

Abstract
A method called complete hypothetical scanning Monte Carlo has been introduced for calculating the absolute entropy, S, and free energy, F, of fluids. Here, the method is extended to peptide chains in vacuum. Thus, S is calculated from a given sample by reconstructing each conformation step-by-step by using transition probabilities (TPs); at each step, part of the chain coordinates have already been determined (the “frozen past”), and the TP is obtained from a Monte Carlo simulation of the (future) part of the chain whose TPs as yet have not been calculated. Very accurate results for S and F are obtained for the helix, extended, and hairpin microstates of a simplified model of decaglycine (Gly)10 and (Gly)16. These results agree well with results obtained by the quasiharmonic approximation and the local states method. The complete HSMC method can be applied to a macromolecule with any degree of flexibility, ranging from local fluctuations to a random coil. Also, the difference in stability, ΔFmn = Fm - Fn between significantly different microstates m and n can be obtained from two simulations only without the need to resort to thermodynamic integration. Our long-term goal is to extend this method to any peptide and apply it to a peptide immersed in a box with explicit water.
In ref. 1, White and Meirovitch discuss the importance and difficulties of calculating the absolute free energy, F, and entropy, S; however, their role in computational structural biology should be further emphasized. The energy surface of a protein, commonly defined by a force field, is highly rugged, consisting of a tremendous number of local minima (2), where the native structure corresponds to the localized energy well with the lowest F. However, molecular dynamics simulations have shown (3, 4) that even a protein with a well defined structure fluctuates significantly within a region called wide microstate (e.g., the conformational region of an α-helix of a peptide) that typically consists of many localized energy wells. A peptide or protein, or protein segments such as surface loops, can exhibit an intermediate flexibility, where several wide microstates are populated significantly at thermodynamic equilibrium. It is essential to be able to identify these wide microstates, m, and to calculate Fm, which lead to their relative populations and to weighted averages of various quantities that can be compared with experimental values (5, 6). Fm is useful particularly if m and n differ significantly; then, calculating the difference, ΔFmn = Fm - Fn is straightforward, whereas calculating it by thermodynamic integration might be prohibitive (see refs. 7–12 and references therein).
In ref. 1, the hypothetical scanning (HS) method for calculating the absolute F and S (10) has been further developed and applied to liquid argon and water. This method, named complete hypothetical scanning Monte Carlo (HSMC), is extended here to a peptide in vacuum or peptide described by an implicit solvation. As a first step, we treat a simplified model of decaglycine (Gly)10 simulated by Monte Carlo (MC) (13) at three wide microstates: helix, extended, and hairpin. Each sample conformation is reconstructed gradually by calculating transition probabilities (TP) for the dihedral angles and fixing the related atoms at their positions. A TP is obtained by an MC simulation of the future (yet unfixed) part of the chain and, to avoid the escape of a future sample from the corresponding microstate, we impose restrictions obtained from the local states (LS) method for calculating S (5, 6, 14–18). Therefore, the entire procedure is a hybrid of two techniques and, to test its performance for larger peptides, we also study a 16-residue polyglycine (Gly)16 in the helix and extended wide microstates. The complete HSMC results for S and F are found to be in a very good agreement with results obtained by the LS method and the quasiharmonic (QH) approximation (19, 20). Our long-term goal is to extend the complete HSMC method to any peptide and to apply it to a peptide immersed in a box with explicit water.
Theory and Methodology
The Model and Statistical Mechanics Considerations. We have first studied decaglycine, NH2(Gly)10CONH2, modeled by the amber96 force field in vacuum (21), where the charges of the end groups were neutralized. For simplicity, we denote the dihedral angles ϕi,Ψi, and ωi ordered along the chain by αk, k = 1,3N = 30, where N is the number of residues [the extension for (Gly)16 is straightforward]. The partition function, Z, is an integral over the function exp(-E/kBT) (E is the potential energy and kB, the Boltzmann constant) with respect to the Cartesian coordinates over the whole conformational space, Ω. However, for a stable wide microstate, the integration is carried out only over the limited region Ω0 that defines the wide microstate. To apply the HS or LS methods, one has to change the variables of integration from Cartesian to internal coordinates, which makes the integral dependent also on the Jacobian, J. For a linear chain, J has been shown to be independent of the dihedral angles and is a simple function of the bond angles and bond lengths. Thus, if the potentials of these “hard variables” are strong, their average values can be assigned to J, which to a good approximation can be taken out of the integral (see refs. 19, 22, and 23).
For the same reason, one can assume a more restrictive model (see below), where the bond angles and bond lengths are kept constant at their average values and thus the corresponding exp(-E/kBT) values can be taken out of the integral as well. In particular, notice that, although the contribution of bond stretching to the absolute entropy is not small, it is expected to be similar for different wide microstates of the same molecule. Therefore, to a good approximation, the contribution of bond stretching to the differences ΔSm,n and ΔFm,n between wide microstates m and n cancels out. Assuming that the bond lengths are not correlated with the bond and dihedral angles enables one to carry out the integration over the bond lengths; if a similar assumption is made for the bond angles, the remaining integral becomes a function of the 3N dihedral angles (22, 23) The partition function is
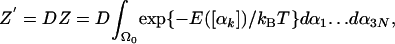 |
[1] |
where the prefactor D is a product of J and the integral over the bond lengths and bond angles; D depends on T and the units in which the bond lengths and bond angles are expressed. For calculating ΔSm,n and ΔFm,n of two wide microstates of the same molecule, lnD cancels and can be ignored (notice, however, that D contributes to the absolute F and S). The probability density corresponding to Z (Eq. 1) is
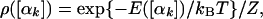 |
[2] |
and the exact entropy (defined up to an additive constant) is
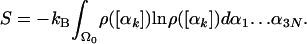 |
[3] |
Thus far we have described the transformation from a peptide model represented by Cartesian coordinates to a model represented by dihedral angles. This is needed for applying both the LS and HS methods. However, we have found MC simulations in Cartesian coordinates to be extremely inefficient; therefore, we have studied a relatively simple model of ployglycine based on the amber96 force field with constant bond lengths and bond angles, an option available in the program tinker (http://dasher.wustl.edu/tinker). Thus, the dihedral angles ϕi, Ψi, and ωi become the variables of an MC procedure significantly more efficient than that based on Cartesian coordinates; for the present model of rigid geometry, the application of the LS and HS methods is therefore direct. Obviously, keeping the bond angles constant is a temporary restriction applied only in this initial study of the complete HSMC method. In what follows, for simplicity, the various methods will be described as applied to this model of polyglycine.
The Exact Scanning Method. The exact scanning method is a step-by-step construction procedure for polymer chains (24) and thus is equivalent to the MC and MD procedures in the sense that large samples generated by all these methods lead to the same averages and fluctuations within the statistical errors. With the exact scanning method an N-residue conformation of polyglycine in the helical region (Ω0), is built by defining the dihedral angles αk step by step with TPs and adding the related atoms; for example, the angle ϕ determines the coordinates of the two hydrogens connected to Cα, and the position of C′ (16, 25). Thus, at step k, k-1 dihedral angles α1,..., αk-1 have already been determined, they and the related structure (the past) are kept constant, and αk should be defined with the exact TP density ρ(αk|αk-1... α1),
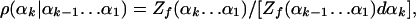 |
[4] |
where dαk is a small segment centered at αk, and Zf(αk... α1) is a future partition function defined over the helical region Ω0 by integrating over the future conformations defined by αk+1... dα3N (within Ω0), where the past angles, α1... αk, are held fixed,
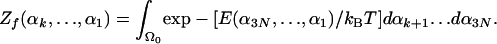 |
[5] |
The probability density of the entire conformation is
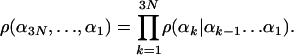 |
[6] |
Because of the equivalence between the MC and the scanning method mentioned above, one can assume that a given MC sample has rather been generated by the scanning method, which enables one to reconstruct each conformation and to calculate the TP densities that hypothetically were used to create it step-by-step. This idea can be implemented in two different ways, by the LS and HS methods. Because some elements of the LS method are implemented within the framework of the complete HSMC method, we describe the LS method first.
The LS Method. In the first step, the MC sample (of a given wide microstate) is visited, and the variability range Δαk is calculated (5, 6, 14–18).
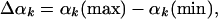 |
[7] |
where αk(max) and αk(min) are the maximum and minimum values of αk found in the sample, respectively. Next, the ranges Δαk are divided into l equal segments, where l is the discretization parameter. We denote these segments by νk, (νk = 1, l). Thus, an angle αk is now represented by the segment νk to which it belongs, and a conformation i is expressed by the corresponding vector of segments [ν1(i), ν2(i),..., ν3N(i)]. Under this discretization approximation, ρ(αk|αk-1... α1) can be estimated by
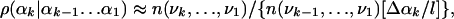 |
[8] |
where n(νk,..., ν1) is the number of times the LS [i.e., the partial vector (νk,...,ν1) representing (αk,...,α1)] appears in the sample. Because the number of local states increases exponentially with k, one has to resort to approximations based on smaller LSs that consist of νk and the b angles preceding it along the chain, i.e., the vector (νk, νk-1,..., νk-b); b is called the correlation parameter. The sample is visited for the second time, and for a given b, one calculates the number of occurrences n(νk, νk-1,..., νk-b) of all of the local states from which a set of TP ρ(νk|νk-1,..., νk-b) are defined. The sample is then visited for the third time, and for each member i of the sample, one determines the 3N local states and the corresponding TP, whose product defines an approximate probability density ρi(b, l) for conformation i:
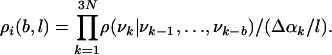 |
[9] |
The larger are b and l, the better the approximation (given enough statistics). ρi(b, l) allows defining an approximate entropy and free energy functional, SA and FA, which constitute rigorous upper and lower bounds for the correct values, respectively (25),
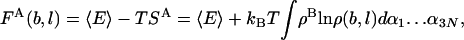 |
[10] |
where 〈E〉 is the Boltzmann average of the potential energy, estimated from the MC sample, and ρB is the Boltzmann probability density with which the sample was created.
SA is estimated from a Boltzmann sample of size n by S̄A:
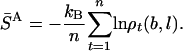 |
[11] |
As discussed in ref. 1, the fluctuation ΔF of the correct free energy is zero, whereas the approximate FA has finite fluctuation, ΔFA (estimated by 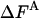 ), which is expected to decrease as the approximation improves (17).
), which is expected to decrease as the approximation improves (17).
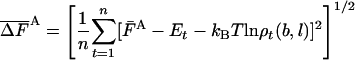 |
[12] |
The LS method can be applied to any chain flexibility, i.e., it is not limited to harmonic or QH fluctuations (19, 20, 22, 23, 26). Thus, free energy differences between wide microstates with significant structural differences can be calculated, which is a difficult task with methods based on thermodynamic integration.
Approximate HS Method. The idea of the HS method is to reconstruct each sample conformation step by step, obtaining the TP density of each αk (Eq. 4) by calculating the future partition functions Zf. However, a systematic integration of Zf within the limits of Ω0 is difficult and becomes impractical for a large peptide where Ω0 is unknown; therefore, thus far, HS was applied only to self-avoiding walks (SAWs) on a lattice, where Ω0 is the entire space and Zf is calculated approximately by enumerating only future SAWs of f steps (i.e., αk,..., αk+f-1), rather than of N - k + 1 steps (27, 28).
The Complete HSMC Method. With the complete HSMC method applied to peptides (like for fluids), one calculates at each reconstruction step k of conformation i the TP density, ρ(αk|αk-1... α1), from nf MC steps (trials) (13), where the entire future of the peptide can move by changing the future angles αk,..., α3N, whereas the dihedral angles α1,..., αk-1 (defining the past) are kept fixed at their values in conformation i. A small segment (bin) δαk (see Eq. 4) is centered at αk, and the number of MC visits to this bin during the simulation, nvisit, is calculated; one obtains
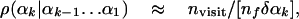 |
[13] |
where the relation becomes exact for a very large nf. The product of these TP densities leads to the probability density of the entire chain (Eqs. 6 and 10). Notice that, unlike the systematic calculation of Zf, where the limits of Ω0 are in practice unknown, with the complete HSMC procedure, the future structures generated by MC at each step k remain in general within the limits of the wide microstate Ω0 defined by the analyzed MC sample. In some cases, however, the future samples were found to escape from this region; therefore, before applying the complete HSMC method, the LS method is applied to the analyzed sample and the αk(min) and αk(max) values (Eq. 7) are calculated; they are then used to keep the future structures within Ω0 by rejecting MC moves with angle values beyond those of αk(min) and αk(max). Although complete HSMC considers the entire future, in practice ρ(αk|αk-1... α1) (Eq. 13) will be somewhat approximate due to insufficient future sampling, imperfect random number generator, etc.; therefore, the corresponding free energy, FA (Eqs. 10 and 11), will underestimate slightly the correct value, where its fluctuation ΔFA (Eq. 12) does not vanish.
Results and Discussion
We have first studied three wide microstates of (Gly)10: helix, hairpin, and extended. Samples of these wide microstates were generated by the Metropolis MC procedure (13) at 100 K where a trial structure is obtained by randomly changing all of the 30 dihedral angles, each within ±1° of its current value. These simulations were started from helical, extended, and hairpin structures obtained by minimizing the energy of the corresponding structures, ϕk = Ψk = -55°, and ωk = 180°, ϕk = Ψk = ωk = 180°, and two extended strands of four residues connected by a type I′ turn. The first 5,000 MC steps were used for equilibration, and from the following 50,000 steps, after every 100 steps the current structure was retained for future analysis; in this way, three equal samples of 500 structures were generated. It should be pointed out that preliminary simulations at 300 K resulted in unstable samples (i.e., the structures escaped from their wide microstates); therefore, the temperature was decreased to 100 K, where the helix and extended simulations were found to be very stable, whereas the hairpin sample remained stable only up to the first 50,000 MC steps. The corresponding Δαk values (Eq. 7) are relatively small (see Table 1), representing relatively concentrated samples. Notice, however, that due to correlations, each wide microstate is significantly smaller than the corresponding region, Δα1×Δα2×.... ×Δα30.
Table 1.
The differences (in degrees) between the minimum and maximum values of the dihedral angles of (Gly)10 in three MC samples of 500 structures (Eq. 4)
| Extended
|
Helix
|
Hairpin
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Number | Δυ | Δυ | Δω | Δυ | Δψ | Δω | Δυ | Δψ | Δω |
| 1 | 47 | 142 | 23 | 43 | 48 | 23 | 35 | 57 | 21 |
| 2 | 61 | 55 | 23 | 20 | 43 | 21 | 37 | 30 | 21 |
| 3 | 57 | 43 | 23 | 28 | 33 | 22 | 37 | 36 | 26 |
| 4 | 68 | 51 | 26 | 22 | 25 | 21 | 54 | 89 | 21 |
| 5 | 58 | 46 | 25 | 30 | 35 | 19 | 59 | 65 | 24 |
| 6 | 68 | 46 | 24 | 25 | 29 | 20 | 31 | 27 | 18 |
| 7 | 61 | 45 | 23 | 22 | 46 | 16 | 30 | 43 | 32 |
| 8 | 66 | 42 | 25 | 27 | 34 | 19 | 39 | 31 | 22 |
| 9 | 60 | 54 | 25 | 30 | 48 | 19 | 41 | 37 | 23 |
| 10 | 65 | 47 | 26 | 49 | 360 | 26 | 56 | 32 | 28 |
To apply the complete HSMC method, each conformation was reconstructed step by step by TPs obtained from MC simulations of the future part carried out in the same way as described above with the additional restriction that a trial conformation with αk larger than αk(max) or smaller than αk(min) is rejected. To check the convergence of the results, they were calculated for four future sample sizes, nf = 20,000, 40,000, 80,000, and 160,000. For the same reason, we studied for each dihedral angle four bin sizes, δ = Δαk/15, Δαk/10, Δαk/5, and 20° centered at αk. Notice that as for the LS method, the bin size is proportional to Δαk. If the counts of the smallest bin are smaller than 50, the bin size is increased to the next size and, if necessary, to the next one (δ = Δαk/5); the same is applied to the second size bin. In the case of zero counts, nvisit is taken to be 1; notice, however, that zero counts is a very rare event.
Results for the entropy (TS) appear in Table 2 for various nf values and bin sizes; the results for the largest bin (of 20°) are not provided, because they are significantly worse than those obtained for the smaller bins. All of the HSMC results are based on samples of 400 structures (of the entire samples of 500 conformations), and the statistical errors were obtained from the fluctuations and results based on partial samples. The accuracy of complete HSMC can always be improved by decreasing the bin size and increasing the future sample size, meaning that correspondingly SA (Eq. 11) is expected to decrease [provided that the probability density is defined on the same conformational space (i.e., the wide microstate) as the Boltzmann probability density used to generate the sample]. Indeed, for each bin, the entropy decreases (or remain constant) as nf increases, where the only exception is the entropy for the helix based on the smallest sample, nf = 20,000, which is smaller than the entropies of the larger samples; this probably stems from an HSMC probability density that is defined on only a partial region of the helical wide microstate due to insufficient sampling.
Table 2.
Entropy, TSA (T = 100 K) in kcal/mol (Eqs. 10 and 11) for (Gly)10 for various bin sizes (Eq. 4) and future sample sizes, nf, obtained with the complete HSMC method
| Bin size | nf | Extended | Helix | Hairpin |
|---|---|---|---|---|
| Δαk/5 | 20,000 | 20.30 | 16.33 | 18.32 |
| 40,000 | 20.14 | 16.36 | 18.15 | |
| 80,000 | 20.06 | 16.34 | 18.02 | |
| 160,000 | 20.03 | 16.35 | 17.97 | |
| Δαk/10 | 20,000 | 20.24 | 16.08 | 18.17 |
| 40,000 | 20.11 | 16.16 | 18.03 | |
| 80,000 | 20.04 | 16.16 | 17.90 | |
| 160,000 | 20.01 | 16.16 | 17.86 | |
| Δαk/15 | 20,000 | 20.23 | 16.01 | 18.14 |
| 40,000 | 20.11 | 16.10 | 18.02 | |
| 80,000 | 20.04 | 16.10 | 17.89 | |
| 160,000 | 20.01 | 16.11 | 17.85 | |
| QH | 19.83 | 16.13 | 17.76 | |
| LS | 20.05 | 17.50 | 19.29 |
Δαk is defined in Eq. 7. The HSMC results are based on a sample of 400 conformations. The statistical errors are not larger than ±0.05 kcal/mol for the HSMC and QH results (Eq. 14) and are not larger than ±0.03 kcal/mol the LS results (b = 1, l = 10). The entropy is defined up to an additive constant.
The entropy results for the extended microstate for a given nf are the same for the different bin sizes, and the results for nf = 80,000 and 160,000 are converged within the error bars. The same applies to the hairpin results for the two smallest bins. The helix results behave differently, where for each bin they are constant for the three largest nf values, whereas they decrease with decreasing the bin size and probably have not yet completely converged. However, within the accuracy of the usual force fields, entropy and free energy differences smaller than 0.1 kcal/mol are in general ignored; therefore, even the helix results can be considered as converged.
It is of interest to compare the complete HSMC results with those obtained by other methods. For that, we increased the samples of the three wide microstates from 500 to 30,000 structures by imposing the restriction on the MC procedure that a trial conformation with αk larger than αk(max) or smaller than αk(min) obtained for the initial sample (Table 1) is rejected. We applied the QH approximation (19, 20) to a subsample of 4,000 conformations, where
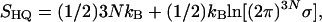 |
[14] |
and σ is the determinant of the covariance matrix of the 3N dihedral angles. We also applied the LS method (with correlation parameter, b = 1 and l = 10) to the entire increased sample. The QH results presented in Table 2 are very close to the complete HSMC values, probably because the three samples are approximately QH. The LS and HSMC entropies are equal for the extended microstate, because the angular correlations along the chain are short, and b = 1 already captures most of them. On the other hand, the range of these correlations increases for the helix and the hairpin, and the LS entropies, as expected, become slightly larger (upper bounds) than the HSMC values.
In Table 3, complete HSMC results are presented for the free energy, which is defined by FA (Eq. 10), as discussed for the entropy above. These results are given only for the smallest bin, because the free energies for the other bins can be obtained from the entropies of Table 2; as expected, the free energy increases as the approximation improves (i.e., as nf is increased). Again, the QH results are close to the HSMC values, and the LS result is close to the HSMC value for the extended microstate and smaller for the other two microstates. Notice, however, that the energy components of QH and LS are calculated from the corresponding larger samples (see previous paragraph). We also provide in Table 3 results for the average energy (obtained from 400 structures) and the fluctuations of the energy and free energy. As expected, the free energy fluctuations decrease as nf is increased, and for nf = 160,000, they are four times smaller than the corresponding fluctuations of the energy, except for the hairpin where the ratio is ≈2.
Table 3.
Results for the free energy, FA (Eqs. 10 and 11) and its fluctuation 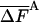 (Eq. 12) obtained for (Gly)10 by the complete HSMC method
(Eq. 12) obtained for (Gly)10 by the complete HSMC method
| Extended
|
Helix
|
Hairpin
|
||||
|---|---|---|---|---|---|---|
| HSMC/nf | —FA | 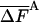 |
—FA | 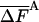 |
—FA | 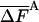 |
| 20,000 | 74.75 (4) | 0.61 (3) | 98.48 (3) | 0.50 (4) | 84.57 (3) | 0.84 (4) |
| 40,000 | 74.64 (4) | 0.39 (3) | 98.57 (2) | 0.32 (4) | 84.45 (3) | 0.63 (3) |
| 80,000 | 74.57 (3) | 0.26 (3) | 98.58 (2) | 0.23 (4) | 84.32 (3) | 0.45 (3) |
| 160,000 | 74.53 (2) | 0.18 (3) | 98.59 (2) | 0.18 (4) | 84.27 (3) | 0.36 (3) |
| QH | 74.48 (8) | 98.69 (8) | 84.64 (8) | |||
| LS | 74.68 (1) | 100.08 (1) | 86.14 (1) | |||
| —Energy | 54.53 (7) | 0.73 (4) | 82.48 (5) | 0.80 (5) | 66.43 (7) | 0.68 (5) |
All results are in kcal/mol. T = 100 K. The first result in the last line is for the average energy, and the second is for its fluctuation. The HSMC results are presented only for the smallest bin size, δ = Δαk/15, but for all the future samples sizes, nf. F is defined up to an additive constant. The free energy obtained with the QH approximation (Eq. 14) and the LS method is based on larger samples. The statistical error is given in parentheses, e.g., 82.48 (5) = 82.48 ± 0.05.
The main interest in this study is to determine the relative stability of the three wide microstates. In the upper part of Table 4, we present results for the differences, TΔS, ΔF, and ΔE between these microstates for (Gly)10. Within their uncertainty of 0.1–0.2 kcal/mol, the differences are very stable for the three bin sizes, for nf = 40,000–160,000, for samples as small as 200 conformations, and for the helix-extended differences also for 100 conformations. This demonstrates that, in practice, complete HSMC can be quite efficient. For the model studied, the helix is the most stable, where its free energy is lower by 14.3 and 24.1 kcal/mol than that of the hairpin and extended microstates, respectively. These differences are mostly governed by the energy differences, 11.9 and 27.95 kcal/mol, where the TΔS values are only 1.7 and 3.9 kcal/mol, respectively.
Table 4. Differences in the entropy, TΔSA, the free energy, ΔFA, and the energy, ΔE, among the three wide microstates (these properties are denoted R).
| TΔSA | ΔFA | ΔE | |
|---|---|---|---|
| (Gly)10 | |||
| R(extended) — R(hairpin) | 2.2 (1) | 9.7 (1) | 16.1 (1) |
| R(extended) — R(helix) | 3.9 (1) | 24.1 (1) | 27.95 (6) |
| R(hairpin) — R(helix) | 1.7 (2) | 14.3 (2) | 11.9 (1) |
| (Gly)16 | |||
| R(extended) — R(helix) | 7.1 (2) | 55.7 (2) | 62.9 (3) |
Results are in kcal/mol. TΔSA and ΔFA were obtained by the complete HSMC method at T = 100 K. The statistical error is defined in Table 3.
It is of interest to test the performance of complete HSMC for larger peptides, and we therefore also applied it to (Gly)16. Two samples of size 600 each spanning the extended and helical wide microstates were generated by MC [as described for (Gly)10], where 400 and 600 conformations of them were reconstructed by HSMC, respectively. The dihedral angle values of these samples are concentrated around their canonical values with deviations Δαk (Eq. 4) very close to those obtained for (Gly)10 in Table 1, where significant differences exist only for ΔΨ of the first and last residues. Results for TSA, FA, and its fluctuation, ΔFA, appear in Tables 5 and 6, which are structured as Tables 2 and 3. The corresponding results for TSA are basically unchanged (i.e., converged) as bin size decreases, i.e., in going from Δαk/5 (results not shown) to Δαk/10, and to Δαk/15. Within each bin size, the helix results are constant as well, meaning that already a future sample size of nf = 40,000 is sufficient. On the other hand, for the extended microstate, the results for each bin size decrease as nf is increased and larger nf is needed to reach convergence; for nf = 320,000, the expected extrapolated result is TSA = 31.90, which is used in calculating the differences for (Gly)16 in Table 4.
Table 5. Results for the entropy, TSA, of (Gly)16.
| Extended
|
Helix
|
||
|---|---|---|---|
| Bin size | nf | TSA | TSA |
| Δαk/10 | 20,000 | 33.26 (8) | 24.54 (4) |
| 40,000 | 32.48 (6) | 24.73 (5) | |
| 80,000 | 32.13 (6) | 24.74 (5) | |
| 160,000 | 31.96 (5) | 24.73 (5) | |
| Δαk/15 | 20,000 | 33.26 (8) | 24.50 (4) |
| 40,000 | 32.52 (6) | 24.72 (5) | |
| 80,000 | 32.15 (6) | 24.74 (5) | |
| 160,000 | 31.97 (5) | 24.73 (5) | |
| QH | 32.5 (2) | 25.8 (2) | |
| LS | 32.80 (4) | 26.90 (6) |
Table 6. Results for the free energy, FA, of (Gly)16.
| Extended
|
Helix
|
||||
|---|---|---|---|---|---|
| Bin size | nf | —FA | 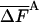 |
—FA | 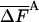 |
| Δαk/15 | 20,000 | 100.70 (6) | 1.20 (6) | 154.81 (3) | 0.69 (7) |
| 40,000 | 99.96 (4) | 0.81 (4) | 155.03 (2) | 0.47 (6) | |
| 80,000 | 99.59 (3) | 0.57 (3) | 155.05 (2) | 0.33 (4) | |
| 160,000 | 99.41 (2) | 0.42 (3) | 154.03 (1) | 0.24 (2) | |
| QH | 99.8 (1) | 155.2 (1) | |||
| LS | 100.34 (4) | 157.60 (7) | |||
| —Energy | 67.44 (6) | 0.94 (10) | 130.31 (8) | 1.13 (10) | |
As expected, for both microstates, the LS results (b = 1, l = 10) slightly overestimate the HSMC values, whereas the QH results are equal to the HSMC values within a relatively large statistical error. The free energy fluctuations, as expected, decrease monotonically as the approximation improves, and they are smaller than the energy fluctuations by a factor of 4.7 and 2.2 for the helix and extended microstates, respectively. The LS and QH results were obtained from relatively large samples of 5·104 and 25·103 conformations, respectively, hence the corresponding energies are slightly different from those based on the smaller HSMC samples; thus, whereas the FA(LS) and F(QH) are close to FA(HSMC), a strict comparison is not straightforward. In Table 4 the differences, TΔS, ΔF, and ΔE for the extended and helix microstates are presented with acceptable errors of 0.2–0.3 kcal/mol (see above). It should be pointed out that the results for SA and the energy of the helix scale with increasing peptide size, whereas the energy of the extended state does not; therefore, ΔE, and ΔF, do not scale in going from (Gly)10 to (Gly)16.
At this stage of development of the complete HSMC method, reconstructing a single conformation of (Gly)10 based on nf = 160,000 requires ≈90-min central processing unit (CPU) time on a 2.6-GHz Athlon processor, meaning that a nf = 40,000 run, which is sufficient for providing the 0.1–0.2 kcal/mol accuracy, requires 23-min CPU time; for (Gly)16, the time increases by a factor of ≈2.2. However, one can increase the efficiency further by decreasing the amount of sampling (nf) for the smaller future peptides and using importance sampling methods to enhance the number of counts.
Summary
We have introduced here the complete HSMC method for a peptide chain in vacuum. In this initial study, we sought to treat a simple model with minimal degrees of freedom and therefore chose a polyglycine model with constant bond lengths and bond angles described by the amber force field. Although ignoring the contribution of the bond lengths to differences in entropy is a valid approximation, the contribution of the bond angles is significant and should not be ignored; however, adding this contribution is straightforward and is currently being studied by us. To be able to generate stable wide microstates around helix, extended, and hairpin structures, the temperature was decreased to 100 K; the corresponding samples were approximately QH, which allowed comparing the HSMC results with those obtained by the QH method. However, unlike the QH method, the complete HSMC is general, in the sense that it can be applied to any chain flexibility, where side chains visit all of the available rotamers, for example. Complete HSMC is probably the only method for calculating the absolute entropy of peptide chains that, practically, is exact.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 GM66090 and in part by National Institutes of Health Grant R01 GM61916.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HSMC, hypothetical scanning Monte Carlo; TP, transition probabilities; LS, local states; QH, quasiharmonic; HS, hypothetical scanning; MC, Monte Carlo.
References
- 1.White, R. P. & Meirovitch, H. (2004) Proc. Natl. Acad. Sci. USA 101, 9235-9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vásquez, M., Némethy, G. & Scheraga, H.A. (1994) Chem. Rev. 94, 2183-2239. [Google Scholar]
- 3.Stillinger, F. H. & Weber, T. A. (1984) Science 225, 983-989. [DOI] [PubMed] [Google Scholar]
- 4.Elber, R. & Karplus, M. (1987) Science 235, 318-321. [DOI] [PubMed] [Google Scholar]
- 5.Meirovitch, H. & Meirovitch, E. (1996) J. Phys. Chem. 100, 5123-5133. [Google Scholar]
- 6.Baysal, C. & Meirovitch, H. (1999) Biopolymers 50, 329-344. [DOI] [PubMed] [Google Scholar]
- 7.Beveridge, D. L. & DiCapua, F. M. (1989) Annu. Rev. Biophys. Biophys. Chem. 18, 431-492. [DOI] [PubMed] [Google Scholar]
- 8.Kollman, P. A. (1993) Chem. Rev. 93, 2395-2417. [Google Scholar]
- 9.Jorgensen, .W. L. (1989) Acc. Chem. Res. 22, 184-189. [Google Scholar]
- 10.Meirovitch, H. (1998) in Reviews in Computational Chemistry, eds. Lipkowitz, K. B. & Boyd, D. B. (Wiley, New York), Vol. 12, pp. 1-74. [Google Scholar]
- 11.Szarecka, A., White, R. P. & Meirovitch, H. (2003) J. Chem. Phys. 119, 12084-12095. [Google Scholar]
- 12.White, R. P. & Meirovitch, H. (2003) J. Chem. Phys. 119, 12096-12105. [Google Scholar]
- 13.Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H. & Teller, E. (1953) J. Chem. Phys. 21, 1087-1092. [Google Scholar]
- 14.Meirovitch, H. (1977) Chem. Phys. Lett. 45, 389-392. [Google Scholar]
- 15.Meirovitch, H. (1983) J. Stat. Phys. 30, 681-698. [Google Scholar]
- 16.Meirovitch, H., Vásquez, M. & Scheraga, H. A. (1987) Biopolymers 26, 651-671. [DOI] [PubMed] [Google Scholar]
- 17.Meirovitch, H., Kitson, D. H. & Hagler, A. T. (1992) J. Am. Chem. Soc. 114, 5386-5399. [Google Scholar]
- 18.Meirovitch, H. (1999) J. Chem. Phys. 111, 7215-7224. [Google Scholar]
- 19.Karplus, M. & Kushick, J. N. (1981) Macromolecules 14, 325-332. [Google Scholar]
- 20.Rojas, O. L., Levy, R. M. & Szabo, A. (1986) J. Chem. Phys. 85, 1037-1049. [Google Scholar]
- 21.Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Jr., Ferguson, D. M., Spellmeyer, D. C., Fox, T., Caldwell, J. W. & Kollman, P. A. (1995) J. Am. Chem. Soc. 117, 5179-5197. [Google Scholar]
- 22.Gō, N. & Scheraga, H. A. (1969) J. Chem. Phys. 51, 4751-4767. [Google Scholar]
- 23.Gō, N. & Scheraga, H. A. (1976) Macromolecules 9, 535-542. [Google Scholar]
- 24.Meirovitch, H. (1988) J. Chem. Phys. 89, 2514-2522. [Google Scholar]
- 25.Meirovitch, H., Vásquez, M. & Scheraga, H. A. (1988) Biopolymers 27, 1189-1204. [DOI] [PubMed] [Google Scholar]
- 26.Hagler, A. T., Stern, P. S., Sharon, R., Becker, J. M. & Naider, F. (1979) J. Am. Chem. Soc. 101, 6842-6852. [Google Scholar]
- 27.Meirovitch, H. (1985) Phys. Rev. A 32, 3709-3715. [DOI] [PubMed] [Google Scholar]
- 28.Meirovitch, H. (1992) J. Chem. Phys. 97, 5816-5823. [Google Scholar]