodriguez et al., 2008) and further post-translational modifications (Martin et al., 2011). Exactly how tau phosphorylation might affect its physiological function at the synapse, however, remains to be determined.
One of the emerging physiological roles of tau at the synapse is in synaptic plasticity, the molecular mechanisms by which synaptic connections are dynamically modulated, and the process thought to underlie learning and memory (Mayford et al., 2012). Tau has been identified to be required for long-term depression (LTD) of synaptic transmission, which describes the weakening of a synaptic connection (Kimura et al., 2014). Glycogen synthase kinase-3 (GSK-3β), a prominent tau kinase (Mandelkow et al., 1992; Ishiguro et al., 1993), has previously been identified as an important signaling component of NMDA receptor (NMDAR)-dependent LTD (Peineau et al., 2007) and its activation is required for AMPAR internalization (Du et al., 2010). Our previous study (Kimura et al., 2014) suggests that GSK-3β activity causes tau phosphorylation during LTD, suggesting that tau is likely a downstream substrate of GSK-3β in LTD. This raises the question as to the possible significance of these tau phosphorylation events.
Here we demonstrate that tau has an important mediatory role in regulating the interaction between protein kinase C binding protein 1 (PICK1) and GluA2, a molecular mechanism fundamental to AMPAR internalization. Using biochemical and electrophysiological assays, we show that site-specific phosphorylation at 396 (S396) of tau is associated with and required for LTD. In addition, we show that the absence of tau results in the reversal of spatial memory deficits, underscoring the importance of the role of tau in hippocampal LTD.
Materials and Methods
Animals.
All procedures involving animals were performed in accordance with the UK Animals (Scientific Procedures) Act, 1986. Male Wistar rats (Charles River) were used to prepare organotypic (7-d-old rats) and acute hippocampal slices (2- to 3-week-old rats). Male B6.129-Mapttm1Hnd/J mice (Tau KO, The Jackson Laboratory) and wild-type (WT) C57BL/6J counterparts were used to prepare acute hippocampal slices (2–3 weeks old) and for behavioral tests (4–5 months old), as approved by the Institutional Animal Care and Use Committee of Kyung Hee University (Seoul, Republic of Korea). Mice or rats were housed four or five per cage, and were allowed access to water and food ad libitum. The cages were maintained at a constant temperature (23 ± 1°C) and relative humidity (60 ± 10%) under a 12-h light/dark cycle (lights on from 7:30 A.M. to 7:30 P.M.).
Acute hippocampal slice preparation.
Animals were killed by cervical dislocation and were decapitated. Following this, the brain was rapidly removed and placed into ice-cold artificial CSF (aCSF; continuously bubbled with 95% O2/5% CO2) containing 124 mm NaCl, 3 mm KCl, 26 mm NaHCO3, 1.25 mm NaH2PO4, 2 mm CaCl2, 1 mm MgSO4, and 10 mm d-glucose. Hippocampi were extracted and transverse hippocampal slices (400 μm thickness) were cut using a McIlwain tissue chopper. Following manual separation, the slices were then submerged in aCSF for a minimum of 1 h before experiments commenced.
Organotypic hippocampal slice preparation.
Organotypic slices were cultured based upon a method previously described by Stoppini et al. (1991). Rats were decapitated, and their brains were rapidly removed and placed into ice-cold dissecting medium containing the following: 238 mm sucrose, 2.5 mm KCl, 26 mm NaHCO3, 1 mm NaH2PO4, 5 mm MgCl2, 11 mm d-glucose, and 1 mm CaCl2. Hippocampi were extracted, and transverse hippocampal slices (350 μm thickness) were cut and placed upon sterile, semi-porous membranes. These were stored and maintained at the interface between air and culture medium (containing the following: 78.8% Mimimum Essential Medium, 20% heat-inactivated horse serum, 30 mm HEPES, 26 mm d-glucose, 5.8 mm NaHCO3, 2 mm CaCl2, 2 mm MgSO4, 70 μm ascorbic acid, and 1 μg/ml insulin, pH adjusted to 7.3 and 320–330 mOsm/kg) inside a humidified incubator at 35°C with a 5% CO2-enriched atmosphere. Culture medium was refreshed every 2 d, and slices were used for whole-cell recording at 6–8 din vitro (DIV).
Biolistic transfection and plasmid constructs.
DNA-coated microcarriers for biolistic transfection of organotypic hippocampal slices were prepared based on previously described methods (Lo et al., 1994). At DIV 3–5, neurons were transfected with plasmids expressing shRNA against rat tau protein (OriGene Technologies). A mixture of four different tau shRNA constructs (1:1:1:1, in pGFP-V-RS vector) was used for tau silencing. Phosphorylation-null (serine residues mutated to alanine) human tau constructs of the AT8 [S199, S202, threonine 205 (T205)] and PHF-1 (S396, S404) epitopes, in pCI-neo vectors, were provided by Dr. A. Takashima (Department of Aging Neurobiology, National Center for Geriatrics and Gerontology, Obu, Japan). Individual mutations to the PHF-1 epitope (residues S396 or S404) were generated by site-directed mutagenesis (Agilent Technologies) of 2N4R human tau.
Electrophysiology.
For whole-cell recordings from organotypic slices, the recording chamber was perfused with a buffer solution containing 119 mm NaCl, 2.5 mm KCl, 4 mm CaCl2, 4 mm MgCl2, 26 mm NaHCO3, 1 mm NaH2PO4, 11 mm glucose, 0.02 mm picrotoxin, and 0.01 mm 2-chloroadenosine. The buffer solution was maintained at 29–30°C and saturated with 95% O2/5% CO2. Bipolar stimulating electrodes were placed on the Schaffer collateral pathway and on the subiculum–CA1 input. Recording electrodes (5–6 MΩ) containing CsMeSO4 filling solution (comprising 130 mm CsMeSO4, 8 mm NaCl, 4 mm Mg-ATP, 0.3 mm Na-GTP, 0.5 mm EGTA, 10 mm HEPES, and 6 mm QX-314, pH 7.2–7.3 and 270–290 mOsm/kg) were used to patch and voltage clamp CA1 pyramidal neurons. EPSCs were recorded at a holding voltage of −70 mV, and only cells that had an initial Rs (series resistance) < 20 MΩ that was maintained at a level within 20% of that value from start to finish were included in final data analysis. To induce LTD, depolarization of the cell to a holding voltage of −40 mV was paired with 200 pulses of 1 Hz stimulation to the Schaffer collateral input. For LTP experiments, depolarization of the cell to 0 mV was paired with 200 pulses of 2 Hz stimulation to the Schaffer collateral input. In both cases, the change in peak amplitude of the EPSC, relative to baseline, was used to assess the effects of these stimulation protocols on synaptic efficacy. On-line electrophysiology data acquisition and analysis were performed using WinLTP software (Anderson and Collingridge, 2007).
LTD induction and microdissection.
LTD was induced in hippocampal slices using low-frequency stimulation (LFS; slices were stimulated with 900 pulses at 1 Hz by a stimulation electrode placed in the Schaffer collateral–commissural fiber pathway). The CA1 dendritic region was microdissected immediately after treatment, as previously described (Kimura et al., 2014), and homogenized in lysis buffer containing 1% SDS, 5 mm NaF, 1 mm EDTA, 0.3 m sucrose, 1× protease inhibitor cocktail (Roche), and 1× phosphatase inhibitor cocktail 3 (Sigma-Aldrich).
Western blotting and antibodies.
Soluble protein lysates were resolved by SDS-PAGE and transferred to PVDF membranes. The following antibodies were used for probing: Tau-5, mouse monoclonal (1:500; Invitrogen); AT8, mouse monoclonal (1:200; Thermo Scientific); PHF-1, mouse monoclonal (1:1000; provided by Dr. P. Davies, Department of Pathology, Albert Einstein College of Medicine, Bronx, NY); pS396, rabbit polyclonal (1:1000; Life Technologies); pS404, rabbit polyclonal (1:1000; Life Technologies); and β-actin, mouse monoclonal (1:10,000; Abcam). Epitopes of phosphor (p)-tau antibodies are denoted by their position on the longest isoform (2N4R) of human tau (441 residues in length). Immunoreactive bands were imaged using the G:BOX XT4 imaging system (Syngene), and optical densities were quantified using NIH ImageJ software and normalized to relative control protein levels.
Coimmunoprecipitation.
Hippocampal slices from wild-type/tau KO mice or rats were treated with NMDA (25 μm NMDA for 3 min) and in some cases were pretreated with CT-99021 (1 μm). Crude cellular lysates were prepared in lysis/binding buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 15 μm CaCl2, and a protease inhibitor cocktail (Roche), and were precleared with washed protein G Sepharose beads for 1 h at 4°C. Aliquots (300 μg) of precleared lysates were subjected to immunoprecipitation with 3 μg of rabbit polyclonal anti-PICK1 H-300 antibody (Santa Cruz Biotechnology) for 4 h at 4°C, followed by immunocapture on washed protein G Sepharose beads for 2 h at 4°C. The immunoprecipitate complexes were washed three times with lysis/binding buffer followed by one wash with Tris-buffered saline and eluted with SDS sample buffer. Proteins were resolved by SDS-PAGE, and Western blotting was performed with the following antibodies: mouse monoclonal anti-GluA2 (1:1000; Millipore) and goat polyclonal anti-PICK1 C-20 (1:500; Santa Cruz Biotechnology). Optical densities of immunoreactive bands were quantified using NIH ImageJ software. GluA2 immunoreactivities were normalized to PICK1 band intensity.
Behavioral studies.
To assess spatial learning and memory, a Barnes circular maze test was performed as described below. The Barnes circular maze is a planar, round, white Plexiglas platform (90 cm in diameter), 1 m above the floor, with 20 evenly spaced holes (7 cm diameter) located 5 cm from the perimeter. A black escape box (15 × 7 × 7 cm) was placed under one hole. Spatial cues with distinct patterns and shapes were placed on the wall of the testing room. A 60 W light was turned on during the trial. An experimenter remained in the same place with minimal movement throughout the trials. The platform and the escape box were cleaned thoroughly with 70% ethanol and paper towels between each trial to remove olfactory cues. One day before the training trials began, test mice were habituated in the target box for 3 min. The training trials were repeated for 4 consecutive days, and three trials per day were performed with 20 min intertrial intervals. At the beginning of each trial, the mouse was placed in a cylindrical holding chamber (10 cm in diameter) that was located in the center of the maze. After 10 s of holding time, the mouse was allowed to search for the target hole for 3 min. If the mouse failed to find the target hole in 3 min, it was gently guided into the target hole by the experimenter's hands. When the mouse entered the escape box, the light was turned off, and the mouse remained undisturbed for 1 min. The movement of the mouse was recorded, and the latency to find the target hole was measured during the training trials by video tracking software (Nodulus). On day 5, the probe trial was performed with each mouse. The escape box was removed, and the test mouse was allowed to find the target hole freely for 90 s. During the probe trial, the percentage of time spent in each quadrant, the total distance traveled, and speeds were measured using the video tracking software. In reversal learning, the escape box was placed at the new quadrant, and the mice were then retrained over 4 d (on days 6–9). On day 10, each mouse was subjected to a single 90 s probe trial.
Statistics.
Data were analyzed from one slice per rat or mouse for biochemical experiments (n = number of slices = number of rats/mice), from individual cells for electrophysiology (n = number of cells) and from individual mice for behavioral data (n = number of mice). All data are expressed as the mean ± SEM. For synaptic plasticity experiments, the effects of the stimulation protocol were measured ∼30 min after LTD/LTP induction. Data are expressed relative to a normalized baseline or averaged control (100% = no change). Significance (p < 0.05) from baseline or control, or between mice, was tested using two-tailed paired or unpaired t tests or one-way ANOVAs, with post hoc pairwise Bonferroni correction where appropriate, as indicated in the figure legends. All experimental powers were calculated using SigmaPlot data analysis software and were above the recommended threshold of 0.8.
Results
Tau is required for an AMPAR endocytosis signaling interaction
Molecular processes of AMPAR endocytosis underpin the LTD-associated removal of AMPARs from the postsynaptic membrane. These processes are regulated, in part, by protein interactions between the C terminus of the GluA2 subunit of AMPARs and PICK1 (Hanley and Henley, 2005; Lu and Ziff, 2005; Terashima et al., 2008). Enhanced binding between PICK1 and GluA2 evokes AMPAR endocytosis following NMDAR activation and Ca2+ influx (Hanley and Henley, 2005), and preventing this interaction blocks AMPAR internalization and hippocampal LTD (Terashima et al., 2008).
Given that tau is required for hippocampal LTD and is phosphorylated by GSK-3β—a key regulator of AMPAR internalization (Du et al., 2010; Kimura et al., 2014)—during this process, we wondered whether tau itself might also regulate AMPAR internalization. Using a coimmunoprecipitation assay, we therefore tested the hypothesis that tau is an important regulator of an AMPAR endocytosis signaling mechanism, namely, the GluA2 and PICK1 interaction. NMDA treatment induces a chemical form of LTD (chem-LTD, data not shown) that is considered to be analogous to LTD induced by activity (Lee et al., 1998). Following NMDA treatment of hippocampal slices (25 μm, 3 min), we found an enhanced protein association between GluA2-PICK1 in WT mice but not in tau knock-out (KO) mice (NMDA in WT mice: 438 ± 85% of non-NMDA treatment; p = 0.016; n = 4; NMDA in tau KO mice: 104 ± 46% of non-NMDA treatment; p = 0.947; n = 4; Fig. 1A,B).
Figure 1.

NMDA leads to tau-dependent GluA2–PICK1 association. A, NMDA treatment (25 μm NMDA for 3 min) of WT hippocampal slices causes an increase in GluA2–PICK1 association that was not observed in tau KO hippocampal slices. B, Pooled data after NMDA treatment show significant increases in GluA2–PICK1 association in WT slices, with no significant change being observed in tau KO hippocampal slices. C, Representative Coimmunoprecipitation experiment showing that NMDA treatment of WT mouse hippocampal slices causes an increase in GluA2–PICK1 association that is attenuated by pretreatment with the GSK-3β inhibitor, CT-99021. D, Pooled data showing that CT-99021 blocks NMDA-induced changes in GluA2–PICK1 association in WT mice. E, Representative Coimmunoprecipitation experiment showing that NMDA treatment of WT rat hippocampal slices causes an increase in GluA2–PICK1 association that is attenuated by pretreatment with the GSK-3β inhibitor CT-99021. F, Pooled data showing that CT-99021 blocks NMDA-induced changes in GluA2–PICK1 association in WT rat. All bars represent the mean ± SEM. Significance (*p < 0.05, **p < 0.01, ***p < 0.001) was determined using an unpaired t test (B) comparing data to the relative control groups (non-NMDA), or a one-way ANOVA with post hoc pairwise Bonferroni test (D, F). IP, Immunoprecipitation.
The observation that tau is required for the GluA2–PICK1 interaction during LTD is intriguing, but could reflect nonspecific dysregulation of protein interactions in tau KO mice. To support the hypothesis that this role for tau is LTD specific, we tested whether blocking tau phosphorylation during LTD could also prevent the GluA2–PICK interaction. We used the GSK-3β inhibitor CT-99021 to prevent GSK-3β-mediated phosphorylation of tau during NMDA treatment. Critically, the observed GluA2–PICK1 enhancement after NMDA treatment in hippocampal slices from WT mice was significantly inhibited by pretreatment with CT-99021 (NMDA: 366.84 ± 51.7%; NMDA plus CT-99021: 142.76 ± 36.68%; p = 0.002; n = 8; Fig. 1C,D), while no significant modulation of the basal interaction was observed when CT-99021 was administered alone (control: 100 ± 28.24%; n = 8; CT-99021: 113.51 ± 29.96%; n = 4; p = 1.0). Previous studies from our laboratory and other laboratories have extensively investigated the mechanisms of LTD using rat hippocampus brain slices (Lee et al., 1998; Zhu et al., 2002; Dickinson et al., 2009; Li et al., 2010; Nicolas et al., 2012; Kimura et al., 2014). We therefore further verified the above findings in rat hippocampal slices treated in the same manner (NMDA: 153.2 ± 6.98%; NMDA plus CT-99021: 75.7 ± 10.45%; p = 0.008; n = 4; Fig. 1E,F). Treatment with CT-99021 alone in this model also had no effect on the basal interaction (control: 100 ± 10.95%; CT-99021: 70.39 ± 6.79%; p = 0.231; n = 4). Combined, these results suggest that dynamic phosphorylation of tau by GSK-3β during LTD processes specifically regulates the GluA2-PICK1 AMPAR internalization signaling interaction in two independent species.
NMDAR activation induces tau phosphorylation at the PHF-1 epitope
Though the exact function of phosphorylation of tau has yet to be fully characterized, the numerous phosphorylation sites of tau have been extensively examined (for review, see Hanger and Noble, 2011). We were therefore interested in determining whether tau was subject to specific phosphorylation events during LTD. LFS was delivered to Schaffer collateral axons of acutely dissected rat hippocampal slices to induce LTD at CA1 synapses (Dudek and Bear, 1992). Following LFS, we measured the phosphorylation status of tau in the microdissected CA1 region of the rat hippocampal slices, as previously described (Zhu et al., 2002; Peineau et al., 2007; Kimura et al., 2014), using the antibodies PHF-1 (an antibody that recognizes phosphorylation at residues S396 and S404 of tau; Otvos et al., 1994) and AT8 (an antibody that recognizes phosphorylation at S202 and T205 residues of tau; Goedert et al., 1995). PHF-1 and AT8 sites were chosen specifically as they are substrates for GSK-3β (Hanger and Noble, 2011), and their phosphorylation can confer distinct changes to tau protein folding (Jeganathan et al., 2008; Bibow et al., 2011) and localization (Pooler et al., 2012), both of which could alter the ability of tau to interact with proteins relevant to AMPAR trafficking. We observed a dramatic increase in the phosphorylation of tau using PHF-1 (174 ± 24% of averaged control; p = 0.005 vs non-LFS; n = 4; Fig. 2A,B). In contrast, no changes were found with AT8 (93 ± 13% of averaged control; p = 0.535; n = 4; Fig. 2A,B). It is well established that this form of LTD induced by LFS at hippocampal CA1 synapses is reliant upon the activation of postsynaptic NMDARs (Dudek and Bear, 1992; Mulkey and Malenka, 1992). Consistent with the phosphorylation of tau being of relevance to this form of LTD, the NMDAR antagonist AP5 blocked the LFS-induced phosphorylation of tau detected by PHF-1 (PHF-1: 111 ± 13% of averaged control; p = 0.045 vs LFS; Fig. 2A,B).
Figure 2.

NMDAR LTD stimulation evokes specific tau phosphorylation. A, Representative Western blot showing LFS induces an increase in the phosphorylation of tau at the PHF-1 epitope, and at individual S396 and S404 residues. These changes are prevented by treatment with the NMDAR antagonist AP5 (50 μm). No changes were observed in phosphorylation at the AT8 epitope or in total levels of tau (tau-5). B, Quantification of tau PHF-1 and AT8 phosphorylation levels normalized to tau-5 following LFS in the presence and absence of AP5. C, Quantification of tau S396 and S404 phosphorylation levels normalized to tau-5 following LFS in the presence and absence of AP5. D, Quantification of total tau levels normalized to β-actin. All bars represent the mean ± SEM. Significance (*p < 0.05; **p < 0.01) was determined using a one-way ANOVA with post hoc Bonferroni test.
In agreement with the above findings, antibodies recognizing phosphorylation at the individual S396 and S404 residues (Bramblett et al., 1993; Singer et al., 2005) also showed enhanced expression following LFS (pS396: 184 ± 7% of averaged control; p = 0.012 vs non-LFS; n = 4; pS404: 179 ± 19% of averaged control; p = 0.016 vs non-LFS; n = 6; Fig. 2A,C). This LFS-induced phosphorylation was similarly blocked by AP5 (pS396: 85 ± 16% of averaged control; p = 0.004 vs LFS; pS404: 98 ± 19% of averaged control; p = 0.014 vs LFS; Fig. 2A,C). No difference in the total levels of tau protein were found using Tau-5 (LFS: 105 ± 10% of averaged control; LFS plus AP5: 93 ± 20% of averaged control, n = 4; p = 0.844; Fig. 2A,D), an antibody that recognizes all tau independent of phosphorylation status (LoPresti et al., 1995). Together, these data suggest that specific tau phosphorylation is associated with NMDAR activity-dependent key molecular mechanisms of LTD.
Tau phosphorylation at serine 396 is required for LTD
So far, we have demonstrated that tau phosphorylation at specific serine residues within the PHF-1 epitope is associated with LTD. This observation raises a fundamental question: are these phosphorylation events functionally related to LTD, or are they simply a byproduct of enhanced activity of LTD signaling molecules such as GSK-3β (Peineau et al., 2007)? To answer this, we generated mutants of human tau that were phosphorylation null at particular sites, by mutating the serine/threonine residues to alanine (see Materials and Methods). Next, we biolistically cotransfected constructs containing rat tau-shRNA and the phosphomutant human tau (not targeted by rat tau-shRNA) into cultured hippocampal slice neurons. Delivering a 1 Hz, 200 pulse [holding potential (Vh) = −40 mV] stimulation routinely induced LTD in untransfected neurons (49.7 ± 4.2% of baseline; p = 0.006 vs baseline, n = 6; Fig. 3A). However, in neurons expressing S396/404A tau (phosphorylation null at the PHF-1 epitope), LTD was inhibited (92.5 ± 6.5% of baseline; p = 0.0001 vs untransfected neurons; n = 9; Fig. 3A). In contrast, LTD was robustly induced in neurons expressing S/T199/202/205A tau (phosphorylation null at the AT8 epitope; S/T199/202/205A: 43.9 ± 6.7% of baseline; n = 6; neighboring untransfected neurons: 40.6 ± 5.9% of baseline; n = 6; p = 0.721; Fig. 3C). In both sets of tau mutant neurons, LTP was assayed by pairing a 2 Hz, 200 pulse stimulation protocol with strong postsynaptic depolarization (Vh = 0 mV). LTP was present in both sets of neurons transfected with S396/404A (S396/404A: 164 ± 24% of baseline; n = 6; neighboring untransfected neurons: 164 ± 11%; n = 6; p = 0.866; Fig. 3B) or S/T199/202/205A tau (S/T199/202/205A: 163 ± 24% of baseline; n = 6; neighboring untransfected neurons: 175 ± 17% of baseline; n = 6; p = 0.662; Fig. 3D). These data suggest that phosphorylation at the PHF-1 epitope of tau is required for LTD, but not for LTP.
Figure 3.

Tau phosphorylation at serine 396 is required for LTD. A, LFS (black bar; 200 pulses at 1 Hz, holding voltage −40 mV) induces LTD in untransfected neurons (open circles) but not in neighboring neurons transfected with rat tau-shRNA plus S396/404A phosphomutant human tau (closed circles). B, A total of 200 pulses at 2 Hz (holding voltage 0 mV; black square) induces LTP in untransfected neurons (open circles) and in neurons transfected with S396/404A mutant tau (closed circles). C, LFS induces LTD in untransfected neurons (open circles) and in neighboring neurons transfected with rat tau-shRNA plus S/T199/202/205A phosphomutant human tau (closed circles). D, LTP is induced in both untransfected (open circles) and tau S/T199/202/205A-transfected neurons (closed circles). E, LTD is induced in untransfected neurons (open circles) but not in neighboring neurons transfected with rat tau-shRNA plus S396A phosphomutant human tau (closed circles). F, LTD is induced in both untransfected neurons (open circles) and neurons transfected with rat tau-shRNA plus S404A phosphomutant human tau (closed circles). Insets are representative traces of EPSCs recorded and averaged at time points indicated by the numbers on the graphs. All circles represent the mean ± SEM. Significance (***p < 0.001) was determined using unpaired t tests comparing transfected and untransfected neurons.
To further test the specificity of LTD inhibition by the mutant S396/404A tau, we generated phosphorylation-null mutants for the individual residues S396 and S404. We found that LTD was significantly impaired in neurons transfected with S396A mutant tau (S396A: 88.8 ± 11.1% of baseline; n = 6; neighboring untransfected neurons: 50.0 ± 5.4% of baseline; n = 6; p = 0.0002; Fig. 3E) but remained intact in neurons expressing S404A mutant tau (S404A: 68.7 ± 6.1% of baseline; n = 6; neighboring untransfected neurons: 51.4 ± 2.2% of baseline; n = 6; p = 0.370; Fig. 3F). These data highlight that phosphorylation of tau at the individual S396 residue is a critical molecular event during LTD.
Tau is required for reversal learning memory
Previous behavioral assays of tau KO mice have largely revealed few or no differences in cognition when compared with WT mice (Roberson et al., 2007; Ittner et al., 2010; Lei et al., 2012). However, as a specific role for tau in hippocampal LTD, but not LTP, has now been identified (Kimura et al., 2014), we tested a behavioral paradigm where LTD expression has been deemed important: hippocampus-dependent spatial reversal learning (Kim et al., 2011; Dong et al., 2013). We used the Barnes maze to test spatial learning and reversal memory function. No significant differences were observed in the total distance traveled by the mice (Fig. 4A; p = 0.136) or in their average velocity (Fig. 4B; p = 0.269) during a probe trial on day 5. Both WT and tau KO mice showed a similar ability to learn the location of the escape hole following successive trials on days 1–4 (day 4 escape latency: tau KO mice, 38 ± 10 s; n = 8; WT mice, 24 ± 6 s; n = 9; p = 0.126; Fig. 4C). Similarly, during a probe trial on day 5, both mice spent similar durations in the quadrant of the Barnes maze containing the escape hole [percentage of time spent in the target quadrant (TQ): tau KO mice, 58 ± 6%; WT mice, 44 ± 4%; p = 0.093; Fig. 4D]. However, upon repositioning the escape hole to a different quadrant on day 6, tau KO mice showed a deficit in learning the new location, when compared with WT mice, during subsequent trials on days 7–9 (escape latency on day 9: tau KO mice, 37 ± 9 s; WT mice, 11 ± 3 s; p = 0.014; Fig. 4E). Accordingly, during a probe trial on day 10 tau KO mice spent significantly less time in the new TQ compared with WT mice (percentage of time spent in TQ: tau KO mice, 38 ± 4%; WT mice, 58 ± 8%; p = 0.049; Fig. 4F). Interestingly, this was accompanied by the observation that tau KO mice spent a significantly longer time in the quadrant that was formerly occupied by the escape hole (percentage of time spent in the previous quadrant (PQ): tau KO mice, 25 ± 2%; WT mice, 14 ± 4%; p = 0.031; Fig. 4F). Together, these data indicate a selective deficit in spatial reversal learning as a result of genetic tau ablation in these mice.
Figure 4.
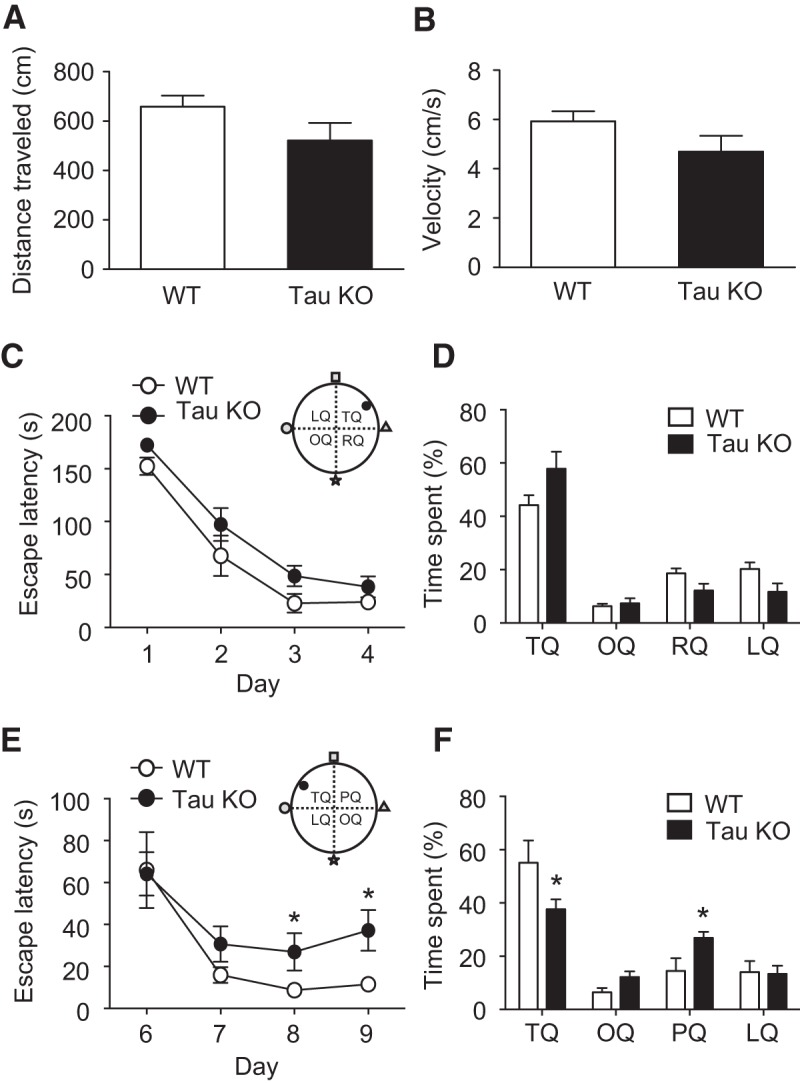
Reversal learning memory is impaired in tau KO mice. A, B, Total distance moved (A) and average velocity (B) in probe trials on day 5 of Barnes maze test are similar across both mice. C, Time taken to reach the escape hole in the training trials shows similar learning ability in WT and tau KO mice. D, Both WT and tau KO mice spend a similar percentage of time in the target quadrant during a probe trial on day 5 of the experiment. E, Time taken to reach the escape hole in the new training trials of a reversal Barnes maze test shows reduced learning in tau KO mice. F, The percentage of time spent in each quadrant during the probe trial on day 10 of the experiment is significantly different between WT and tau KO mice. OQ, opposite quadrant; RQ, right quadrant; LQ, left quadrant. All data are presented as the mean ± SEM. Significance (*p < 0.05) was determined using a two-tailed paired t test comparing tau KO and WT data.
Discussion
Tau phosphorylation is ordinarily considered in terms of aberrant hyperphosphorylation and aggregation under pathological neurotoxic conditions in the brain (Grundke-Iqbal et al., 1986; Ihara et al., 1986). Consequently, limited progress has been made in determining how specific tau phosphorylation events can affect neurons in a physiological sense. Tau can be phosphorylated at multiple epitopes, and there is some evidence to suggest that phosphorylation at particular sites can confer specific attributes to the tau protein (Noble et al., 2013). Here we have shown that tau is robustly phosphorylated at both the S396 and S404 residues following the activation of NMDARs. However, while S396 phosphorylation was required for LTD, S404 phosphorylation was not. The reasons for this discrepancy are unclear, but likely represent the shared tendency for both residues to become phosphorylated together (Li and Paudel, 2006), which is a specific mechanistic attribute of the S396 residue for LTD.
It has been suggested that phosphorylation at S396 contributes to reduced tau–microtubule binding (Bramblett et al., 1993), and is also correlated with C-terminal tau cleavage (Mondragón-Rodríguez, et al., 2014) and distinct conformational changes (Jeganathan et al., 2008). How these effects upon tau could be related to LTD is not yet apparent, but could potentially affect its synaptic translocation and its interaction with synaptic proteins such as PSD-95, Fyn kinase, or F-actin, which can alter synaptic strength (Ittner et al., 2010; Mondragón-Rodríguez et al., 2012). Nevertheless, our finding that a site-specific phosphorylation event on tau can confer a distinct physiological function at the level of synaptic modulation lends weight to the notion that future studies ought to examine more closely the phosphorylation code of tau, rather than broad levels of tau phosphorylation and hyperphosphorylation per se. We recognize that another study (Mondragón-Rodríguez et al., 2012) has shown that PHF-1, AT8, and AT180 epitopes all show enhanced phosphorylation after a chem-LTD stimulation. This differs somewhat from our findings. The reasons for the differences in observations are unclear, although the difference in LTD stimulation protocol is likely a significant factor. Interestingly, their study shows progressive changes in phosphorylation patterns of tau at different time points after LTD induction. Therefore, it requires further investigation to explain the dynamics of tau phosphorylation between the induction and maintenance of LTD in the future.
Another unanswered question is how tau might regulate AMPAR trafficking. It is clear from our GluA2–PICK1 coimmunoprecipitation data that the presence of tau is necessary for the occurrence of a molecular association that promotes AMPAR internalization and/or intracellular AMPAR retention (Lu and Ziff, 2005; Terashima et al., 2008). Notably, basal levels of GluA2–PICK1 interactions do not differ between WT and tau KO mice, rather the deficit is noticed only upon treatment with NMDA. This is consistent with an effector role for tau in the activity-driven internalization of GluA2-containing AMPARs mediated by PICK1 (Hanley and Henley, 2005; Terashima et al., 2008; Nakamura et al., 2011), rather than in constitutive GluA2 recycling. The knowledge of the full repertoire of molecules involved in AMPAR internalization and LTD is far from complete but likely involves hundreds of different proteins that are capable of regulating endocytotic molecules and scaffolding proteins, many of which could potentially be affected by tau. There is emerging evidence that alterations in microtubule dynamics play an important part in the expression of LTD (Kapitein et al., 2011). Conceivably then, through its interactions with both the microtubule and actin networks, tau may provide a mechanistic link between these cytoskeletal elements in the trafficking of AMPARs at the synapse.
Finally, in our study we address the potential and hypothetical behavioral relevance of the role of tau in LTD. Tau KO mice appear to exhibit normal cognition in spatial learning tasks (Roberson et al., 2007; Ittner et al., 2010; Lei et al., 2012). What, then, is the underlying relevance of the finding that tau is required for LTD? The answer appears to stem from the physiological relevance of hippocampal LTD itself. Conceptually, LTD-like physiological synapse weakening is thought to contribute to information encoding and discrimination (Kemp and Manahan-Vaughan, 2007), and is considered to be a fundamental cellular mechanism in certain cognitive processes such as novelty discrimination and tasks requiring behavioral flexibility (Manahan-Vaughan and Braunewell, 1999; Nicholls et al., 2008). In particular, a number of studies now link spatial reversal learning to the molecular mechanisms of hippocampal NMDAR-dependent LTD (Nicholls et al., 2008; Kim et al., 2011; Dong et al., 2013). Our novel finding of a deficit in spatial reversal learning memory in tau KO mice may therefore reflect the role for LTD-like signaling in certain cognitive functions.
In summary, we have shed further light on a physiological role for tau at the synapse. Tau joins a plethora of dynamically interacting synaptic proteins, which interact in a spatially and temporally distinct manner to ensure specificity of synaptic events. Phosphorylation is an important regulator of such specificity, and it is already established that phosphorylation at specific residues of other proteins, such as serine 845 of GluA1 (Esteban et al., 2003) or serine 880 of GluA2 (Seidenman et al., 2003) are important signals for the modulation of AMPAR trafficking during synaptic plasticity. Here we have shown that the internalization of AMPARs is associated with changes in tau function induced by phosphorylation at the individual serine 396 residue. This appears to be a critical event during a serial LTD signaling cascade and paves the way for further examination of tau phosphorylation in a physiological, rather than a pathological, sense.
Footnotes
K.C. and D.J.W. were supported by the UK Wellcome Trust-Medical Research Council (MRC) Neurodegenerative Disease Initiative. D.-H.K. and J.-H.Y. were supported by the Korea-UK Alzheimer's Research Consortium Programme under the auspices of the Korean Ministry of Health and Welfare. P.R. was supported by the MRC. D.J.W. was supported by the Royal Society, London. K.C. was supported by the Wolfson Research Merit Award and Royal Society, London.
The authors declare no competing financial interests.
References
- Anderson WW, Collingridge GL. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–356. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bibow S, Ozenne V, Biernat J, Blackledge M, Mandelkow E, Zweckstetter M. Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J Am Chem Soc. 2011;133:15842–15845. doi: 10.1021/ja205836j. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-X. [DOI] [PubMed] [Google Scholar]
- Cáceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343:461–463. doi: 10.1038/343461a0. [DOI] [PubMed] [Google Scholar]
- Dickinson BA, Jo J, Seok H, Son GH, Whitcomb DJ, Davies CH, Sheng M, Collingridge GL, Cho K. A novel mechanism of hippocampal LTD involving muscarinic receptor-triggered interactions between AMPARs, GRIP and liprin-alpha. Mol Brain. 2009;2:18. doi: 10.1186/1756-6606-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Bai Y, Wu X, Li H, Gong B, Howland JG, Huang Y, He W, Li T, Wang YT. Hippocampal long-term depression mediates spatial reversal learning in the Morris water maze. Neuropharmacology. 2013;64:65–73. doi: 10.1016/j.neuropharm.2012.06.027. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Feinstein SC, Shooter EM, Kirschner MW. Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol. 1985;101:1799–1807. doi: 10.1083/jcb.101.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Wei Y, Liu L, Wang Y, Khairova R, Blumenthal R, Tragon T, Hunsberger JG, Machado-Vieira R, Drevets W, Wang YT, Manji HK. A kinesin signaling complex mediates the ability of GSK-3β to affect mood-associated behaviors. Proc Natl Acad Sci U S A. 2010;107:11573–11578. doi: 10.1073/pnas.0913138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci U S A. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, Mandelkow E, Zweckstetter M. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48:10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-E. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger DP, Noble W. Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation. Int J Alzheimers Dis. 2011;2011:352805. doi: 10.4061/2011/352805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara Y, Nukina N, Miura R, Ogawara M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimers-disease. J Biochem. 1986;99:1807–1810. doi: 10.1093/oxfordjournals.jbchem.a135662. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325:167–172. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Yau KW, Gouveia SM, van der Zwan WA, Wulf PS, Keijzer N, Demmers J, Jaworski J, Akhmanova A, Hoogenraad CC. NMDA receptor activation suppresses microtubule growth and spine entry. J Neurosci. 2011;31:8194–8209. doi: 10.1523/JNEUROSCI.6215-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp A, Manahan-Vaughan D. Hippocampal long-term depression: master or minion in declarative memory processes? Trends Neurosci. 2007;30:111–118. doi: 10.1016/j.tins.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Kim JI, Lee HR, Sim SE, Baek J, Yu NK, Choi JH, Ko HG, Lee YS, Park SW, Kwak C, Ahn SJ, Choi SY, Kim H, Kim KH, Backx PH, Bradley CA, Kim E, Jang DJ, Lee K, Kim SJ, et al. PI3Kγ is required for NMDA receptor-dependent long-term depression and behavioral flexibility. Nat Neurosci. 2011;14:1447–1454. doi: 10.1038/nn.2937. [DOI] [PubMed] [Google Scholar]
- Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo S, Brown C, Hashikawa T, Murayama M, Seok H, Sotiropoulos I, Kim E, Collingridge GL, Takashima A, Cho K. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130144. doi: 10.1098/rstb.2013.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer's disease. Ann Neurol. 1987;22:639–643. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/S0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BXW, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- Li T, Paudel HK. Glycogen synthase kinase 3beta phosphorylates Alzheimer's disease-specific Ser396 of microtubule-associated protein tau by a sequential mechanism. Biochemistry. 2006;45:3125–3133. doi: 10.1021/bi051634r. [DOI] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo DC, McAllister AK, Katz LC. Neuronal transfection in brain slices using particle-mediated gene transfer. Neuron. 1994;13:1263–1268. doi: 10.1016/0896-6273(94)90412-X. [DOI] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci U S A. 1995;92:10369–10373. doi: 10.1073/pnas.92.22.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47:407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Manahan-Vaughan D, Braunewell KH. Novelty acquisition is associated with induction of hippocampal long-term depression. Proc Natl Acad Sci U S A. 1999;96:8739–8744. doi: 10.1073/pnas.96.15.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochem Int. 2011;58:458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Mayford M, Siegelbaum SA, Kandel ER. Synapses and memory storage. Cold Spring Harb Perspect Biol. 2012;4:a005751. doi: 10.1101/cshperspect.a005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragón-Rodriguez S, Basurto-Islas G, Santa-Maria I, Mena R, Binder LI, Avila J, Smith MA, Perry G, García-Sierra F. Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer's disease. Int J Exp Pathol. 2008;89:81–90. doi: 10.1111/j.1365-2613.2007.00568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem. 2012;287:32040–32053. doi: 10.1074/jbc.M112.401240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragon-Rodríguez S, Perry G, Luna-Muñoz J, Acevedo-Aquino MC, Williams S. Phosphorylation of tau protein at sites Ser(396–404) is one of the earliest events in Alzheimer's disease and Down syndrome. Neuropathol Appl Neurobiol. 2014;40:121–135. doi: 10.1111/nan.12084. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-C. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Wood CL, Patton AP, Jaafari N, Henley JM, Mellor JR, Hanley JG. PICK1 inhibition of the Arp2/3 complex controls dendritic spine size and synaptic plasticity. EMBO J. 2011;30:719–730. doi: 10.1038/emboj.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S, Kandel ER. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008;58:104–117. doi: 10.1016/j.neuron.2008.01.039. [DOI] [PubMed] [Google Scholar]
- Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, Sim SE, Bradley C, Lee K, Zhuo M, Kaang BK, Gressens P, Dournaud P, Fitzjohn SM, Bortolotto ZA, Cho K, et al. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374–390. doi: 10.1016/j.neuron.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W, Hanger DP, Miller CC, Lovestone S. The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol. 2013;4:83. doi: 10.3389/fneur.2013.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL. LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, Hanger DP. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging. 2012;33:431.e27–e38. doi: 10.1016/j.neurobiolaging.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Reynolds CH, Garwood CJ, Wray S, Price C, Kellie S, Perera T, Zvelebil M, Yang A, Sheppard PW, Varndell IM, Hanger DP, Anderton BH. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J Biol Chem. 2008;283:18177–18186. doi: 10.1074/jbc.M709715200. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23:9220–9228. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer D, Volke D, Hoffmann R. Characterization of phosphorylation dependent antibodies to study the phosphorylation status of the tau protein. Int J Pept Res Ther. 2005;11:279–289. doi: 10.1007/s10989-005-9269-4. [DOI] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-M. [DOI] [PubMed] [Google Scholar]
- Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JT. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008;57:872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usardi A, Pooler AM, Seereeram A, Reynolds CH, Derkinderen P, Anderton B, Hanger DP, Noble W, Williamson R. Tyrosine phosphorylation of tau regulates its interactions with Fyn SH2 domains, but not SH3 domains, altering the cellular localization of tau. FEBS J. 2011;278:2927–2937. doi: 10.1111/j.1742-4658.2011.08218.x. [DOI] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/S0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]