

Abstract
Two recombinant myxoma viruses (MYXV expressing a fluorescent protein [MYXV-Tred] and MYXV-Tred encoding murine interleukin-15 [MYXV-IL15]) were evaluated for therapeutic effects in an aggressive B16F10 melanoma model in immunocompetent mice. It was hypothesized that continuous expression of IL-15 within a tumor would recruit cytotoxic effector cells to induce an antitumor immune response and improve treatment efficacy. Weekly intratumoral injections were given to evaluate the effect of treatment on the median survival time of C57BL/6 mice bearing established B16F10 melanomas. Mice that received MYXV-Tred or MYXV-IL15 lived significantly longer than mice given treatment controls. Unexpectedly, the median survival time of MYXV-IL15-treated mice was similar to that of MYXV-treated mice. At 1, 2, and 4 days postinoculation, viral plaque assays detected replicating MYXV-Tred and MYXV-IL15 within treated tumors. At these time points in MYXV-IL15-treated tumors, IL-15 concentration, lymphocyte grades, and cluster of differentiation-3+ cell counts were significantly increased when compared to other treatment groups. However, viral titers, recombinant protein expression, and lymphocyte numbers within the tumors diminished rapidly at 7 days postinoculation. These data indicate that treatment with recombinant MYXV should be repeated at least every 4 days to maintain recombinant protein expression within a murine tumor. Additionally, neutrophilic inflammation was significantly increased in MYXV-Tred- and MYXV-IL15-treated tumors at early time points. It is speculated that neutrophilic inflammation induced by intratumoral replication of recombinant MXYV contributes to the antitumoral effect of MYXV treatment in this melanoma model. These findings support the inclusion of neutrophil chemotaxins in recombinant poxvirus oncolytic virotherapy.
Keywords: cancer, interleukin-15, melanoma, myxoma virus, neutrophils, oncolytic virus
Introduction
Despite small annual decreases in the overall cancer rate in humans, the incidence of melanoma is increasing by more than 2% each year.1 This drives the need for novel melanoma treatments that are targeted, safe, and effective. Treatment with an oncolytic virus, such as myxoma virus (MYXV), is a promising new strategy that may eliminate tumors by direct lysis of infected tumor cells and by indirect induction of a strong immune response against cancer cells.
MYXV is a poxvirus that is nonpathogenic for all nonrabbit vertebrate species tested.2–7 Yet, MYXV productively infects cultured cancer cells from several species.8–11 Although MYXV-encoded protein expression has been observed in noncancerous cells after intracranial injection of recombinant MYXV, protein expression is transient and no clinical disease is observed.12 One intratumoral injection of MYXV has been shown to eliminate a significant percentage of tumors in nude mice bearing intracranial glioma or medulloblastoma xenografts.13,14 However, in rodent allograft models of melanoma and glioma, multiple injections of MYXV, concurrent immunosuppressive therapy with rapamycin, and/or concurrent T-cell immunotherapy were required to improve outcome.15–17
Ideally, genetic modification of an oncolytic virus could eliminate the necessity of concurrent immunosuppressive treatments. Several studies correlate increased numbers of intratumoral cytotoxic lymphocytes with improved survival of cancer patients or decreased metastases to lymph nodes and distal organs.18–21 It was hypothesized that intratumoral injection of MYXV expressing interleukin-15 (IL-15), a lymphotrophic cytokine, would enhance recruitment of a cell-mediated immune response to the tumor bed and thereby improve the therapeutic efficacy of MYXV.
MYXV was modified to coexpress murine IL-15 and tandem dimer Tomato red fluorescent protein (MYXV-IL15). This virus (previously designated vMyx-IL-15-tdTr) is attenuated in European rabbits, further improving its safety profile.22 Immunocompetent mice were injected subcutaneously with B16F10 cells to establish melanomas. Weekly intratumoral injections of MYXV expressing tandem dimer Tomato red fluorescent protein (MYXV-Tred) or MYXV-IL15 were initiated when tumor length measured 5 mm. Although MYXV-IL15 treatment did not impart a survival benefit when compared to MYXV-Tred treatment, weekly injection of recombinant MYXV significantly prolonged survival when compared to controls. A detailed histologic evaluation of oncolytic MYXV therapy in this subcutaneous B16F10 model of melanoma indicated that neutrophilic inflammation within tumors was significantly increased following intratumoral administration of replicating recombinant MYXV.
Material and methods
Cell lines
The murine melanoma cell line, B16F10, was obtained from American Type Culture Collection (Manassas, VA, USA). Rabbit kidney epithelial (RK13) cells were a gift from Dr Richard Moyer (University of Florida, Gainesville, FL). Cell lines were maintained at 37°C, 5% carbon dioxide, and 100% humidity in minimum essential medium with Earle’s salts (Corning Inc, Corning, NY) supplemented with 2 mM glutamine, 50 U/mL penicillin G, 50 μg/mL streptomycin, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids (MEM-C), and HyClone® 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA).
Viruses
The Lausanne strain of MYXV was used to create recombinant MYXV-Tred and MYXV-IL15 as previously described.22 Ultraviolet-inactivated virus (UV-MYXV) was prepared by exposing MYXV-Tred to a G30T8 germicidal UV light that produced irradiation at 254 nm for 24 hours. Viral inactivity was confirmed by the lack of cytopathic effect, viral plaques, and fluorescence when cultured on RK13 cells.
Viral replication in cell culture
Cells were seeded into Nunc® six-well cell culture plates (Thermo Fisher) and allowed to grow in MEM-C with 10% FBS until they reached 90%–95% confluency. Cells were inoculated with recombinant MYXV diluted in 400 μL MEM-C and were incubated at 37°C in 5% carbon dioxide for 1 hour, rocking every 15 minutes. For multi-step growth curves, cells were inoculated at a multiplicity of infection (moi) of 0.1 recombinant MYXV plaque-forming units (pfu) per cell, and then incubated for 1 hour at 37°C and 5% carbon dioxide. The inoculum was removed, cell monolayers were washed with phosphate-buffered saline (PBS), and 1.5 mL MEM-C with 10% FBS was added to each well. Viral inoculations of each cell line were performed in triplicate. Cells were scraped into the media and collected at several time points postinoculation. Cells were pelleted at 500 g for 5 minutes, supernatant was removed, and then cells were resuspended in 0.5 mL PBS and stored at −80°C. Collected cells were disrupted by three freeze/thaw cycles and sonication in order to release virus from the cells. Samples were titered in duplicate.
Titering consisted of placing ten-fold serial dilutions of the samples in supplemented MEM-C onto RK13 cellular monolayers in six-well cell culture plates. The inoculated cells were incubated for 1 hour at 37°C and 5% carbon dioxide, and then the inoculum was removed and the RK13 cells were overlaid with equal amounts of 1% agarose (Lonza, Basel, Switzerland) and 2 × MEM-C with 20% FBS. Plaques were visualized as small white foci (red foci under fluorescent light) and counted at 7 days postinoculation (dpi).
Immunoblotting
Western blot analysis was performed to evaluate phosphorylation of Akt in B16F10 cells. Cells were grown in 35-mm diameter plates to 95% confluency, inoculated with MYXV-Tred (moi = 5), and then collected at 0 or 4 hours postinoculation (hpi) in cell lysis buffer with Pierce® Halt protease and phosphatase inhibitor (Thermo Fisher). Total protein concentration was determined with a Bradford assay, and 50 μg of protein from each cell lysate was analyzed via sodium dodecyl sulfate polyacrylamide gel electrophoresis (10%). Proteins were transferred to a nitrocellulose membrane, which was incubated in blocking buffer overnight at 4°C, then incubated with rabbit antiphospho-Akt (serine 473) monoclonal antibody (Cell Signaling Technology, Danvers, MA) in accordance with the manufacturer’s protocol. Membranes were incubated for 1 hour at room temperature with a horseradish peroxidase-conjugate goat antirabbit immunoglobulin G polyclonal antibody (EMD Millipore, Billerica, MA) diluted 1:2000 in blocking buffer. A chemiluminescent Western blot detection kit (GE Healthcare, Little Chalfont, UK) was used to detect antigen–antibody complexes. Membranes were stripped in accordance with the protocol provided by the manufacturer of the detection kit, then incubated with rabbit antiAkt monoclonal antibody (Cell Signaling Technology) following the manufacturers’ instructions, and antigen–antibody complexes indicating total Akt in the cell lysates were detected.
Cell viability assays
A trypan blue exclusion assay was used to assess death of B16F10 cells following inoculation with MYXV-Tred. Cells were infected with MYXV-Tred (moi = 10) or mock-infected (by inoculating cell with MEM-C lacking virus) and collected at 0, 24, and 48 hpi. Cells were stained by combining cells suspended in media with 0.4% trypan blue solution (Thermo Fisher) at a 1:10 ratio. Clear cells and blue cells were counted on a hemocytometer to calculate the percentage of dead (blue) cells. Data was analyzed with Student’s t-test for independent samples using Statistica software (version 7) (StatSoft, Tulsa, OK). Any nonparametric data was compared using a Mann–Whitney U test. Significance was set at P ≤ 0.05.
A CellTiter-Blue® cell viability assay (Promega Corporation, Fitchburg, WI) was performed to assess cell viability of B16F10 cells following inoculation with MYXV. This assay detects the conversion of resazurin, a redox dye, into a fluorescent product (resorufin) by viable cells. Fifty-thousand cells were added to each well of a 96-well plate, centrifuged at 200 g for 5 minutes, and incubated for 4 hours in media with 10% FBS to allow the cells to adhere to the plate. Media was then removed and cells were mock-infected or infected with MYXV (moi = 10) in 50 μL of media without serum as previously described. The experiment was performed in triplicate. Wild-type MYXV was used so as to avoid assay interference by MYXV-Tred fluorescence. Virus inocula were removed, 100 μL of media with 10% FBS was added, and cells were incubated for 24–48 hours. Cell titer-blue reagent (20 μL) was added 3 hours before analysis. Analysis was performed using SpectraMax® Gemini™ EM microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA) with excitation at 544 nm and emission at 590 nm. Data was analyzed with Student’s t-test for independent samples using GraphPad Prism® 5.0 (GraphPad Software, Inc, La Jolla, CA). Significance was set at P ≤ 0.05.
Measurement of cytokine concentrations
The concentrations of interferon-α (IFNα), IFNβ, tumor necrosis factor-α (TNFα), and IL-15 in cell cultures and tissue lysates were determined using commercially available enzyme-linked immunosorbent assays (ELISAs). Samples were prepared by infecting cells with MYXV recombinants (moi = 5 for the IFNα, IFNβ, and TNFα assays; moi = 1 for the IL-15 assay), or mock-infecting cells, as previously described.
Infected cell culture supernatants were collected at 24 and 48 hpi, as well as at 72, 96, and 168 hpi for IL-15. In addition, tumors from mice treated with MYXV-IL15 were collected and homogenized for detection of IL-15 within the treated tumors. All samples were concentrated using Amicon® Ultra-4 centrifugal filter units (EMD Millipore), from initial sample volumes of 1.5–4 mL, to a final volume of approximately 450 μL, by centrifuging at 3220 g at room temperature for 5–10 minutes as needed. IFNα and IFNβ ELISAs (mouse IFNα ELISA kits; PBL Biomedical Laboratories, Piscataway, NJ), TNFα ELISA (mouse TNFα ELISA Ready-SET-Go!®; eBioscience, San Diego, CA), and the IL-15 ELISA (mouse IL-15 ELISA Ready-SET-Go!; eBioscience) were performed according to the manufacturer’s instructions. Each kit included a purified protein standard which was used to establish a standard curve. Positive controls for each ELISA were obtained by adding known quantities of standard from the ELISA kits to additional cell lysates from the cell lines, while negative controls were obtained from wells that contained media but did not contain any cells. Negative controls were subtracted from the test samples in order to compensate for background. A SpectraMax Plus384 microplate spectrophotometer (Molecular Devices) was used to detect absorbance at 450 nm.
Mice
C57BL/6 mice obtained from Jackson Laboratory (Bar Harbor, ME) were used because they are regarded as a type one helper T-cell-dominant mouse strain;23,24 and the B16F10 cells used in this study were originally derived from a C57BL/6 mouse.25 Mice were housed in sterilized ventilated cages and maintained under biosafety level two conditions in a Division of Animal Resources facility at the University of Illinois (Urbana, IL). All procedures were approved by the Institutional Animal Care and Use Committee. Six-week-old female mice were allowed to acclimate for 7 days before tumor inoculation.
To establish melanoma allografts, mice were anesthetized with isoflurane gas and then 5 × 105 B16F10 cells diluted in 100 μL PBS were injected subcutaneously into the left flank. Once tumors reached 5 mm in length, the tumors were directly injected with 106 pfu of sucrose-pad purified MYXV-Tred or MYXV-IL15 diluted in 100 μL PBS. Controls were established by injecting tumors with UV-MYXV diluted in 100 μL PBS or with 100 μL PBS alone. Injections were repeated weekly and the inoculum was injected into several sites in the larger tumors in order to spread the virus throughout the mass.
Mice were observed and weighed every 3 days until tumors were detected, and then mice were weighed daily. Tumors were measured daily using calipers until a criterion of euthanasia was met. Euthanasia criteria included: tumor length ≥ 3 cm, lethargy, significant dermal ulceration over the tumor site, or 25% baseline body weight loss. Samples from five to seven mice in each treatment group were collected at 1, 2, 4, and 7 dpi, and twelve to fourteen mice in each treatment group were allowed to reach a criterion of euthanasia. Survival data were recorded from the time of the initial viral injection until euthanasia and were plotted using a Kaplan–Meier curve. The Cox proportional hazards model was used to describe survival time distribution. Data were analyzed using the PHREG procedure in SAS software (version 9.3) (SAS Institute Inc, Cary, NC).
Detection of viral neutralizing antibodies
Plaque reduction neutralization tests (PRNT) were performed to evaluate anti-MYXV antibody production in mice after multiple weekly treatments with MYXV-Tred, MYXV-IL15, or UV-MYXV. PRNT50 values represent the reciprocal of the serum dilution that reduced the viral titer by 50%. Serum samples that did not reduce the viral titer by 50% were assigned a PRNT50 value of zero to indicate that an antiviral antibody response was not detected. The 50% endpoint titer was defined as half of the average titer recovered from virus inoculum incubated with serum from PBS-treated mice.
To collect serum, blood was removed from mice by cardiac puncture at the time of euthanasia, placed into red-topped tubes, and centrifuged. Serum samples from PBS-treated mice were added to equal volumes of diluted MYXV-Tred creating a 1:2 dilution of the serum, which was used to inoculate confluent RK13 cellular monolayers (100 pfu/well). Serum samples from mice treated with MYXV-Tred, MYXV-IL15, or UV-MYXV were serially diluted in MEM-C. The diluted samples were incubated with MYXV-Tred (100 pfu/well) on RK13 cells in 24-well cell culture plates (TPP Techno Plastic Products AG, Trasadingen, Switzerland) for 1 hour. Cells were then overlaid with a 1:1 mixture of 1% agarose and 2 × MEM-C with 20% FBS. Cells were examined by fluorescent microscopy for viral plaques at 7 dpi. Virus titers (pfu/mL of diluted sample) were plotted versus the reciprocal of the sample dilution of serum from MYXV-Tred-, MYXV-IL15-, and UV-MYXV-treated mice. Using Microsoft Excel® 2010 (Microsoft Corporation, Redmond, WA), the sample dilution of the 50% endpoint was determined by linear interpolation of the dilutions above and below the 50% endpoint titer as previously described.26
To determine statistical significance of these data, an analysis of variance model was expressed as: yijk = μ∞ + trti + dayj + (trt * day)ij + εijk where yijk represents the measurement of a particular variable for kth mouse on jth day in ith treatment (control) group, μ∞ is a constant, trti represents ith treatment (control) effect, dayj represents jth day effect, and (trt * day)ij represents the interaction between treatment (control) effect and time effect; εijk are independent N (0, σ2); i = 1,2,3,4; j = 1,2,3,4,5. The analysis of variance was performed for each variable to test whether there was difference among the means of the four groups (two treatments and two controls), or equivalently, whether there was a treatment effect of replicating MYXV as compared to control treatments. Duncan’s test was employed as a multiple comparison test.
Tissue collection
Mice were anesthetized with isoflurane gas prior to euthanasia by intraperitoneal injection of pentobarital (100–150 mg/kg). All major internal organs were grossly examined in all mice in order to check for metastases, evidence of viral infection, or other pathology. Major organs and tumors were collected and placed in 10% buffered formalin for examination by light microscopy (histopathology) using an Olympus DP70 microscope (Center Valley, PA, USA). A section of tumor from most mice and sections of several internal organs (brain, eye, salivary gland, lung, thymus, heart, liver, kidney, spleen, gastrointestinal tract, uterus, ovary, and bladder) from at least one mouse per treatment group were frozen and stored in liquid nitrogen to determine the viral titer in the tissue. A section of the tumor from most mice was also placed in optimal cutting temperature compound (Sakura Finetek, Torrance, CA), flash-frozen, and then stored in liquid nitrogen for subsequent immunohistochemical examination.
Viral isolation from tissues
Tumors and organs from mice in all treatment groups were individually weighed and homogenized in 400 μL MEM-C. The tissue homogenates were titered as described above and the amount of virus isolated per gram of tissue was calculated.
MYXV-Tred fluorescence in vivo
MYXV-Tred fluorescence was evaluated in all flash-frozen tumor sections prior to treatment with Vectashield® mounting medium (Vector Laboratory Inc, Burlingame, CA). The presence of red fluorescence was recorded as zero or one, indicating absence or presence of the virus, respectively. Colocalization of the virus with any foci of inflammation was noted and photomicrographs were taken. The presence of virus was compared between treatment groups using Fisher’s exact test. Significance was set at P ≤ 0.05.
Histopathology
Tumors and several organs (brain, eye, lung, heart, liver, kidney, spleen, uterus, ovary, bladder, adrenal gland, and lymph nodes) were trimmed into cassettes, paraffinized, sectioned, stained with hematoxylin and eosin, and then evaluated by Dr R A Doty using light microscopy. Tumor pathology was graded on percent necrosis, neutrophil infiltration, and lymphocyte infiltration (Table 1). Representative photomicrographs of graded sections are shown in Figure S1. For the inflammatory cell infiltrates, sites of cellular infiltration (within the tumor, at the junction of the tumor and surrounding tissue, within the surrounding tissue, and within areas of tumor necrosis) were evaluated and the 400× magnified field with the highest number of inflammatory cells was used for grading each area. Organs also were examined for evidence of metastases or inflammation, although these findings did not contribute to the grading system used.
Table 1.
Grading schemes for tumor histology sections
| Grade 0 | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
|---|---|---|---|---|---|
| Percent of necrosis per section | 0 | 1–15 | 16–49 | 50–74 | 75–100 |
| Neutrophils per 400× field† | 0–10 | 11–25 | 26–49 | >50 | >150 |
| Lymphocytes per 400× field‡ | 0–5 | 6–15 | 16–30 | >30 | >150 |
Notes:
The microscopic field with the most severe neutrophilic infiltrate was utilized;
the microscopic field with the most severe lymphocytic infiltrate was utilized.
Immunohistochemistry
Tissues flash-frozen in an optimal cutting temperature compound were trimmed to 6 μm sections on a cryostat, placed on a slide, rinsed with PBS, and blocked with 10% goat serum for 1 hour at room temperature. Serial sections were analyzed for infiltrating leukocytes. Tissues were incubated with either rat antimouse Ly-6G (Gr-1) monoclonal antibodies (eBioscience) to detect neutrophils, rat antimouse major histocompatibility complex class II 1A + 1E monoclonal antibody (Abcam, Cambridge, MA) to detect antigen presenting cells, rat antimouse cluster of differentiation-3 (CD3) monoclonal antibody (Abcam) to detect T-cells, rat antimouse CD4 monoclonal antibody (Becton, Dickinson, and Company, Franklin Lakes, NJ) to detect helper T-cells, rat antimouse CD8a monoclonal antibody (Becton Dickinson) to detect cytotoxic lymphocytes, or rabbit antimouse granzyme B polyclonal antibody (Abcam) to detect natural killer cells and cytotoxic lymphocytes. The antibodies were diluted in PBS and placed on the tissues for 1 hour at room temperature. Tissues were then washed with PBS and incubated with goat antirat polyclonal antibody conjugated to fluorescein isothiocyanate (FITC; Jackson ImmunoResearch Laboratories, West Grove, PA) to detect rat-derived antibodies or with FITC-conjugated goat antirabbit immunoglobulin G polyclonal antibody (Abcam) for detection of granzyme B. Slides were rinsed with PBS, covered with Vectashield mounting medium that contained 4′,6-diamidino-2-phenylindole (Vector Laboratory), and then coverslipped.
Immunohistochemistry was performed on at least three tumors per treatment group at each time point. The entire tumor was reviewed and the highest numbers of FITC+ cells in a 400× magnified field were counted in four areas of each tumor section: within the tumor, at the junction of the tumor and the surrounding tissue, within the surrounding tissue, and within areas of tumor necrosis. The numbers of FITC+ cells in these four areas were added together for each sample and then values from at least three mice for each time point and treatment group were averaged. Differences in average cell numbers over time and between treatment groups were compared as described for the PRNT50 data.
Results
Recombinant MYXV replicated in B16F10 melanoma cells, induced significant cell death, and expressed recombinant proteins
Previous reports that B16F10 cells support replication of MYXV were confirmed by microscopic visualization of cytopathic effects and red fluorescent protein expression after inoculation of cells with MYXV-Tred (Figure 1A) as well as by determination of viral growth kinetics of MYXV-Tred and MYXV-IL15 (Figure 1B).16,17 Viral titers were similar in B16F10 and RK13 cells at all time points confirming that MYXV-Tred and MYXV-IL15 replicate equally well in RK13 and B16F10 cells. Likewise, the presence of phosphorylated Akt in B16F10 cells, a property of cells permissive to MYXV infection,16,27 was confirmed via Western blot (Figure 1C).
Figure 1.
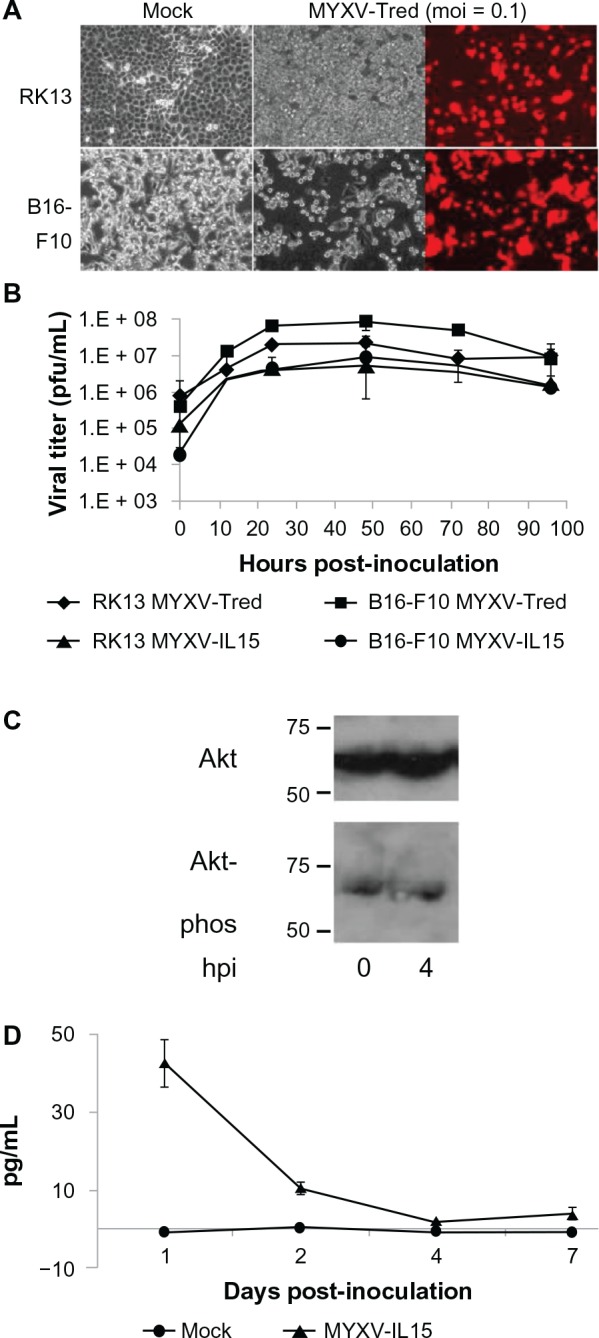
Effects of recombinant MYXV infection of B16F10 cell cultures. (A) Cytopathic effects. Cell lines were mock-infected or infected with MYXV-Tred (moi = 0.1). Both RK13 and B16F10 cells showed significant cytopathic effects at 48 hpi. Fluorescent photomicrographs were obtained using a 560/40 nm bandpass excitation filter and an exposure time of 200 milliseconds. (B) Viral replication kinetics after inoculation of RK13 or B16F10 cells with MYXV-Tred or MYXV-IL15 (moi = 0.1). The average viral titers isolated from three experiments are shown. (C) Western blot detection of total Akt and phosphorylated Akt in MYXV-Tred-infected B16F10 cells at 0 and 4 hpi (moi = 5). (D) Average concentrations of IL-15 (pg/mL) in MYXV-IL15-infected B16F10 cell lysates (moi = 1) were detected at 1, 2, 4, and 7 dpi by enzyme-linked immunosorbent assay.
Notes: Error bars represent standard error of the mean. RK13 are rabbit kidney control cells and B16F10 is a murine melanoma cell line.
Abbreviations: dpi, days postinoculation; hpi, hours postinoculation; IL-15, interleukin-15; moi, multiplicity of infection; MYXV, myxoma virus; MYXV-IL15, MYXV coexpressing murine IL-15 and tandem dimer Tomato red fluorescent protein; MYXV-Tred, MYXV expressing tandem dimer Tomato red fluorescent protein; pfu, plaque-forming units.
As expected in cells permissive to MYXV infection,10,28 B16F10 cells did not produce detectable concentrations of IFNα, IFNβ, or TNFα following recombination MYXV inoculation (data not shown). Importantly, the production of murine IL-15 in MYXV-IL15-infected B16F10 cells was confirmed (Figure 1D).
The ability of MYXV-Tred to kill murine B16F10 cells was observed using a trypan blue assay (Figure 2A). Similar results were observed when cellular viability was measured using a cell viability assay (Figure 2B). The marked reduction of IL-15 between 24 and 48 hpi (Figure 1D) correlated with a significant increase in virus-induced cellular death at 48 hpi (Figure 2).
Figure 2.
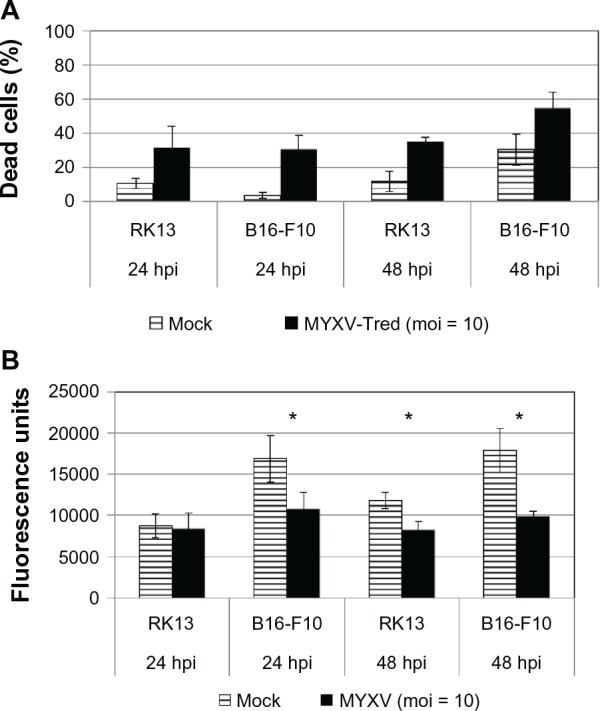
Viability of B16F10 melanoma cells after inoculation with MYXV. (A) Cell death assay. Trypan blue analyses were performed with RK13 and B16F10 cells at 24 or 48 hpi with MYXV-Tred (moi = 10). Data points indicate the average percentage of dead cells from four experiments. Significantly increased numbers of dead cells were observed at 24 and 48 hpi with MYXV-Tred when compared to mock-infected cells (P ≤ 0.0183). (B) Cell viability assay. Promega CellTiter-Blue assays were performed at 24 and 48 hpi with MYXV (moi = 10).
Notes: Higher fluorescence unit values indicate the presence of larger numbers of live cells. *Indicates statistical significance (P ≤ 0.0017); error bars represent standard error of the mean; RK13 are rabbit kidney control cells and B16F10 is a murine melanoma cell line.
Abbreviations: hpi, hours postinoculation; moi, multiplicity of infection; MYXV, myxoma virus; MYXV-Tred, MYXV expressing tandem dimer Tomato red fluorescent protein.
Repeated intratumoral recombinant MYXV treatments were safe in C57BL/6 mice
Safety is one of the most critical properties of a new cancer therapeutic. Mice in this study did not have any observable adverse effects after intratumoral injection of any of the treatments; they continued to eat, drink, behave, and gain weight as expected.29 Histological review of internal organs did not reveal any significant pathological changes that could be attributed to treatment.
Portions of multiple organs were homogenized and inoculated on RK13 cellular monolayers to determine if there was unwanted viral spread outside of the treated tumor bed. Viral infection of organs was generally absent, with the exception of detection of the virus in lung homogenates from two MYXV-Tred-treated mice that were euthanized 1 dpi (106 and 108 pfu/g, respectively) and one viral colony in an eye homogenate from a MYXV-Tred-treated mouse euthanized 4 dpi (53 pfu/g). The fact that the virus was not detected in noncancerous tissues at later time points indicates that the virus was not able to productively infect the normal cells of the body.
MYXV-Tred and MYXV-IL15 treatments resulted in significantly increased survival times
This study asked whether IL-15 expression by MYXV would increase the number of tumor-infiltrating lymphocytes, which is hypothesized to increase the median survival time as compared to mice treated with MYXV-Tred. To test this, intratumoral injections were repeated weekly until mice with established subcutaneous melanomas exhibited a criterion of euthanasia. Mice received between one and four injections before criteria of euthanasia were met. Groups of mice that were treated with replicating virus (MYXV-Tred or MYXV-IL15) had a significantly longer median survival time than control mice treated with UV-MYXV or PBS (P = 0.0067; Figure 3A), indicating that repeated inoculation with recombinant MYXV is an effective stand-alone treatment in this aggressive primary melanoma tumor model. However, expression of IL-15 by MYXV did not result in a statistically significant increase in median survival time when compared to MYXV-Tred treatment.
Figure 3.
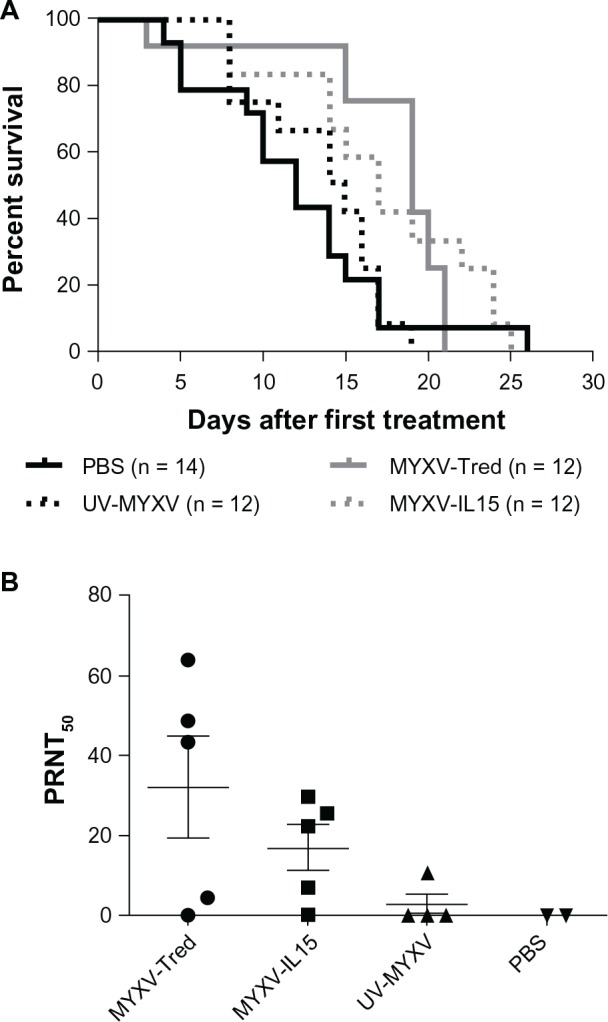
Long-term outcomes in C57BL/6 mice bearing B16F10 melanomas after initiation of weekly intratumoral treatment injections with MYXV-Tred, MYXV-IL15, UV-MYXV, or PBS. (A) Kaplan–Meyer survival curves. (B) Development of anti-MYXV antibodies.
Notes: Plaque reduction neutralization tests were used to calculate the serum antibody titer that caused a 50% reduction of MYXV-Tred plaque formation on RK13 cellular monolayers. RK13 are rabbit kidney control cells and B16F10 is a murine melanoma cell line.
Abbreviations: MYXV, myxoma virus; MYXV-IL15, MYXV coexpressing murine interleukin-15 and tandem dimer Tomato red fluorescent protein; MYXV-Tred, MYXV expressing tandem dimer Tomato red fluorescent protein; PBS, phosphate-buffered saline; PRNT50, plaque reduction neutralization test value representing the reciprocal of the serum dilution that reduced the viral titer by 50%; UV-MYXV, ultraviolet-inactivated MYXV.
It is possible that MYXV-IL15 was eliminated from the tumor more rapidly than MYXV-Tred, preventing MYXV-IL15 from improving therapeutic efficacy. The clearance of MYXV-Tred and MYXV-IL15 from the tumor was evaluated in several ways: (1) antiviral antibody production was measured, (2) tumors were examined for the presence of virus, (3) recombinant protein expression within tumors was evaluated, and (4) cellular infiltrates in the tumor microenvironment were quantified (see below).
An anti-MYXV serum antibody response was detected in some virus-treated mice
To determine if mice acquired an adaptive B-cell response to repeated recombinant MYXV inoculation, blood was collected at the time of euthanasia from a subset of mice at survival endpoints and the ability of their serum to neutralize MYXV-Tred was determined with a PRNT. An antibody response capable of reducing the formation of MYXV-Tred plaques by more than 50% (when compared to serum from PBS-treated mice) was observed in four of five (80%) MYXV-Tred-treated mice, four of five (80%) MYXV-IL15-treated mice, and one of four (25%) UV-MYXV-treated mice (Figure 3B). These results were not statistically significant between groups. When data from mice treated with MYXV-Tred or MYXV-IL15 were grouped and compared to data from UV-MYXV-treated controls, mice treated with a replicating virus had a significantly greater mean PRNT50 (P = 0.0282).
Viral particles and expression of recombinant genes were detected at 1, 2, and 4 dpi, but decreased by 7 dpi
To determine how long the virus persisted in the treated subcutaneous melanoma tumors, the presence of virus was detected by plaque assay to isolate infectious viral particles (Figure 4A) and/or by fluorescent microscopy to observe tandem dimer Tomato red protein expression (Figure 4B). The virus was detected by plaque assay and/or fluorescent imaging in all of the tumors treated with either MYXV-Tred or MYXV-IL15 that were collected at 1, 2, and 4 dpi, but the frequency of detection as well as the viral titer significantly decreased by 7 dpi. Similarly, IL-15 expression was detectable by ELISA in MYXV-IL15-treated tumors collected at 1, 2, and 4 dpi, but dropped to very low levels by 7 dpi (Figure 4C).
Figure 4.
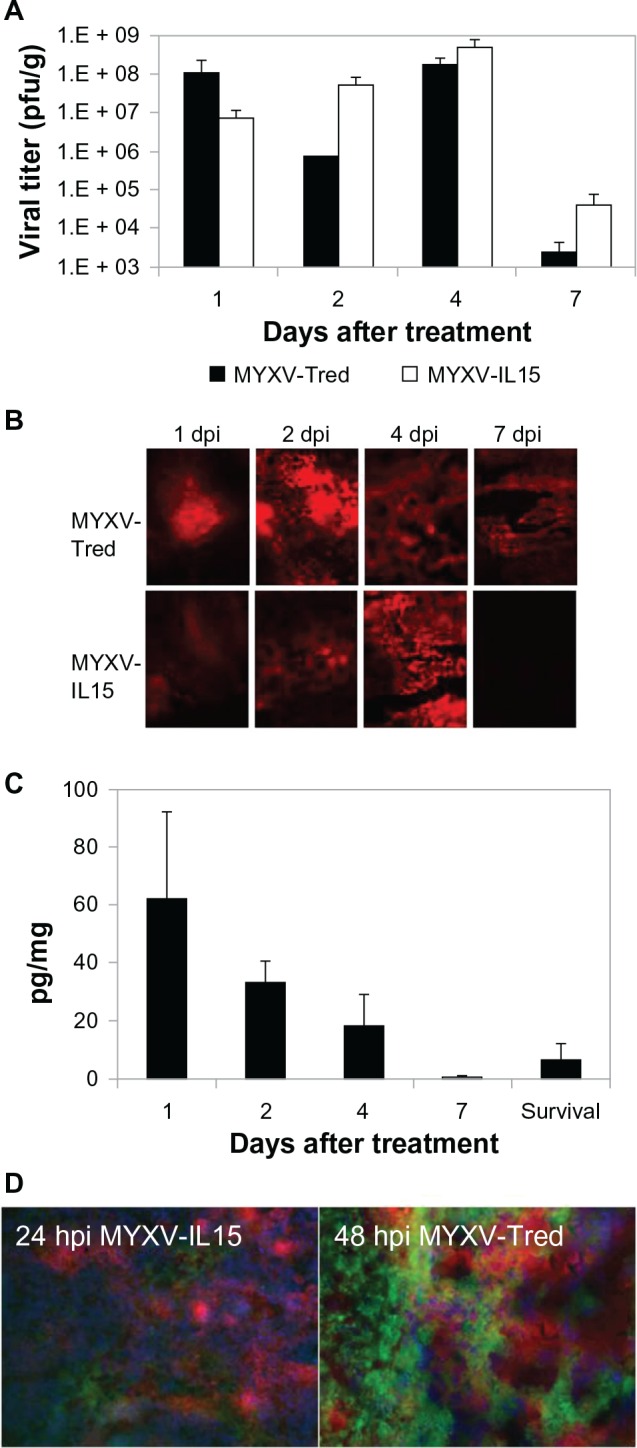
Virus and recombinant protein detection in tumor sections. (A) Replicating virus was detected by plaque assay from one to six tumors per group. Data indicate the average viral titer (pfu/g). (B) Representative images of red fluorescence observed in flash-frozen tumor sections. The fluorescence resulted from tandem dimer Tomato red expression during virus infection (200×). (C) Average IL-15 concentration within tumor sections (pg/mg tissue). Error bars represent standard error of the mean. (D) Merged fluorescent images of B16F10 cells infected with MYXV-IL15 24 hpi (100×) or MYXV-Tred 48 hpi (400×).
Notes: Both tandem dimer Tomato red fluorescent protein expression by recombinant MYXVs and fluorescein isothiocyanate-labeled Gr-1+ neutrophils were located in areas of necrosis within the tumors. B16F10 is a murine melanoma cell line.
Abbreviations: dpi, days postinoculation; hpi, hours postinoculation; IL-15, interleukin-15; MYXV, myxoma virus; MYXV-IL15, MYXV coexpressing murine IL-15 and tandem dimer Tomato red fluorescent protein; MYXV-Tred, MYXV expressing tandem dimer Tomato red fluorescent protein; pfu, plaque-forming units.
At 7 dpi, three of seven (43%) mice treated with MYXV-Tred and two of six (33%) mice treated with MYXV-IL15 had detectable virus, indicating that viral clearance was not significantly different in MYXV-Tred- and MYXV-IL15-treated tumors. In mice used to collect survival data, the isolation of virus, visualization of fluorescence, or detection of IL-15 depended upon the time between the last treatment injection and euthanasia. When fluorescence was present in tumor sections, it was typically observed within areas of tumor necrosis and colocalized with tumor infiltrating neutrophils and macrophages (Figure 4D). No virus was isolated from and no fluorescence was observed in tumors of UV-MYXV- or PBS-treated mice. These results show that, while recombinant MYXV can persist for 7 days after treatment, the virus is more consistently present in higher amounts at earlier time points after inoculation.
Histology of subcutaneous B16F10 melanomas following intratumoral injection
Subcutaneous B16F10 melanoma tumors collected from affected mice in each treatment group were examined histologically to evaluate tumor architecture, aggressiveness, and leukocyte infiltrates and determine if any differences were evident between the treatment groups. On gross examination, no significant differences were observed between treatment groups. In all mice, melanotic tumors could be observed through the thin muscle of the abdominal wall; however, the tumors did not infiltrate all of the way through the body wall. Subgross examination of tumors revealed an unencapsulated, moderately well-demarcated, invasive, and highly cellular tumor within the dermis (Figure 5A), which occasionally invaded the lumbar and gluteal skeletal muscle deep to the mass (Figure 5B).
Figure 5.

Representative B16F10 melanoma histology (hematoxylin and eosin). Images were captured from all treatment groups. No significant differences in morphology of the tumors or metastases were observed between treatment groups. (A) Subgross image of an expansive subcutaneous melanoma (20×). (B) Invasion of tumor cells into skeletal muscle (200×). (C) Neoplastic cells within lymphatic vessels (200×). (D) Edema with mixed leukocyte infiltrates (400×). (E) Necrotic edges of the tumor (200×). (F) Granulation tissue surrounding the tumor (200×). (G) Necrosis of the skin over the tumor (400×). (H) Gross metastases in the renal lymph node (left kidney and uterus have been moved right across the midline to improve visualization of the renal lymph node). (I) Metastases in the renal lymph node (50×). (J) Gross metastases to the lungs. (K) Micrometastasis in the lung (400×). (L) Metastasis within a blood vessel in the lung (400×).
Note: B16F10 is a murine melanoma cell line.
Microscopically, the tumor cells were large round cells with distinct cell margins and abundant basophilic cytoplasm that occasionally had a fine to moderate dusting with brown melanin granules. The nuclei were large and round to oval with marginated chromatin and one to three large nucleoli. Marked anisocytosis and anisokaryosis were observed. Mitotic figures averaged six per 400× magnified field. Individual or small rafts of neoplastic cells could be observed within surrounding lymphatics in some tumors (Figure 5C).
The tumors contained multifocal to coalescing areas of necrosis that ranged from 5%–85% of the tumor, and tended to be located centrally. The multifocal distribution of necrosis was due to remaining viable blood vessels, which were surrounded by a thick cuff of viable neoplastic cells, while cells further away from the blood vessels were necrotic. When large areas of necrosis were present, the vessels in the area were also necrotic.
Tumors from the mice euthanized at 1, 2, and 4 dpi were often surrounded by mild to marked dermal edema that contained mixed leukocytes, including macrophages, lymphocytes, and neutrophils (Figures 5D, 7A, and 8A, respectively). The outer edge of the tumors was lined by a thin band of necrosis (Figure 5E) and was surrounded by granulation tissue (Figure 5F). The epidermis overlying the tumors was occasionally absent due to ulceration, but was more often hypereosinophilic with loss of cellular detail (necrotic) and variable infiltration by neutrophils (serocellular crusts; Figure 5G).
Figure 7.
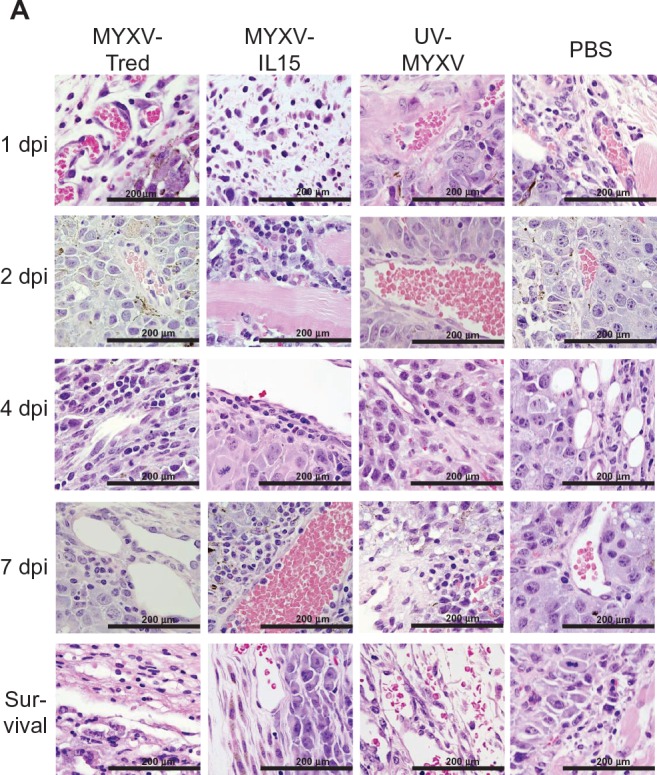
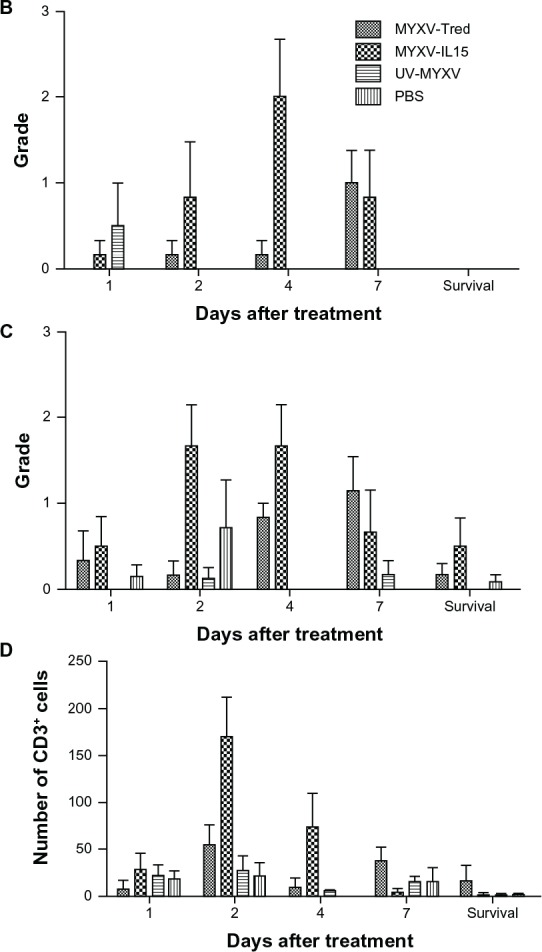
Lymphocyte infiltrates in and around melanoma tumors. (A) Histopathology of representative tumor sections at the indicated time points after intratumoral treatment (hematoxylin and eosin, 400×). In all treatment groups at early time points, tumors often were surrounded by dermal edema that contained mixed leukocytes (eg, MYXV-Tred and MYXV-IL15 1 dpi) and occasionally invaded the lumbar and gluteal skeletal muscle deep to the mass (eg, MYXV-IL15 2 dpi). Lymphocytes were often located surrounding vessels in the tumor (eg, MYXV-IL15 4 and 7 dpi; UV-MYXV 2 and 7 dpi; PBS 7 dpi), in the dermis (eg, MYXV-Tred 1 dpi; PBS 1 dpi), and at the junction of the tumor and the dermis (eg, MYXV-Tred 7 dpi; MYXV-IL15 survival; UV-MYXV survival; PBS 4 dpi and survival). The average grade of lymphocyte infiltrates (B) within tumors and (C) surrounding the tumors indicate an increase in lymphocyte numbers within MYXV-IL15-treated tumors 2 and 4 dpi. (D) The average combined number of CD3+ cells observed in the highest amounts within the tumor, at the junction of the tumor and the surrounding tissue, and within the surrounding tissue is shown.
Notes: Increased numbers of CD3+ cells are present within MYXV-IL15-treated tumors 2 and 4 dpi. Error bars represent standard error of the mean of three to twelve samples.
Abbreviations: CD3+, cluster of differentiation-3+; dpi, days postinoculation; MYXV-IL15, myxoma virus coexpressing murine interleukin-15 and tandem dimer Tomato red fluorescent protein; MYXV-Tred, myxoma virus expressing tandem dimer Tomato red fluorescent protein; PBS, phosphate-buffered saline; UV-MYXV, ultraviolet-inactivated myxoma virus.
Figure 8.


Neutrophil infiltrates in and around melanoma tumors. (A) Histopathology of representative tumor sections at the indicated time points after intratumoral treatment (hematoxylin and eosin, 400×). Tumors from the mice euthanized 1, 2, and 4 dpi were often surrounded by mild to marked dermal edema that contained mixed leukocytes, including lymphocytes, macrophages, and neutrophils (eg, PBS 1 and 2 dpi; MYXV-Tred 4 dpi). The epidermis overlying the tumors was often necrotic with infiltration by neutrophils (eg, PBS survival). Neutrophils were in highest numbers in areas of necrosis (eg, MYXV-Tred 1, 2, and 7 dpi; MYXV-IL15 1, 2, and 7 dpi and survival; UV-MYXV 2 and 4 dpi) and in ulcerated areas (eg, PBS 4 dpi and survival). Neutrophils were more prevalent in areas surrounding the tumor (eg, MYXV-Tred 4 dpi; UV-MYXV survival; PBS 1, 2, and 7 dpi) than in viable areas of the tumors, which only contained neutrophils during early time points. The average grade of neutrophil infiltrates (B) within the tumor, (C) surrounding the tumor, and (D) within areas of tumor necrosis are shown graphically. (E) The average combined number of Gr-1+ cells observed in the highest amounts within the surrounding tissue, the tumor, and areas of tumor necrosis indicate there are increased numbers of neutrophils in MYXV-Tred- and MYXV-IL15-treated tumors between 1 and 4 dpi.
Note: Error bars represent standard error of the mean of three to twelve samples.
Abbreviations: dpi, days postinoculation; MYXV-IL15, myxoma virus coexpressing murine interleukin-15 and tandem dimer Tomato red fluorescent protein; MYXV-Tred, myxoma virus expressing tandem dimer Tomato red fluorescent protein; PBS, phosphate-buffered saline; UV-MYXV, ultraviolet-inactivated myxoma virus.
No overt differences in tumor architecture or aggressiveness were observed between groups of mice at each time point after treatment. However, at early time points, tumor-infiltrating leukocytes were increased in mice treated with MYXV-Tred or MYXV-IL15 when compared to mice treated with UV-MYXV or PBS (see below). Neutrophils were in highest numbers in areas of necrosis and in ulcerated areas, while lymphocytes were often located surrounding vessels in the tumor or in the dermis, or at the junction of the tumor and the dermis. In every treatment group, tumors became larger over time (Figure 6A).
Figure 6.
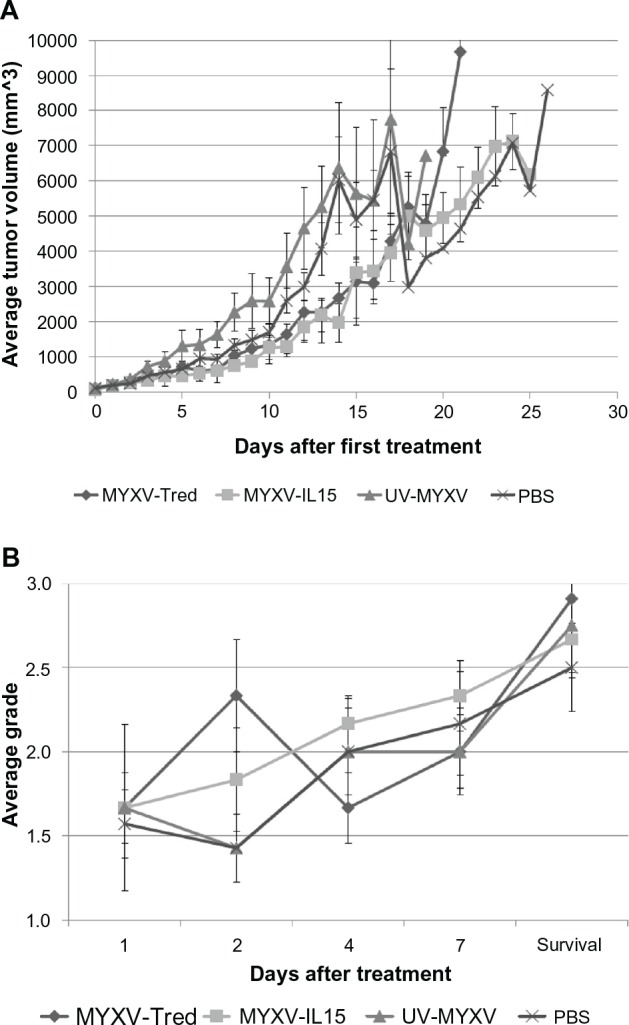
Tumor characteristics. (A) The average tumor volume determined each day after the initial treatment injection. Marked decreases in average tumor volume at 15 and 18 days after initial treatment with UV-MYXV and PBS reflect the deaths of mice with large tumors in these two groups. (B) Average histologic grade of necrosis observed in tumors from mice at 1, 2, 4, and 7 days after treatment and at the survival endpoint.
Note: Error bars represent standard error of the mean.
Abbreviations: MYXV-IL15, myxoma virus coexpressing murine interleukin-15 and tandem dimer Tomato red fluorescent protein; MYXV-Tred, myxoma virus expressing tandem dimer Tomato red fluorescent protein; PBS, phosphate-buffered saline; UV-MYXV, ultraviolet-inactivated myxoma virus.
B16F10 melanomas metastasized to the draining (renal) lymph node and the lungs
To determine if treatment affected the rate or localization of B16F10 metastatic lesions, mice were examined after euthanasia for grossly apparent metastases and organs were examined microscopically for micrometastases. No decrease in the rate of metastasis was observed after recombinant MYXV treatment. In all treatment groups, metastases were most often located in the lymph node medial to the kidney (Figure 5H and I). The second most common area to find metastases was the lungs where tumors often could be observed grossly (Figure 5J), but sometimes were only detectable by light microscopy (Figure 5K). Microscopic metastases commonly were adjacent to small vessels in the lungs or along the pleural surface. Micrometastases adjacent to small vessels indicate that cells likely spread to the lungs by the hematogenous route. Additionally, melanoma cells rarely were observed in blood vessels within the lung (Figure 5L).
Invasion of the renal lymph node by melanoma cells was observed in 13 of 48 (27%) mice at survival endpoints, while metastases were detected in the lungs in twelve of 48 (25%) mice at survival endpoints. Metastases were more common in the renal lymph node and lung of MYXV-Tred-treated mice (five of twelve and three of twelve, respectively) and MYXV-IL15-treated mice (five of twelve and five of twelve, respectively) than in UV-MYXV-treated mice (one of twelve and two of twelve, respectively) and PBS-treated mice (two of twelve and two of twelve, respectively), possibly due to the longer survival times obtained by the mice treated with live virus. Other organs where metastases were found included the spleen (one MYXV-IL15-treated mouse at 7 dpi) and the diaphragm (one PBS-treated mouse at 15 dpi).
Tumor necrosis increased over time in all treatment groups
Tumor cell death is a desired outcome of oncolytic viral therapy. As an indicator of cellular death within the tumors from each treatment group, histological tumor sections were graded and compared as described in the Materials and Methods section (Table 1). While the degree of necrosis increased over time in all treatment groups (P < 0.0001), there was not a statistically significant difference in the amount of necrosis between individual treatment groups (P = 0.5793; Figure 6B) or between replicating virus treatments when compared to control treatments (P = 0.1662). This suggests that virus inoculation does not induce significant tumor necrosis. Instead, the tumor expansion and concurrent lack of blood supply, which was observed histologically, appears to be the main cause of necrosis in this tumor model.
Lymphocytes and CD3+ cells increased in MYXV-IL15-treated tumors
One purpose of this study was to determine if intratumoral MYXV-IL15 treatment increases the number of lymphocytes that infiltrate a solid tumor. To accomplish this goal, tumors were graded histologically on the highest number of lymphocytes as described in the Materials and Methods section (Table 1). Histologic images of representative tumors at 1, 2, 4, and 7 dpi and at survival endpoints are shown in Figure 7A. The average lymphocyte grades within the tumor (Figure 7B) or surrounding the tumor (Figure 7C) were compared at the same time points. Notably, no lymphocytes were observed within areas of tumor necrosis at any time point or treatment group. This is expected, as lymphocytes do not typically infiltrate necrotic tissue. Analysis of variance and multiple comparison of treatment groups over all time points indicate that, as predicted, the average lymphocyte grades within and surrounding MYXV-IL15-treated tumors were higher than any of the other treatment groups (P < 0.0001). Increased lymphocyte grades in MYXV-IL15-treated mice were most evident at 2 and 4 dpi (Figure 7B and C).
Immunohistochemistry was used to evaluate the lymphocyte subsets infiltrating the tumors as described in the Material and Methods section. Based upon the known functions of IL-15, infection with MYXV-IL15 was predicted to increase CD3+, CD8+, and granzyme B+ cells within the tumor and surrounding microenvironment. As predicted, MYXV-IL15-treated tumors were found to have more CD3+ cells than any other treatment group (P = 0.0005; Figure 7D) with the greatest numbers of CD3+ cells present at 2 and 4 dpi. This suggests that intratumoral IL-15 expression by MYXV-IL15 induces T-cell infiltration into the tumor microenvironment. However, no statistical differences were observed in the numbers of infiltrating cells expressing CD8, granzyme B, CD4, or major histocompatibility complex class II.
In summary, MYXV-IL15 treatment effectively increased the numbers of intratumoral CD3+ T-cells above and beyond MYXV-Tred therapy alone. This directly correlated with the increased numbers of lymphocytes observed in MYXV-IL15-treated tumor histology, but did not impart a survival advantage for MYXV-IL15-treated mice with melanoma.
Neutrophils were increased within tumors treated with virus
Neutrophilic inflammation in and around melanoma tumors was graded as described in the Materials and Methods section (Table 1) to better evaluate this important component of innate immunity. The neutrophil grade within viable areas of the tumor, surrounding the tumor, and within areas of tumor necrosis were compared between treatment groups at 1, 2, 4, and 7 dpi, as well as at survival endpoints (Figure 8A). Overall, neutrophils were more prevalent in areas surrounding the tumor and in necrotic tissue than in viable areas of the tumors, which only contained neutrophils during early time points. Figure 8B graphically depicts the average neutrophil grades within viable areas of the tumors, in which mice treated with replicating viruses had significantly higher numbers of neutrophils than other treatment groups (P = 0.0013). In areas surrounding the tumor (Figure 8C), PBS-treated mice had significantly fewer neutrophils than UV-MYXV-, MYXV-Tred-, or MYXV-IL15-treated mice (P = 0.0280). Lastly, in areas of tumor necrosis (Figure 8D), MYXV-Tred and MYXV-IL15 each had significantly increased numbers of neutrophils when compared with PBS- or UV-MYXV-treated mice (P < 0.0001).
Neutrophils were also examined by immunohistochemistry using an anti-Gr-1 antibody and were evaluated as described in the Materials and Methods section (Figure 8E). MYXV-Tred and MYXV-IL15 treatments caused a significant infiltrate of Gr-1+ cells when compared to UV-MYXV treatment (P = 0.0081). When replicating viruses were grouped and compared to control treatment groups, significantly more Gr-1+ cells were detected (P = 0.0004). These results were similar to those obtained by histopathologic analysis of neutrophil grades which were highest in MYXV-Tred- and MYXV-IL15-treated tumors at early time points postinoculation. The authors speculate that the increase in neutrophils contributed to the mildly increased median survival time of mice with melanomas treated with MYXV-Tred or MYXV-IL15.
Discussion
The absence of “danger signals,” such as inflammatory cytokines, and the lack of cellular and molecular T-cell activation have been considered the main causes of poor tumor immunity.30,31 Oncolytic virotherapy has the potential to change this by causing inflammation specifically in the tumor microenvironment. This study indicated that weekly intratumoral injection with MYXV-Tred or MYXV-IL15 caused a statistically significant, albeit modest, increase in the median survival time of mice with aggressive subcutaneous B16F10 melanomas. Additionally, MYXV-Tred and MYXV-IL15 treatments induced neutrophilic inflammation at the site of injection. Neutrophilic inflammation has been recognized as a key component of oncolytic viral therapy in several models.32–35 In support of these findings, studies have demonstrated increased antitumoral effects using oncolytic viruses that express the neutrophil chemotaxin, granulocyte– macrophage colony-stimulating factor.36–39
Although there is increasing evidence that neutrophils contribute to antitumor immunity, strong cytotoxic lymphocyte and natural killer cell responses are thought to be important components of immunological elimination of cancerous cells. The expression of IL-15 within a tumor is expected to induce proliferation and activation of cytotoxic effector cells and monocytes, thereby enhancing this aspect of antitumor immunity.40 Delivery of the gene for IL-15 directly into a tumor using a viral expression vector (such as MYXV-IL15) can provide continuous production of IL-15 every time the virus replicates. The benefit of viral expression of IL-15 has been shown in murine models of human cervical cancer and human breast cancer,41,42 as well as in a murine colon adenocarcinoma model.43 In these models, the level of IL-15 expression within the tumors may be higher than the amount that was detected in mice after intratumoral treatment of MYXV-IL15; however, intratumoral IL-15 concentrations cannot be directly compared due to the inability to calculate IL-15 (in pg) per mg of tissue from the previous studies.
The number of tumor-infiltrating CD3+ T-lymphocytes was increased following MYXV-IL15 treatment in this cancer model. However, the increase of tumor-infiltrating lymphocytes did not correspond to an increased median survival time or a decreased number of metastatic lesions. It is possible that the function of the tumor-infiltrating lymphocytes was impaired in this melanoma model. Assessing the function of the lymphocytes was beyond the scope of this study, but several publications suggest that antitumoral lymphocyte and natural killer cell functions can be improved by coexpressing IL-15 receptor-α with IL-15 during cancer treatment.44–46 Therefore, experiments evaluating the combined delivery of IL-15 and IL-15 receptor-α by MYXV in murine tumors are ongoing. The more likely cause for the lack of an effective antitumoral lymphocyte response in this model was the failure to maintain high numbers of intratumoral lymphocytes after 4 dpi. The decrease in lymphocyte numbers corresponded to the decrease in MYXV-IL15 titer and IL-15 concentration in the tumors after 4 dpi. Indeed, booster injections of recombinant MYXV twice weekly may be beneficial to maintain increased expression of recombinant proteins within a tumor.
One of the potential complications with repeated treatment of viral therapy, that is not an issue with conventional cancer therapies, is the development of an antibody response aimed against the virus. In this study, only a portion of the mice treated with recombinant MYXV developed an antibody response. The development of an antibody response in these mice did not correspond with a difference in median survival time, decrease in viral titer, or reduction in the number of infiltrating leukocytes. This is similar to findings in human patients with colorectal cancer that received recombinant vaccinia virus treatment.47 These data suggest that the benefits of poxvirus oncolytic virotherapy will not be limited in cancer patients with previous exposure to the virus.
An extensive histological characterization of the effects of intratumoral MYXV treatment in a subcutaneous murine B16F10 melanoma model was performed and revealed slight differences from previously published literature. Interestingly, in the current model, metastases rarely were found in the axillary lymph nodes (which drain the trunk), a location sited in previous studies.48,49 Instead, the primary sight of metastatic disease was the renal lymph node, which drains the lumbar area, kidney, and perirenal area, as well as the iliac and paraaortic lymph nodes that drain the gluteal area.48
Also surprisingly, recombinant MXYV was detected outside of the tumor in the lungs of two mice and the eye of one mouse. Most likely, recombinant MYXV spread to the lung hematogenously during, or shortly after, treatment injection. The one viral colony isolated from the eye may have spread there by contact inoculation with an ulcerated MYXV-Tred-infected mass. Previous studies using recombinant MYXV as an oncolytic agent have not reported viral replication outside of the tumor bed, although recombinant protein expression was observed in ependymal and subventricular cells after intracranial injection of recombinant MYXV in healthy mice.12 In this study, as in previously published experiments, no long-term complications were observed in any mice treated with recombinant MYXV. Additionally, extratumoral virus was not detected in any organ of any mouse after 4 dpi. These findings indicate that recombinant MYXV replication outside of the tumor bed occurs, but is self-limiting, and suggests that simple precautions (including bandaging ulcerated lesions) should be taken when administering recombinant MYXV.
Conclusion
In summary, IL-15 expression and transient lymphocyte infiltration induced by MYXV-IL15 did not provide a survival advantage over MYXV-Tred treatment alone in this B16F10 melanoma model in immunocompetent mice. The decreases in intratumoral viral titer, IL-15 expression, and lymphocyte numbers after 4 dpi indicate that recombinant MYXV injections should be administered every 4 days to maintain recombinant protein expression within a murine tumor. Importantly, weekly intratumoral treatment with replication-competent recombinant MYXV significantly increased median survival time and neutrophilic inflammation in mice with melanomas when compared to UV-MYXV- and PBS-treated mice. These data suggest that virus replication and neutrophil infiltration contribute to the oncolytic effects of MYXV in this murine model of melanoma and support the inclusion of neutrophil chemotaxins, such as granulocyte–macrophage colony-stimulating factor, in novel cancer therapeutic approaches.
Supplementary figure
Grading schemes for tumor histology sections (hematoxylin and eosin). Images are representative examples of the grades assigned to the severity of necrosis (40×), and neutrophil and lymphocyte infiltration (400×) within a tumor section.
Acknowledgments
The authors greatly appreciate the scientific input of Dr Joanna Shisler and the statistical expertise of the University of Illinois Department of Statistics for their evaluations of these data. Funding for this project was provided by a Pfizer Animal Health Veterinary Scholars Fellowship, University of Illinois start-up funds, and University of Illinois Urbana Campus Research Board Award 11015. The GM laboratory is supported by NIH R01 AI080607, R01 CA138541, and R21 CA149869.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Kohler BA, Ward E, McCarthy BJ, et al. Annual report to the nation on the status of cancer, 1975–2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst. 2011;103(9):714–736. doi: 10.1093/jnci/djr077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrewes CH, Harisijades S. Propagation of myxoma virus in one-day old mice. Br J Exp Pathol. 1955;36(1):18–21. [PMC free article] [PubMed] [Google Scholar]
- 3.Burnet FM. Myxomatosis as a method of biological control against the Australian rabbit. Am J Public Health Nations Health. 1952;42(12):1522–1526. doi: 10.2105/ajph.42.12.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fenner F. Adventures with poxviruses of vertebrates. FEMS Microbiol Rev. 2000;24(2):123–133. doi: 10.1016/S0168-6445(00)00027-9. [DOI] [PubMed] [Google Scholar]
- 5.Gorski J, Mizak B, Chrobocinska M. Control of rabbit myxomatosis in Poland. Rev Sci Tech. 1994;13(3):869–879. doi: 10.20506/rst.13.3.803. [DOI] [PubMed] [Google Scholar]
- 6.Jackson EW, Dorn CR, Saito JK, McKercher DG. Absence of serological evidence of myxoma virus infection in humans exposed during an outbreak of myxomatosis. Nature. 1966;211(5046):313–314. doi: 10.1038/211313a0. [DOI] [PubMed] [Google Scholar]
- 7.Pignolet B, Boullier S, Gelfi J, et al. Safety and immunogenicity of myxoma virus as a new viral vector for small ruminants. J Gen Virol. 2008;89(Pt 6):1371–1379. doi: 10.1099/vir.0.83595-0. [DOI] [PubMed] [Google Scholar]
- 8.MacNeill AL, Moldenhauer T, Doty R, Mann T. Myxoma virus induces apoptosis in cultured feline carcinoma cells. Res Vet Sci. 2012;93(2):1036–1038. doi: 10.1016/j.rvsc.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Sypula J, Wang F, Ma Y, Bell J, McFadden G. Myxoma virus tropism in human tumor cells. Gene Ther Mol Biol. 2004;8:103–114. [Google Scholar]
- 10.Wang F, Ma Y, Barrett JW, et al. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5(12):1266–1274. doi: 10.1038/ni1132. [DOI] [PubMed] [Google Scholar]
- 11.Woo Y, Kelly KJ, Stanford MM, et al. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann Surg Oncol. 2008;15(8):2329–2335. doi: 10.1245/s10434-008-9924-z. [DOI] [PubMed] [Google Scholar]
- 12.France MR, Thomas DL, Liu J, McFadden G, MacNeill AL, Roy EJ. Intraventricular injection of myxoma virus results in transient expression of viral protein in mouse brain ependymal and subventricular cells. J Gen Virol. 2011;92(Pt 1):195–199. doi: 10.1099/vir.0.026690-0. [DOI] [PubMed] [Google Scholar]
- 13.Lun X, Yang W, Alain T, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65(21):9982–9990. doi: 10.1158/0008-5472.CAN-05-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lun XQ, Zhou H, Alain T, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67(18):8818–8827. doi: 10.1158/0008-5472.CAN-07-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lun X, Alain T, Zemp FJ, et al. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010;70(2):598–608. doi: 10.1158/0008-5472.CAN-09-1510. [DOI] [PubMed] [Google Scholar]
- 16.Stanford MM, Shaban M, Barrett JW, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther. 2008;16(1):52–59. doi: 10.1038/sj.mt.6300348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas DL, Doty R, Tosic V, et al. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol Immunother. 2011;60(10):1461–1472. doi: 10.1007/s00262-011-1045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coca S, Perez-Piqueras J, Martinez D, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79(12):2320–2328. doi: 10.1002/(sici)1097-0142(19970615)79:12<2320::aid-cncr5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 19.Ishigami S, Natsugoe S, Tokuda K, et al. Clinical impact of intratumoral natural killer cell and dendritic cell infiltration in gastric cancer. Cancer Lett. 2000;159(1):103–108. doi: 10.1016/s0304-3835(00)00542-5. [DOI] [PubMed] [Google Scholar]
- 20.Pages F, Berger A, Camus M, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 21.Villegas FR, Coca S, Villarrubia VG, et al. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer. 2002;35(1):23–28. doi: 10.1016/s0169-5002(01)00292-6. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Wennier S, Reinhard M, Roy E, MacNeill A, McFadden G. Myxoma virus expressing interleukin-15 fails to cause lethal myxomatosis in European rabbits. J Virol. 2009;83(11):5933–5938. doi: 10.1128/JVI.00204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivera J, Tessarollo L. Genetic background and the dilemma of translating mouse studies to humans. Immunity. 2008;28(1):1–4. doi: 10.1016/j.immuni.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe H, Numata K, Ito T, Takagi K, Matsukawa A. Innate immune response in Th1- and Th2-dominant mouse strains. Shock. 2004;22(5):460–466. doi: 10.1097/01.shk.0000142249.08135.e9. [DOI] [PubMed] [Google Scholar]
- 25.Fidler IJ, Nicolson GL. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J Natl Cancer Inst. 1976;57(5):1199–1202. doi: 10.1093/jnci/57.5.1199. [DOI] [PubMed] [Google Scholar]
- 26.Borges MB, Kato SE, Damaso CR, et al. Accuracy and repeatability of a micro plaque reduction neutralization test for vaccinia antibodies. Biologicals. 2008;36(2):105–110. doi: 10.1016/j.biologicals.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Wang G, Barrett JW, Stanford M, et al. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc Natl Acad Sci U S A. 2006;103(12):4640–4645. doi: 10.1073/pnas.0509341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartee E, McFadden G. Human cancer cells have specifically lost the ability to induce the synergistic state caused by tumor necrosis factor plus interferon-beta. Cytokine. 2009;47(3):199–205. doi: 10.1016/j.cyto.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Jackson Laboratory JAX® mice strain C57BL/6J (000664) phenotype and body weight information. 2012. [Accessed April 16, 2012]. Available from: http://jaxmice.jax.org/strain/000664.html.
- 30.Fuchs EJ, Matzinger P. Is cancer dangerous to the immune system? Semin Immunol. 1996;8(5):271–280. doi: 10.1006/smim.1996.0035. [DOI] [PubMed] [Google Scholar]
- 31.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 32.Breitbach CJ, Paterson JM, Lemay CG, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther. 2007;15(9):1686–1693. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- 33.Fu X, Tao L, Rivera A, Xu H, Zhang X. Virotherapy induces massive infiltration of neutrophils in a subset of tumors defined by a strong endogenous interferon response activity. Cancer Gene Ther. 2011;18(11):785–794. doi: 10.1038/cgt.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grote D, Cattaneo R, Fielding AK. Neutrophils contribute to the measles virus-induced antitumor effect: enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res. 2003;63(19):6463–6468. [PubMed] [Google Scholar]
- 35.Zhang Y, Patel B, Dey A, et al. Attenuated, oncolytic, but not wild-type measles virus infection has pleiotropic effects on human neutrophil function. J Immunol. 2012;188(3):1002–1010. doi: 10.4049/jimmunol.1102262. [DOI] [PubMed] [Google Scholar]
- 36.Kim JH, Oh JY, Park BH, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther. 2006;14(3):361–370. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 37.Lee JH, Roh MS, Lee YK, et al. Oncolytic and immunostimulatory efficacy of a targeted oncolytic poxvirus expressing human GM-CSF following intravenous administration in a rabbit tumor model. Cancer Gene Ther. 2010;17(2):73–79. doi: 10.1038/cgt.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16(9):1637–1642. doi: 10.1038/mt.2008.143. [DOI] [PubMed] [Google Scholar]
- 39.Lun X, Chan J, Zhou H, et al. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther. 2010;18(11):1927–1936. doi: 10.1038/mt.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vera M, Razquin N, Prieto J, Melero I, Fortes P, Gonzalez-Aseguinolaza G. Intratumoral injection of dendritic cells transduced by an SV40-based vector expressing interleukin-15 induces curative immunity mediated by CD8+ T lymphocytes and NK cells. Mol Ther. 2005;12(5):950–959. doi: 10.1016/j.ymthe.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 41.Yiang GT, Harn HJ, Yu YL, et al. Immunotherapy: rAAV2 expressing interleukin-15 inhibits HeLa cell tumor growth in mice. J Biomed Sci. 2009;16:47. doi: 10.1186/1423-0127-16-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu YL, Wei CW, Chen YL, Chen MH, Yiang GT. Immunotherapy of breast cancer by single delivery with rAAV2-mediated interleukin-15 expression. Int J Oncol. 2010;36(2):365–370. [PubMed] [Google Scholar]
- 43.Stephenson KB, Barra NG, Davies E, Ashkar AA, Lichty BD. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012;19(4):238–246. doi: 10.1038/cgt.2011.81. [DOI] [PubMed] [Google Scholar]
- 44.Epardaud M, Elpek KG, Rubinstein MP, et al. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res. 2008;68(8):2972–2983. doi: 10.1158/0008-5472.CAN-08-0045. [DOI] [PubMed] [Google Scholar]
- 45.Steel JC, Ramlogan CA, Yu P, et al. Interleukin-15 and its receptor augment dendritic cell vaccination against the neu oncogene through the induction of antibodies partially independent of CD4 help. Cancer Res. 2010;70(3):1072–1081. doi: 10.1158/0008-5472.CAN-09-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Z, Xu Y. IL-15R alpha-IgG1-Fc enhances IL-2 and IL-15 anti-tumor action through NK and CD8+ T cells proliferation and activation. J Mol Cell Biol. 2010;2(4):217–222. doi: 10.1093/jmcb/mjq012. [DOI] [PubMed] [Google Scholar]
- 47.Harrop R, Drury N, Shingler W, et al. Vaccination of colorectal cancer patients with modified vaccinia ankara encoding the tumor antigen 5T4 (TroVax) given alongside chemotherapy induces potent immune responses. Clin Cancer Res. 2007;13(15 Pt 1):4487–4494. doi: 10.1158/1078-0432.CCR-07-0704. [DOI] [PubMed] [Google Scholar]
- 48.Tilney NL. Patterns of lymphatic drainage in the adult laboratory rat. J Anat. 1971;109(Pt 3):369–383. [PMC free article] [PubMed] [Google Scholar]
- 49.Van den Broeck W, Derore A, Simoens P. Anatomy and nomenclature of murine lymph nodes: descriptive study and nomenclatory standardization in BALB/cAnNCrl mice. J Immunol Methods. 2006;312(1–2):12–19. doi: 10.1016/j.jim.2006.01.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Grading schemes for tumor histology sections (hematoxylin and eosin). Images are representative examples of the grades assigned to the severity of necrosis (40×), and neutrophil and lymphocyte infiltration (400×) within a tumor section.