

Abstract
The hypothetical scanning (HS) method is a general approach for calculating the absolute entropy and free energy by analyzing Boltzmann samples obtained by Monte Carlo (MC) or molecular dynamics techniques. With HS applied to a fluid, each configuration i of the sample is reconstructed by adding its atoms gradually to the initially empty volume, i.e., by placing them in their positions at i using transition probabilities (TPs). At each step of the process, the volume is divided into two parts, the already visited part (the “past”) and the “future” part, where obtaining the TP requires calculating partition functions over the future part in the presence of the frozen past. In recent publications, the TPs were calculated approximately by taking into account only partial future. Here we present a “complete HSMC” procedure, where the TPs are calculated from MC simulations carried out over the complete future. The complete HSMC method is applied to systems of liquid argon and the TIP3P model of water, and very good results for the free energy are obtained, as compared with results obtained by thermodynamic integration.
The absolute entropy, S, is a measure of order and is also a fundamental component of the absolute Helmholtz free energy, F, F = E - TS, where E is the energy and T is the absolute temperature. The free energy is a key quantity as it constitutes the correct criterion of stability, which is mandatory for determining the relative populations of protein structures, for example. However, calculation of S and therefore F of a complex system, such as a peptide or a protein in water by computer simulation, is an extremely difficult problem (1–4). Using any simulation technique, it is relatively easy to calculate the energy, Ei, which is “written” on system configuration i in terms of microscopic interactions (e.g., Lennard–Jones interactions of argon). On the other hand, calculating S ≈ -ln 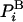 requires knowledge of the value of the Boltzmann probability, which is the sampling probability. However,
requires knowledge of the value of the Boltzmann probability, which is the sampling probability. However, 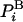 is not provided directly by the commonly used dynamical techniques, Metropolis Monte Carlo (MC) (5) and molecular dynamics (MD) (6–7), therefore, F is unknown as well. In most cases, calculation of F is based on reversible thermodynamic integration (TI) techniques that provide the difference in the free energy, ΔFm,n, between two states m and n, (e.g., helical and hairpin states of a peptide) and only when the absolute entropy of one state is known can that of the other be obtained. Although TI is a robust approach (refs. 1–4, 8, and 9 and references therein), for proteins, such integration is feasible only if the structural variance between the two states is very small; otherwise, the integration path can become prohibitively lengthy and complex. Therefore, it is important to develop methods that provide ln
is not provided directly by the commonly used dynamical techniques, Metropolis Monte Carlo (MC) (5) and molecular dynamics (MD) (6–7), therefore, F is unknown as well. In most cases, calculation of F is based on reversible thermodynamic integration (TI) techniques that provide the difference in the free energy, ΔFm,n, between two states m and n, (e.g., helical and hairpin states of a peptide) and only when the absolute entropy of one state is known can that of the other be obtained. Although TI is a robust approach (refs. 1–4, 8, and 9 and references therein), for proteins, such integration is feasible only if the structural variance between the two states is very small; otherwise, the integration path can become prohibitively lengthy and complex. Therefore, it is important to develop methods that provide ln 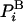 at least approximately, enabling one to calculate the absolute Fm and Fn from two samples of the states m and n; in this case ΔFm,n = Fm - Fn can be calculated even for significantly different states because the integration process is avoided.
at least approximately, enabling one to calculate the absolute Fm and Fn from two samples of the states m and n; in this case ΔFm,n = Fm - Fn can be calculated even for significantly different states because the integration process is avoided.
A unique approach for calculating the absolute entropy has been proposed by Meirovitch, where two related approximate techniques, the local states (10–14) method and the hypothetical scanning (HS) method (15–17), have been developed and applied to magnetic systems, polymers, and peptides. These methods demonstrate that like the energy, 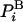 is also “written” on system configuration i in terms of a product of transition probabilities (TPs), where each method provides a different prescription for “reading” these TPs from i. Thus, the absolute entropy can be obtained approximately from a given Boltzmann sample generated by MC or MD, or any other exact technique.
is also “written” on system configuration i in terms of a product of transition probabilities (TPs), where each method provides a different prescription for “reading” these TPs from i. Thus, the absolute entropy can be obtained approximately from a given Boltzmann sample generated by MC or MD, or any other exact technique.
Our long-term goal is to be able to calculate the absolute free energy of a peptide or a surface loop of a protein immersed in explicit water. Therefore, in recent studies, as a first step, the HS method has been extended to liquid argon in two different approximations, one called grand canonical hypothetical scanning (8) and the other hypothetical scanning Monte Carlo (HSMC) (9). Although very good results were obtained, in some cases better accuracy would be needed. An additional issue is that the methods are implemented with boundary conditions that are different from those used to generate the analyzed sample (typically periodic boundary conditions). This inconsistency, along with the way the TPs are calculated in general, makes these methods inconvenient to apply to inhomogeneous systems such as a peptide solvated in a box of water molecules. Therefore, in this paper we develop a method called complete HSMC that leads to the exact entropy for infinite MC sampling; this method is applied to argon systems of different size, and to a system of 64 TIP3P water molecules (18). In the companion paper (19), the complete HSMC method is extended to peptides, which ultimately makes our approach applicable to model peptide-explicit water systems. In what follows, we describe the complete HSMC method as applied to liquids.
Theory and Implementation
Free Energy and its Fluctuations. We start by defining the free energy and discussing some of its properties. For simplicity, we consider a discrete system of configurations, i with energy Ei. The Boltzmann probability, 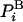 , is
, is
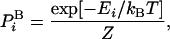 |
[1] |
where kB is the Boltzmann constant, T is the absolute temperature, and Z is the partition function. [For continuum systems, 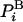 is replaced by the corresponding probability density (see below).] Using
is replaced by the corresponding probability density (see below).] Using 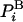 , the entropy, S, and free energy, F, can be formally expressed as ensemble averages:
, the entropy, S, and free energy, F, can be formally expressed as ensemble averages:
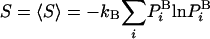 |
[2] |
and
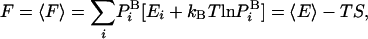 |
[3] |
where 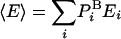 is the average energy. An extremely important property of this representation of F (but not other representations) is that the variance vanishes, Δ2F = 0 (17, 20); indeed, substituting the expression for
is the average energy. An extremely important property of this representation of F (but not other representations) is that the variance vanishes, Δ2F = 0 (17, 20); indeed, substituting the expression for 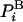 in the brackets (Eq. 3) leads to a constant, -kBT ln Z for any i. This means that the exact free energy can be obtained from a single structure i if ln
in the brackets (Eq. 3) leads to a constant, -kBT ln Z for any i. This means that the exact free energy can be obtained from a single structure i if ln 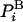 is known! Moreover, although F is an extensive variable, its zero fluctuation property holds for any number of atoms N. This important property is not shared by the entropy and the energy: their fluctuations increase as ≈√N, and therefore it is difficult to estimate them accurately for a large system.
is known! Moreover, although F is an extensive variable, its zero fluctuation property holds for any number of atoms N. This important property is not shared by the entropy and the energy: their fluctuations increase as ≈√N, and therefore it is difficult to estimate them accurately for a large system.
In practice, however, estimation of 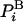 in simulations will always be approximate. In particular, with the HS method described below, the configurations generated with an exact method such as MC (i.e., with
in simulations will always be approximate. In particular, with the HS method described below, the configurations generated with an exact method such as MC (i.e., with 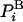 ), are analyzed and their probabilities are estimated approximately by
), are analyzed and their probabilities are estimated approximately by 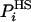 . This procedure can, in principle, be applied to all the ensemble configurations defining thereby approximate entropy and free energy functionals, SA and FA, respectively, over the entire configurational space:
. This procedure can, in principle, be applied to all the ensemble configurations defining thereby approximate entropy and free energy functionals, SA and FA, respectively, over the entire configurational space:
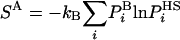 |
[4] |
and
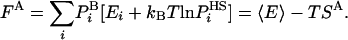 |
[5] |
SA can be shown rigorously to be an upper bound for the correct entropy S (16), thus FA is a lower bound of F. In HS implementations 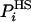 is generally a function of some set of parameters or running conditions (see for example refs. 8 and 9). These parameters effectively determine the level of approximation of
is generally a function of some set of parameters or running conditions (see for example refs. 8 and 9). These parameters effectively determine the level of approximation of 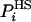 , the better the approximation the smaller is SA (and the larger is FA). For an approximate
, the better the approximation the smaller is SA (and the larger is FA). For an approximate 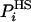 , the fluctuation (variance), Δ2FA is not zero, i.e., [
, the fluctuation (variance), Δ2FA is not zero, i.e., [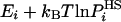 ] is not constant for all i; one can estimate it from a Boltzmann sample of size n, by the arithmetic average,
] is not constant for all i; one can estimate it from a Boltzmann sample of size n, by the arithmetic average,
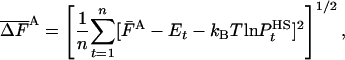 |
[6] |
where the bar over F stands for estimation. However, this fluctuation is expected to decrease as the approximation improves, where for very good approximations of 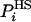 , the free energy can be very accurately determined by averaging [
, the free energy can be very accurately determined by averaging [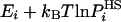 ] over just a handful of configurations (or even a single one). The present complete HSMC method can provide this accuracy, and very good values for the free energy have been obtained from a small number of configurations.
] over just a handful of configurations (or even a single one). The present complete HSMC method can provide this accuracy, and very good values for the free energy have been obtained from a small number of configurations.
Statistical Mechanics of the Liquid Models. In this paper, we study liquid models for argon and water. Argon is represented by the standard Lennard–Jones potential with ε/kB = 119.8 K and σ = 3.405 Å; water is represented by the three-site TIP3P potential (18). We consider N atoms (molecules) enclosed in a box of volume, V, at temperature, T [(NVT) ensemble]. The configurational partition function is given by
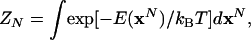 |
[7] |
where E(xN) is the potential energy, xN is the set of Cartesian and orientational (for water) coordinates, and dxN is the corresponding differential (including any necessary Jacobian factors). The integration is carried out over the configurational space, VN for argon and (8π2V)N for water. Using the Boltzmann configurational probability density ρ(xN),
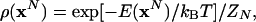 |
[8] |
the total entropy, S, is
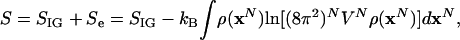 |
[9] |
where SIG is the entropy of the ideal gas at the same temperature and density, and Se is the excess entropy. The factor, (8π2)N, would be replaced by unity for argon. The corresponding excess Helmholtz free energy is
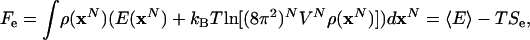 |
[10] |
where 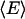 is the average potential energy. For water, we present results for Fe; however, to be consistent with the literature, for argon the configurational free energy, Ac, (21) is provided:
is the average potential energy. For water, we present results for Fe; however, to be consistent with the literature, for argon the configurational free energy, Ac, (21) is provided:
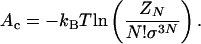 |
[11] |
The Ideal HS Method. It is helpful to introduce the complete HSMC method of this paper by first describing a “perfect” or “limiting” scenario that we will term as ideal HS. For simplicity, we describe the method as applied to argon; the extension for water is straightforward. Assume an NVT argon system with periodic boundary conditions simulated by MC. As discussed in the Introduction, the sample configurations are distributed according the Boltzmann probability density, but its value is not provided by this method, and therefore the absolute entropy, ≈ln ρ (xN), cannot be obtained in a direct manner. However, each argon configuration, in principle, could have been generated by an alternative exact build-up procedure, where argon atoms are added step-by-step to the initially empty volume (box) by using TPs. With the HS method, the given MC sample is assumed to have been generated by this exact build-up procedure, and thus each configuration is reconstructed with this procedure, the TPs are calculated, and their product leads to ρ(xN).
In practice, the box is divided into L3 = L × L × L cubic cells with the maximal size that still guarantees that no more than one center of a spherical argon molecule occupies a cell. During the analysis of configuration i, the cells are visited orderly line-by-line, layer-by-layer starting from one corner of the box until all of them have been treated. The calculation of TPk for the target cell k [which could be a vacant (-) or a populated (+) cell] is outlined as follows. At step k of the process, Nk atoms (i.e., occupied cells) and k - 1 - Nk vacant cells have already been treated, i.e., their TPs have been calculated. These Nk atoms are now positioned at their coordinates of configuration i and together with the already visited vacant cells they define the (frozen) “past”; the L3 - (k - 1) as yet unvisited cells (including target cell k) define the “future volume.” To determine the TP of target cell k, two future canonical partition functions are calculated, Z-(k) and Z+(k) for vacant and occupied cell k, respectively, by scanning all of the possible configurations of the remaining N - Nk (future) atoms in the future volume, whereas the past is excluded, and for Z-(k), the target cell k is excluded as well.
If cell k is vacant, TPk is p(k, -) = Z-(k)/[Z+(k) + Z-(k)]. If cell k is occupied, then the future partition function, Z+(k, x′), is calculated where one of the future atoms is fixed at the position, x′, the exact location at which the atom was exhibited in configuration i. TPk for an occupied cell is the probability density, Z+(k, x′)/{[Z+(k) + Z-(k)]}. After cell k has been treated, it becomes a past cell, empty or occupied according to configuration i. For a periodic system, this means that the images of cell k are also becoming part of the fixed past and will thus affect the TPs of the L3 - k remaining future cells. In this HS procedure, all of the L3 TPs are calculated exactly and their product leads exactly to ρ(xN) (Eq. 8); therefore, we refer to this procedure as the ideal HS method. However, in practice, scanning the entire conformational space to systematically calculate the exact future partition functions is unfeasible.
Transition Probabilities of the Complete HSMC Method. Because the ideal HS method is intractable, one has to resort to approximations. Thus, in two recent papers, the future partition functions were calculated by considering a limited future. In one method, grand canonical HS (8), grand canonical future partition functions based on only four future cells occupied by up to two atoms were calculated in a systematic way. To increase the number of future atoms, in a following paper (9) we replaced the systematic integration of the grand canonical partition functions by canonical MC simulations. Thus, at each step, Nf future atoms were simulated in a specified future volume (in the presence of a frozen past) and the TP was determined from the number of counts of atoms in the target cell. With this HSMC version of HS up to Nf = 40 future atoms were treated. As said earlier, these implementations of HS were carried out in special boundary conditions, different from the periodic boundary conditions used to generate the analyzed sample, which would make it difficult to apply the methods to an inhomogeneous system. Therefore, in this paper we develop an HSMC method where the complete future is considered at each HS step (i.e., all of the future atoms are simulated by MC) and periodic boundary conditions are used in the reconstruction procedure. This method, called complete HSMC, is capable, in principle, of yielding the ideal HS result (described above) in the limit of infinite MC sampling. Indeed, we demonstrate later that the accuracy increases significantly as the sampling is enhanced. In fact, we were able to obtain results that are an order of magnitude more accurate than those obtained with the approximate HSMC (9). Because of this dependence on sampling, we denote the complete HSMC results for the free energy by FA (Eq. 5).
Complete HSMC is conducted as follows: at step k, the previously defined Nk atoms, as well as their associated images, will remain fixed in their assigned positions (in configuration i), whereas all the remaining N - Nk future atoms are allowed to move. An MC trajectory is generated for the N - Nk future atoms (in the presence of the Nk fixed atoms) under standard periodic rules, with the exception that regions inside previously defined cells are excluded. Any trial move that would place a future atom into this previously assigned volume is rejected. A 2D representation of the main simulation box is given in Fig. 1. It is evident that as this treatment proceeds the number of moveable future atoms decreases, and the fully mobile system at the beginning, becomes gradually “frozen down” into the exact configuration i.
Fig. 1.

A 2D illustration of the main simulation box at the kth step of the complete HSMC reconstruction. The 2D volume is divided into cells, where k - 1 of them have already been considered in previous steps (starting from the upper left corner). These k - 1 cells comprise the “past volume” (the region above the heavy lines), which contains previously treated fixed atoms that are denoted by full black circles defined by the van der Waals radius. This region is excluded from the moveable future atoms (denoted by full gray circles), which are thus simulated in the “future volume” below the heavy lines, in the presence of the fixed atoms. The future atoms can visit the target cell k (depicted by dotted lines) and their counts in this cell lead to the transition probability of an empty cell or the transition probability density of an occupied one. Note that for the case of an occupied target cell, counts are actually accumulated for visitations to a smaller region, Vcube (see text) located inside the target cell but not shown in the figure.
The transition probabilities are calculated (from the counts) in the following way. We denote by Mtot the total number of attempted moves in the MC simulation for any reconstruction step k. Mcell is the number of counts for which an atom was observed in the target cell k. The probability for the target cell to be occupied (unoccupied) by an atom is thus given by
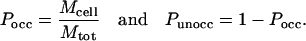 |
[12] |
For the case where the target cell k is vacant in configuration i, the transition probability is TPk = Punocc. For an occupied target cell, one has to calculate the probability density, ρocc, for an atom to be located at the precise location (inside cell k) at which it is found in configuration i. For this we define a much smaller volume (inside cell k), termed a “cube,” which is centered at the exact atom position (in configuration i). We count the visitations of atoms within this cube during the MC simulation and thus estimate the probability density as
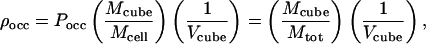 |
[13] |
where Mcube is the number of cube counts and Vcube is the cube volume. For occupied target cells TPk = ρocc. Note, however, that the probability density is assumed to be uniform over the cube volume. To increase the quality of the results, we actually scale ρocc by an ensemble averaged weighting factor (described in ref. 9), which serves to measure the probability density at the atom position more accurately.
The total product of TPk over all L3 cells, a product of N transition probability densities ρocc and L3 - N transition probabilities for empty cells, gives rise to the estimate, ρHS(xN), for the Boltzmann probability density, ρ(xN):
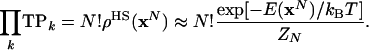 |
[14] |
We use the superscript HS because Eq. 14 holds for any HS approximation, not only for complete HSMC discussed above. Notice that the counting procedure (for the TPk) does not distinguish between labeled atoms, whereas in the integration leading to ZN, all of the labeled arrangements contribute, hence ρ(xN) (Eqs. 8, 9, 10) is a labeled probability density, and N! is required in Eq. 14.
The Choice of Vcube. Provided that the ensemble averaged weighting factor is used, implementations with different values for Vcube will always yield the correct free energy, F, in the limit of very long runs. However, for finite length runs, the quality of the free energy bound, FA, is affected by the size of Vcube. Generally, the ensemble averaged weighting factor (which is a function of cube size) will converge more readily as Vcube is made smaller, but a cube that is too small will lead to statistically unreliable cube counts. Thus, cube sizes at either extreme (too large or too small) will give rise to higher fluctuations and lower (poorer) values of FA.
The following points should be considered when choosing Vcube. The probability density is most sensitive to repulsive van der Waals overlaps; therefore, Vcube should be small on a scale of the molecular size. Still, a considerable range of Vcube values can give acceptable performance. For example, defining VvdW = (4/3)π(σ/2)3 as the molecular size of argon, we have found that values of Vcube/VvdW ranging from 5 × 10-5 to 1 × 10-2 work reasonably well. Although we have not done a systematic optimization, the results reported in this work were generated by using Vcube/VvdW values of ≈2 × 10-3 (see also Table 1). We also used about the same value of Vcube/VvdW for TIP3P water (Vcube is denoted by VCart for water, see below). Here, one can adopt the Lennard–Jones σ value of oxygen or the first peak in the oxygen–oxygen radial distribution function as an approximate molecular diameter. Similar considerations can be used for other molecules.
Table 1. The systems studied and the standard MC and HSMC running conditions.
| N | Density | T, K | Box length, Å | Cell length, Å | Cube length, Å |
|---|---|---|---|---|---|
| Argon | |||||
| 32 | 0.846† | 96.53 | 11.4293 | 2.2859 | 0.3810‡ |
| 64 | 0.846† | 96.53 | 14.4 | 2.40 | 0.3429‡ |
| 125 | 0.846† | 96.53 | 18.0 | 2.25 | 0.3750‡ |
| Water | |||||
| 64 | 1.000 g/cm3 | 298.15 | 12.418 | 2.0697 | 0.2957§ |
Results in reduced density, ρ* = Nσ3/V
We have used a convention such that (cube length) = (cell length)/J, where J is an integer (J = 6, 7, and 6 for N = 32, 64, and 125, respectively); therefore, the cube lengths for the three argon systems are slightly different. This convention is unnecessary, however, and all three systems could have been run at any one of these cube lengths yielding negligible differences in performance compared with the current results
Counts for ρocc (Eq. 13) also require the molecular orientation to be in an angular volume of 0.0125 π2
Enhancements of the Method. Several enhancements to the complete HSMC method have been added to increase the efficiency and accuracy. One of which is the ensemble averaged weighting factor (mentioned above), which gives more accurate transition probability densities. Another addition is MC preferential sampling (22–25), which imposes more frequent trial moves for atoms which are close to the target cell. Additionally, the total MC run length for any particular target cell is based on its estimated sampling difficulty, which is determined during the equilibration period. In other words, more steps are given to cells that would be expected to have low transition probabilities. All of these enhancements are described in more detail in ref. 9. However, as discussed later, the efficiency of the method can still be increased significantly.
Modifications for Water. The implementation described above for argon can be straightforwardly adapted for water. The only fundamental difference is that the molecular orientations must also be taken into account. We take the oxygen as the “center” of the molecule and thus determine Pocc and Punocc as described earlier. However, for the transition probability densities, we must alter the meaning of Vcube in Eq. 13. Thus, we define Vcube = VCartVang, where VCart as before is a small Cartesian cube and Vang is a small orientational region (i.e., a small portion of the total 8π2 molecular angular volume). These Cartesian and angular regions are centered on the exact position and orientation of the actual water molecule in configuration i. Thus in the MC simulation, a (future) water molecule will be located in this “molecular cube” whenever its oxygen is located within the specified Cartesian cube VCart and, simultaneously, the molecular orientation lies in the specified region of angular volume, Vang. With these counts, the transition probability density ρocc can be computed by using Eq. 13, where the above molecular cube, Vcube is used. As for argon, an ensemble averaged weighting factor is also applied to increase the accuracy of the probability density. Although the efficiency of the method does depend on the treatment of Vang, we have found, as for the case of Vcart, that acceptable performance can be obtained over a reasonably wide range of specifications. Vang = 0.0125π2 in this work is ≈0.2% of the total molecular angular volume, 8π2 (see Table 1).
Results and Discussion
The argon systems are comprised of 32, 64, and 125 atoms at T = 96.53 K and reduced density, ρ* = Nσ3/V = 0.846. In all cases, the interactions were spherically truncated at a distance equal to half the box length, and the long-range energy (tail) corrections were added to the results (22). The water system consists of 64 TIP3P water molecules at density 1.000 g/cm3 and temperature T = 298.15 K. Here molecule-based spherical truncation was applied (again, at half the box length), but no long-range correction was added. The details of the MC and HSMC running conditions are summarized in Table 1.
The sample configurations (which are analyzed in the complete HSMC procedure) were generated by using the usual Metropolis MC simulation method (5) in the (NVT) ensemble under standard periodic boundary conditions. Thus, at each MC step an atom (molecule) is selected at random and a random translational trial move is generated within a small Cartesian cube around the atom position. For water, the molecule is also given a small (random) rotation about a randomly chosen axis. Cartesian and rotational step sizes were chosen to give ≈40–50% acceptance. Configurations were recorded at long enough intervals to give an uncorrelated sample. The MC simulations of the future molecules during the HS reconstruction process are very similar to the standard MC simulations. The exceptions (which were discussed above) are: that the system is only partially mobile, the moveable future atoms (molecules) are excluded from the previously treated regions, and atoms (molecules) are selected preferentially for trial moves based on their proximity to the target cell. Additionally, the number of MC steps at each cell, Mtot was not constant but decreased (on average) in the course of the reconstruction process, i.e., with increasing k, and in general was significantly larger for populated cells than for vacant ones. The results in Tables 2 and 3 are given as a function of the average number of MC steps per cell, denoted M̄tot. For the sake of brevity, we shall often refer to the complete HSMC method as the HSMC method.
Table 2. Complete HSMC results for argon.
| Mtot | —Ac/εN(FA) | 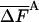 |
n |
|---|---|---|---|
| N = 32 | |||
| 360,000 | 4.1432 (11) | 0.0533 (5) | 2,205 |
| 720,000 | 4.1260 (8) | 0.0369 (5) | 2,001 |
| 1,440,000 | 4.1159 (8) | 0.0284 (5) | 1,410 |
| 3,600,000 | 4.1102 (5) | 0.0171 (5) | 1,127 |
| 7,200,000 | 4.1079 (4) | 0.0117 (3) | 724 |
| TI | 4.105 (1) | ||
| N = 64 | |||
| 720,000 | 4.1321 (14) | 0.0330 (5) | 581 |
| 1,440,000 | 4.1169 (10) | 0.0224 (5) | 495 |
| 2,880,000 | 4.1085 (8) | 0.0167 (5) | 459 |
| 7,200,000 | 4.1046 (5) | 0.0105 (5) | 371 |
| 14,400,000 | 4.1025 (5) | 0.0078 (3) | 244 |
| TI | 4.100 (1) | ||
| N = 125 | |||
| 2,000,000 | 4.1240 (13) | 0.0175 (6) | 179 |
| 4,000,000 | 4.1159 (10) | 0.0110 (10) | 125 |
| 10,000,000 | 4.1124 (6) | 0.0083 (5) | 170 |
| 20,000,000 | 4.1102 (6) | 0.0060 (5) | 99 |
| TI | 4.108 (1) |
Table 3. Complete HSMC results for the TIP3P model of water.
| M̄tot | —Fe (FA), kcal/mol |
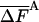 , kcal/mol , kcal/mol |
n |
|---|---|---|---|
| 5,312,000 | 5.74 (1) | 0.064 | 147 |
| 13,280,000 | 5.68 (1) | 0.044 | 77 |
| 26,560,000 | 5.64 (1) | 0.024 | 54 |
| TI | 5.599 (2) |
Several studies of the free energy of argon (21, 26–31) and water (32–38) have been published, most of them for systems that differ in size (as well as other modeling details) from the present ones. Therefore, for an objective evaluation of our results, we also calculated the free energies for our particular systems of argon and water by using TI. The Lennard–Jones interactions (for both argon and water) were scaled by using the shifted scaling potential of Zacharias et al. (39), described in the appendix of ref. 9; the Coulombic interactions (of water) were also distance-shifted in a similar way.
In Table 2, results are presented for the free energy, Ac(FA) (Eq. 11) of argon and their fluctuation, 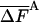 (Eq. 6) as a function of the average number of MC steps per cell, M̄tot; the free energy calculated by TI, which is considered to be exact, and the size of each sample analyzed, n, are provided as well. Table 2 demonstrates that the HSMC results for the free energy depend strongly on M̄tot, the larger is M̄tot the better the approximation, which justifies defining this free energy as FA. For n = 64, the largest M̄tot leads to a result for the free energy that deviates from the TI value by only 0.06%. (The statistical error in the TI result is ≈0.02%.) The worst approximation, based on 20 times smaller M̄tot, still leads to a free energy that is only ≈0.8% lower than the TI value; similar errors are detected for n = 32 and 125. The best results for each system are 1.3 orders of magnitude more accurate than results for Ac(FA) calculated in ref. 9 by using the approximate HSMC. As expected, the free energy fluctuations decrease systematically as the approximation improves and their smallest values, 0.0117, 0.0078, and 0.0060, for n = 32, 64, and 125, respectively, are smaller by a factor of 15.0, 13.2, and 12.5 than their energy counterparts, 0.175, 0.103, and 0.075.
(Eq. 6) as a function of the average number of MC steps per cell, M̄tot; the free energy calculated by TI, which is considered to be exact, and the size of each sample analyzed, n, are provided as well. Table 2 demonstrates that the HSMC results for the free energy depend strongly on M̄tot, the larger is M̄tot the better the approximation, which justifies defining this free energy as FA. For n = 64, the largest M̄tot leads to a result for the free energy that deviates from the TI value by only 0.06%. (The statistical error in the TI result is ≈0.02%.) The worst approximation, based on 20 times smaller M̄tot, still leads to a free energy that is only ≈0.8% lower than the TI value; similar errors are detected for n = 32 and 125. The best results for each system are 1.3 orders of magnitude more accurate than results for Ac(FA) calculated in ref. 9 by using the approximate HSMC. As expected, the free energy fluctuations decrease systematically as the approximation improves and their smallest values, 0.0117, 0.0078, and 0.0060, for n = 32, 64, and 125, respectively, are smaller by a factor of 15.0, 13.2, and 12.5 than their energy counterparts, 0.175, 0.103, and 0.075.
At this stage of development, the complete HSMC method is still significantly less efficient than TI. Thus, reconstructing a single argon configuration of n = 64 and M̄tot = 7,200,000, for example, requires 3.6 h of central processing unit (CPU) time, where because of the 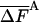 value, this configuration provides the free energy within a small range [-4.115 (-0.35%), -4.094 (+0.15%)] around the correct value 4.100; the TI run for this system required ≈1/3 of this time. However, the complete HSMC program can still be optimized reducing the computer time significantly. Also, as discussed in refs. 8 and 9, based on the correlation between FA and
value, this configuration provides the free energy within a small range [-4.115 (-0.35%), -4.094 (+0.15%)] around the correct value 4.100; the TI run for this system required ≈1/3 of this time. However, the complete HSMC program can still be optimized reducing the computer time significantly. Also, as discussed in refs. 8 and 9, based on the correlation between FA and 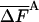 (
(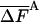 decreases as FA increases), approximate results for these quantities can be extrapolated very close to the correct value. All these topics require further investigation.
decreases as FA increases), approximate results for these quantities can be extrapolated very close to the correct value. All these topics require further investigation.
In Table 3, HSMC results are presented for water. Again the excess free energy (Eq. 10) improves (increases) as M̄tot is increased and the corresponding fluctuations decrease, where the lowest value, 0.024 is 7.8 times smaller than the energy fluctuation, 0.187. The best HSMC result is lower than the TI value by ≈0.7%. The TIP3P model of water is much more complex than the argon system, requiring more sampling and significantly larger computer time for calculating the energy at each MC step. Thus, the reconstruction time of a single configuration is 9.6, 24, and 48 h CPU time for the three approximations in Table 3, compared to ≈6 h for the TI run. Further challenges are manifested by the fact that the transition probabilities for water (as compared to argon) are often much smaller, and therefore more difficult to measure because of the additional degrees of freedom. Strategies designed to remedy these particular issues are required, and subsequent improvement is anticipated.
Summary
The HS method can be applied to fluids in different approximations, as shown in refs. 8 and 9, and in an exact manner, as has been demonstrated in this paper. The efficiency of the complete HSMC method is being enhanced now by applying importance sampling techniques for increasing the number of counts in the occupied cells, and other procedures, such as force bias MC (40). With TI, an ideal gas is integrated reversibly by gradually changing the potential energy parameters to their final values. The complete HSMC method is different: the absolute free energy is obtained, in principle, by reconstructing a single configuration, i.e., adding its atoms (molecules) gradually to an initially empty volume by using transition probabilities. Therefore, complete HSMC constitutes a research tool independent of TI and related methods, which enables one to calculate F by analyzing a given MC or MD sample. In the companion article, the complete HSMC method is extended to peptides, which enables future application of this method to MC or MD samples of a peptide or a surface loop of a protein solvated in explicit water. Thus, the relative stability of two states with a large conformational variance will be obtained by ΔFm,n = Fm - Fn, where the absolute free energies, Fm and Fn, are based on two samples only without the need to resort to procedures that are dependent on complex integration paths.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 GM61916 and in part by National Institutes of Health Grant R01 GM66090.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MC, Monte Carlo; MD, molecular dynamics; TI, thermodynamic integration; HS, hypothetical scanning; TP, transition probability; HSMC, hypothetical scanning Monte Carlo.
References
- 1.Beveridge, D. L. & DiCapua, F. M. (1989) Annu. Rev. Biophys. Biophys. Chem. 18, 431-492. [DOI] [PubMed] [Google Scholar]
- 2.Kollman, P. A. (1993) Chem. Rev. 93, 2395-2417. [Google Scholar]
- 3.Jorgensen, W. L. (1989) Acc. Chem. Res. 22, 184-189. [Google Scholar]
- 4.Meirovitch, H. (1998) Rev. Comput. Chem. 12, 1-74. [Google Scholar]
- 5.Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H. & Teller, E. (1953) J. Chem. Phys. 21, 1087-1092. [Google Scholar]
- 6.Alder, B. J. & Wainwright, T. E. (1959) J. Chem. Phys. 31, 459-466. [Google Scholar]
- 7.McCammon, J. A., Gelin, B. R. & Karplus, M. (1977) Nature 267, 585-590. [DOI] [PubMed] [Google Scholar]
- 8.Szarecka, A., White, R. P. & Meirovitch, H. (2003) J. Chem. Phys. 119, 12084-12095. [Google Scholar]
- 9.White, R. P. & Meirovitch, H. (2003) J. Chem. Phys. 119, 12096-12105. [Google Scholar]
- 10.Meirovitch, H. (1977) Chem. Phys. Lett. 45, 389-392. [Google Scholar]
- 11.Meirovitch, H. (1984) Phys. Rev. B 30, 2866-2874. [Google Scholar]
- 12.Meirovitch, H., Vásquez, M. & Scheraga, H. A. (1987) Biopolymers 26, 651-671. [DOI] [PubMed] [Google Scholar]
- 13.Meirovitch, H., Koerber, S. C., Rivier, J. & Hagler, A. T. (1994) Biopolymers 34, 815-839. [DOI] [PubMed] [Google Scholar]
- 14.Chorin, A. J. (1996) Phys. Fluids 8, 2656-2660. [Google Scholar]
- 15.Meirovitch, H. (1983) J. Phys. A 16, 839-846. [Google Scholar]
- 16.Meirovitch, H. (1985) Phys. Rev. A 32, 3709-3715. [DOI] [PubMed] [Google Scholar]
- 17.Meirovitch, H. (1999) J. Chem. Phys. 111, 7215-7224. [Google Scholar]
- 18.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. (1983) J. Chem. Phys. 79, 926-935. [Google Scholar]
- 19.Cheluvaraja, S. & Meirovitch, H. (2004) Proc. Natl. Acad. Sci. USA 101, 9241-9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meirovitch, H. & Alexandrowicz, Z. (1976) J. Stat. Phys. 15, 123-127. [Google Scholar]
- 21.Li, Z. & Scheraga, H. A. (1988) J. Phys. Chem. 92, 2633-2636. [Google Scholar]
- 22.Allen, M. P. & Tildesley, D. J. (1987) Computer Simulation of Liquids (Clarenden, Oxford).
- 23.Owicki, J. C. (1978) Am. Chem. Soc. Symp. Ser. 86, 159-171. [Google Scholar]
- 24.Jorgensen, W. L. (1983) J. Phys. Chem. 87, 5304-5314. [Google Scholar]
- 25.Owicki, J. C. & Scheraga, H. A. (1977) Chem. Phys. Lett. 47, 600-602. [Google Scholar]
- 26.Torrie, G. M. & Valleau, J. P. (1974) Chem. Phys. Lett. 28, 578-581. [Google Scholar]
- 27.Torrie, G. M. & Valleau, J. P. (1977) J. Comp. Phys. 23, 187-199. [Google Scholar]
- 28.Levesque, D. & Verlet, L. (1969) Phys. Rev. 182, 307-316. [Google Scholar]
- 29.Gosling, E. M. & Singer, K. (1970) Pure Appl. Chem. 22, 303-309. [Google Scholar]
- 30.Mezei, M. (1989) Mol. Simul. 2, 201-207. [Google Scholar]
- 31.Johnson, J. K., Zollweg, J. A & Gubbins, K. E. (1993) Mol. Phys. 78, 591-618. [Google Scholar]
- 32.Mezei, M., Swaminathan, S. & Beveridge, D. L. (1978) J. Am. Chem. Soc. 100, 3255-3256. [Google Scholar]
- 33.Mezei, M. (1982) Mol. Phys. 47, 1307-1315. [Google Scholar]
- 34.Mezei, M. (1987) Mol. Phys. 61, 565-582. [Google Scholar]
- 35.Jorgensen, W. L, Blake, J. F. & Buckner, J. K. (1989) Chem. Phys. 129, 193-200. [Google Scholar]
- 36.Hermans, J., Pathiaseril, A. & Anderson, A. (1988) J. Am. Chem. Soc. 110, 5982-5986. [DOI] [PubMed] [Google Scholar]
- 37.Li, Z. & Scheraga, H. A. (1989) Chem. Phys. Lett. 154, 516-520. [Google Scholar]
- 38.Quintana, J. & Haymet, A. D. J. (1992) Chem. Phys. Lett. 189, 273-277. [Google Scholar]
- 39.Zacharias, M., Straatsma, T. P. & McCammon, J. A. (1994) J. Chem. Phys. 100, 9025-9031. [Google Scholar]
- 40.Pangali, C., Rao, M. & Berne, B. J. (1978) Chem. Phys. Lett. 55, 413-417. [Google Scholar]