

Abstract
Many studies have used DNA microarrays to identify the gene expression signatures of human cancer, yet the critical features of these often unmanageably large signatures remain elusive. To address this, we developed a statistical method, comparative metaprofiling, which identifies and assesses the intersection of multiple gene expression signatures from a diverse collection of microarray data sets. We collected and analyzed 40 published cancer microarray data sets, comprising 38 million gene expression measurements from >3,700 cancer samples. From this, we characterized a common transcriptional profile that is universally activated in most cancer types relative to the normal tissues from which they arose, likely reflecting essential transcriptional features of neoplastic transformation. In addition, we characterized a transcriptional profile that is commonly activated in various types of undifferentiated cancer, suggesting common molecular mechanisms by which cancer cells progress and avoid differentiation. Finally, we validated these transcriptional profiles on independent data sets.
To identify genes potentially important in cancer, scientists have compared the global gene expression profiles of cancer tissue and corresponding normal tissue (1–11). Such analyses usually generate hundreds of genes differentially expressed in cancer relative to normal tissue, making it difficult to distinguish the genes that play a critical role in the neoplastic phenotype from those that represent epiphenomena or are spuriously differentially expressed. Another common experimental design is to compare cancer samples based on their degree of progression, as determined by histological grade, invasiveness, or metastatic potential (2, 11–22). For example, it is known that high-grade undifferentiated-appearing cancers tend to behave more aggressively than their low-grade counterparts, often leading to poorer patient outcomes. To understand the mechanisms by which this progression occurs, many studies have compared the global gene expression profiles of undifferentiated and well differentiated cancers of the same origin. But again, like the “cancer vs. normal” studies, these analyses can also yield hundreds of differentially expressed genes. Thus, it remains a critical problem to elucidate the essential transcriptional features of neoplastic transformation and progression both to direct future research and to define candidate therapeutic targets.
A logical approach for identifying the essential features of a process, given a large set of possibilities observed in a variety of independent systems, is to search for the intersection of observed possibilities across the set of systems, because it is expected that the essential features will be overrepresented and the system-specific, epiphenomenal, and spurious features will be underrepresented. Given the multitude of studies that have attempted to capture the cancer type-specific gene expression programs of neoplastic transformation and progression, we sought to define cancer type-independent, and likely essential, transcriptional features of these important processes. It was initially unclear to us whether such essential features might exist. The complexity in the cellular and molecular origins of cancer might lead one to suspect largely distinct transcriptional programs for independent cancer types, whereas the observation of common phenotypes and behaviors among distinct cancer types might suggest similar transcriptional programs.
In this report, we attempt to identify common transcriptional programs of neoplastic transformation and progression across a wide range of cancer types. To establish a framework for such analysis, we adopted and modified a method, termed meta-analysis of microarrays, which was previously used to validate analogous prostate cancer microarray studies against one another (25). This method avoids many of the pitfalls that complicate the comparison of disparate microarray data sets by comparing statistical measures of differential expression generated independently from each data set rather than actual gene expression measurements. Here, we present a similar method, termed comparative meta-profiling, aimed not at validating analogous data sets, but at comparing and assessing the intersection of many cancer type-specific gene expression data sets, with the goal of identifying cancer type-independent, and likely essential, transcriptional profiles of neoplastic transformation and progression.
Methods
Data Collection, Processing, and Storage. Microarray data sets were downloaded from public web sites or provided by the authors upon request. Data are available at www.oncomine.org/meta. Data were of two general types, two channel ratio data and single channel intensity data, and were usually provided in single composite file format. All available data were included in processing and analysis, except for negative single channel intensity values. All data sets were log transformed and median centered per array, and the standard deviations were normalized to one per array. Studies were named by the following convention: FirstAuthor_TissueTypeProfiled (e.g., Dhanasekaran_Prostate). To facilitate multistudy analysis, microarray features were mapped to Unigene Build 159. Data and initial data analyses were stored in an oracle 8.1 relational database.
Initial Data Analysis. For each of the 40 microarray data sets present in the database, we reviewed the samples profiled. Thirty-four studies had at least four samples corresponding to both classes of one analysis of interest and were further analyzed. Analyses of interest included: cancer versus respective normal tissue, high grade (undifferentiated) cancer versus low grade (differentiated cancer) cancer, poor outcome (metastases, recurrence, or cancer-specific death) cancer versus good outcome (long-term or recurrence-free survival) cancer, metastasis versus primary cancer, and subtype 1 versus subtype 2. After the assignment of samples to classes, each gene was assessed for differential expression with Student's t test using total access statistics 2002 (FM, Vienna, VA). t tests were conducted both as two-sided for differential expression analysis and one-sided for overexpression analysis. To account for multiple hypothesis testing, Q values (26) (estimated false discovery rates) were calculated as
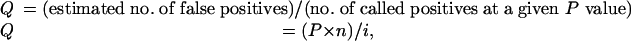 |
where P is P value, n is the total number of genes, and i is the sorted rank of P value.
Meta-Profiling. The purpose of meta-profiling is to address the hypothesis that a selected set of differential expression signatures shares a significant intersection of genes (a meta-signature), thus inferring a biological relatedness. The automated method proceeds as follows: (i) a set of S similar differential expression analyses are selected for meta-profiling; (ii) an overexpression direction (e.g., cancer > normal) and a significance threshold (T) are chosen to define differential expression signatures from the selected analyses (Tdefault = 0.10); (iii) genes are sorted by the number of signatures in which they are present; (iv) the number of genes present in each possible number of signatures is tallied (N0, N1, N2... NS); (v) random permutations are performed (steps iii and iv) in which the actual Q values are randomly assigned to genes per study, so that the genes in each signature change at random, but the number of genes in each signature remain the same. This simulation generates a tally of the number of genes present in each possible number of random signatures (E0, E1, E2... ES); (vi) the significance of intersection among the true signatures is assessed by the minimum meta-false discovery rate (mFDRMIN) calculated as
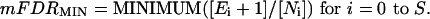 |
(vii)If mFDRMIN < 0.10, a meta-signature is defined as those genes that are significantly differentially expressed (Q < T) in at least j of S analyses, where j is equal to i when mFDRMIN was defined; (viii) if no meta-signature is defined by using Tdefault, steps ii through vii are repeated as T is systematically lowered by 50% at each iteration until either a meta-signature is defined or the number of genes in two or more signatures reaches 0, in which case the result is negative. This assures that a meta-signature is not missed because of an overly liberal Q value threshold. The meta-profiling algorithm was implemented in perl.
Class Prediction. To assess the classification accuracy of the metasignatures, a leave-one-out voting classifier was applied. To predict the class of a particular sample, that sample was removed from the data set, and the remaining samples were used to calculate the two class means for each gene in the signature. The left out sample's gene expression values were compared to the class means. The class mean in which the left out sample's value was closest to received a vote. The votes were tallied, and the prediction was defined as the class with the most votes. A Fisher's exact test was used to assess the significance of the classification. The meta-signatures and class prediction results were visualized by using treeview (27) (http://rana.lbl.gov/eisensoftware.htm).
Results and Discussion
Data and Primary Analysis. As of May 1, 2003, we cataloged information on 152 cancer microarray studies by searching the literature. This catalog and the results from this report are available to explore via our companion web resource, ONCOMINE (www.oncomine.org/meta). Of these published studies, 40 data sets were publicly available and compiled; in total, 37,901,459 gene measurements from 3,762 microarray experiments. Most data sets were of two general formats, either single-channel intensity data, usually corresponding to Affymetrix microarrays, or dual-channel ratio data, usually corresponding to spotted cDNA microarrays, and in the majority of cases, a single composite data file was provided by the study authors and incorporated into our database.
Although many sophisticated analytical and statistical approaches have been applied to microarray normalization and differential expression analysis, we sought a single approach that would be simple in application yet robust to the heterogeneous data formats, experimental platforms, and experimental designs. We first applied a global normalization procedure to all data sets (see Methods). Second, by studying the samples profiled in each of the 40 data sets, we defined potential two class differential expression analyses relevant to the processes of neoplastic transformation and progression. These included cancer versus respective normal tissue, high-grade (undifferentiated) cancer versus low-grade (differentiated cancer) cancer, poor outcome (metastases, recurrence, or cancer-specific death) cancer versus good outcome (long-term or recurrence-free survival) cancer, metastatic cancer versus primary cancer, and cancer subtype 1 (e.g., estrogen receptor positive) versus subtype 2 (e.g., estrogen receptor negative) and were identified in 34 data sets (Fig. 4, which is published as supporting information on the PNAS web site). Based on these classifications, we conducted 81 sets of analyses by defining two classes of samples, calculating a Student's t statistic, P value (false positive rate), and Q value (false discovery rate) for each microarray feature (see Methods) (26). The majority of cancer vs. normal (36 of 40), differentiation (8 of 11), metastases vs. primary (3 of 3), and cancer subtype (15 of 16) analyses identified large sets of differentially expressed genes (Q < 0.10), whereas only 3 of 11 outcome analyses did, two of which compared poor outcome breast cancer with favorable outcome (14, 15), and one which compared poor outcome diffuse large B cell lymphoma with favorable outcome (28). Fig. 4 summarizes the 81 analyses and the number of significant differentially expressed genes identified in each at varying significance thresholds. These data can be analyzed with our companion web resource, ONCOMINE (www.oncomine.org/meta).
Comparative Meta-Profiling Method. Because it is generally agreed that microarray data from distinct experimental platforms, often using distinct reference samples, are not directly comparable, we developed a method that instead compares statistical measures (Q values) generated independently from each data set (25). To compare statistical measures across data sets, our method requires that analogous hypotheses have been tested in each data set (e.g., genes differentially expressed between normal tissue and cancer tissue). To identify and assess the intersection of multiple differential expression signatures, so-called meta-signatures, we applied our automated method, comparative meta-profiling (Fig. 1, see Methods). The method is as follows: (i) a set of analogous differential expression analyses are selected for meta-profiling, (ii) a direction and significance threshold are set to define differential expression signatures from the precomputed differential expression analyses (e.g., overexpressed in cancer relative to normal, Q < 0.10), (iii) genes are sorted based on the number of signatures in which they are present, and (iv) a meta-signature is defined if there are significantly more genes intersecting a given number of signatures than would be expected by chance, as defined by a random simulation. A statistical measure, the minimum meta-false discovery rate (mFDRMIN) is used to assess the degree of intersection among gene expression signatures (see Methods).
Fig. 1.

Comparative meta-profiling flow diagram (see Methods for details).
Meta-Signature of Neoplastic Transformation. We began by metaprofiling 36 neoplastic transformation signatures from 21 data sets (overexpressed in cancer relative to respective normal tissue, Q < 0.10), which span 12 tissue types including breast, prostate, colon, lung, liver, brain, ovary, pancreas, uterus, salivary gland, bladder, and B lymphocytes. We hypothesized that if a meta-signature existed, the genes in the signature would reflect essential transcriptional features of cancer, independent of tissue of origin or initial transforming mechanism. At the significance threshold of Q < 0.10, 183 genes were present in at least 10 of 36 signatures, 67 genes in at least 12 signatures, and one gene in 18 signatures. In a random simulation, in which genes were randomly assigned to signatures while maintaining the number of genes in each signature, no genes were present in 10 or more signatures, indicating that the 183 genes present in at least 10 signatures represented a statistically significant multicancer-type meta-signature (mFDRMIN = 0.0055). Fig. 2A depicts the 67 genes present in at least 12 cancer vs. normal signatures. Many of these genes have previously been associated with cancer; however, often associations have only been made with one specific type of cancer or in cell lines, and not with cancer in general. As defined by the Gene Ontology Consortium (29) (www.geneontology.org), the meta-signature contains genes involved in the cell cyle (CDKN3, CKS2, E2F5, PTMA, PLK, CCT4), invasion (MMP9), transcriptional regulation (E2F5, SOX4, HDAC1, CBX3, SMARCA4), protein folding (HSPD1, HSPE1, CCT4), and the proteasome (PSMA1, PSMC4, PSME2). The genes in this signature can be further explored with ONCOMINE (www.oncomine.org/meta).
Fig. 2.

Meta-signature of neoplastic transformation. (A) Sixty-seven genes overexpressed in cancer relative to normal tissue counterpart in at least 12 of 39 “cancer vs. normal” signatures. Twelve distinct cancer types were selected for the figure. White boxes signify either not present or not significant. Red boxes signify significant overexpression in cancer relative to normal tissue (Q < 0.10), the shade of red indicating the percentage of cancer samples that had an expression value greater than the 90th percentile of normal samples. (B) The signature significantly predicts “cancer vs. normal” status in 32 of 39 analyses. The two bars above each heat map represent the predicted class (P) and the true class (T): red signifies cancer and blue signifies normal tissue. Fisher's exact test was used to assess the significance of classification. In the heat maps, black signifies data not available, white signifies less than or equal to the normal class mean expression level, and red signifies the degree of overexpression relative to the mean normal class expression level. FL, follicular lymphoma; DLBCL, diffuse large B cell lymphoma; SCLC, small cell lung cancer; SqCLC, squamous cell lung cancer; adeno., adenocarcinoma; Prost., prostate; Glio., glioblastoma; Rh, rhabdomyosarcoma; PNET, primitive neuroectodermal tumor.
To assess the universality of the meta-signature, the top 67 genes were used to predict cancer vs. normal status in 39 analyses using a leave-one-out voting classifier (see Methods and Table 1, which is published as supporting information on the PNAS web site). The signature was a significant predictor (P < 0.05) in 29 of 39 analyses (from 19 of 21 data sets), and was marginally predictive (P < 0.10) in 3 of 39 analyses (from 3 of 21 data sets) (Fig. 2B). The seven analyses in which the profile was not an accurate classifier were from a single multicancer data set (30). This data set was the largest in the database, providing 13 of the 39 cancer vs. normal analyses, of which six were predicted significantly. For each of the seven analyses that were not predicted significantly, there was a similar (i.e., same cancer type) analysis from an independent study that was predicted significantly. Taken together, 20 of 21 data sets suggest that the genes in this cancer meta-signature are differentially overexpressed in most, if not all, available cancer types relative to the normal tissue from which they arose.
The existence of a general cancer meta-signature may not be entirely surprising, because all cancer types share the common features of unregulated cell proliferation and invasion, and it would follow that the genes that are essential to these processes would be highly expressed in multiple cancer types. On the other hand, however, it is interesting that a small number of genes are almost universally activated, given the vast array of transforming mechanisms that are known to initiate cancer and the variety of tissue types represented in this analysis. Activation of these genes may represent convergence on the essential transcriptional features of neoplastic transformation. From a clinical standpoint, pharmacological agents that target these essential features of cancer might have broad application. For example, TOP2A, a gene present in 18 cancer vs. normal signatures representing 10 types of cancer, encodes the enzyme topoisomerase II, which is critical for DNA replication and is targeted by numerous chemotherapeutic agents (31). Furthermore, agents targeting the proteasome complex, of which three members were identified in the meta-signature, have also shown promise. These agents are in clinical trials and have been shown to induce apoptosis and sensitize cancer cells to traditional tumoricidal agents (32). The widespread activation of genes that encode successfully targeted proteins suggests that other genes in the meta-signature may play equally critical roles in carcinogenesis, and may serve as novel therapeutic targets.
Meta-Signature of Undifferentiated Cancer. We next sought to identify meta-signatures that characterize cancer progression as defined by histological, pathological, or clinical criteria, similar in concept to a report that identified a metastasis signature common to multiple types of primary tumors (19). As described above, only 3 of 10 outcome-based analyses identified significant differentially expressed genes, and two were of the same cancer type, making it infeasible to attempt to define an outcome meta-signature. However, 8 of 11 differentiation analyses, spanning seven types of cancer, identified significant differential expression signatures (differentially expressed in undifferentiated cancers relative to well differentiated cancers of the same origin, Q < 0.10). Undifferentiated cancers of different tissue types all fail to recapitulate their normal tissue architecture, instead maintaining a disordered state of increased cellular proliferation and invasion. Furthermore, undifferentiated cancers are associated with aggressive behavior and poor patient outcomes. Thus, we hypothesized that if an undifferentiated meta-signature existed, it might suggest common transcriptional mechanisms by which cancer cells avoid differentiation, or dedifferentiate. Meta-profiling was performed on seven “undifferentiated vs. well differentiated” signatures spanning six cancer types (overexpressed in undifferentiated cancers relative to well differentiated cancers, Q < 0.10). Sixty-nine genes were present in at least four of seven signatures, whereas just one gene was significant in four of seven signatures by chance (mFDRMIN = 0.030). Twenty-four genes were present in five signatures, and six genes were present in six of seven signatures, whereas zero genes were significant in five or more by chance, thus defining an undifferentiated meta-signature common to multiple types of cancer. Fig. 3 displays the 69 genes present in at least four of seven signatures. Interestingly, a fraction of genes in this meta-signature overlap with the meta-signature of neoplastic transformation. These genes are predominantly associated with proliferation (TOP2A, MCM3, CDC2, RFC4, etc.), the overlap likely owing to the parallel increase in proliferation in cancer relative to normal tissues and in undifferentiated cancer relative to differentiated cancer. Of note, three genes unique to the undifferentiated metasignature have a demonstrated role in chromatin remodeling and broad spectrum transcriptional regulation, including the polycomb group protein EZH2, which is involved in transcriptional memory (33), and the histone variant proteins, H2AFX and H2AFZ, which are known to control the euchromatin–heterochromatin transition (34). The ability of these genes to modulate the expression of tens or hundreds of genes suggests that they may play a role in maintaining the undifferentiated cellular state of high-grade cancer. Interestingly, our group recently found EZH2 to be involved in the metastatic progression of prostate cancer (13) and in the invasive breast cancer phenotype (35), and another recent study demonstrated amplification of the EZH2 gene locus in several primary tumor types (36). Other genes present in the metasignature whose function suggests a role in the undifferentiated phenotype include MELK, a kinase with a demonstrated role in early mammalian embryogenesis (37), and BIRC5 (survivin), an inhibitor of apoptosis (IAP family), which may allow undifferentiated cancer cells to overcome apoptotic checkpoints favoring aberrant progression through mitosis (38). The genes in this signature can be further explored with ONCOMINE (www.oncomine.org/meta).
Fig. 3.

Meta-signature of undifferentiated cancer. Sixty-nine genes that are overexpressed in undifferentiated cancer relative to well differentiated cancer (Q < 0.10) in at least four of seven signatures representing six types of cancer. See Fig. 2 legend for description.
To assess the generality of the undifferentiated meta-signature, a leave-one-out voting classifier was used to predict “high grade vs. low grade” status in all 11 differentiation analyses (Table 2 and Fig. 5, which are published as supporting information on the PNAS web site). The meta-signature was a significant predictor in six of the seven analyses in which differentially expressed genes were originally identified (P < 0.001). The one analysis that was not predicted accurately was Singh_Prostate (P = 0.75). In the four remaining analyses, in which no significant differentially expressed genes were originally identified, the meta-signature was a significant predictor in one analysis (Welsh_Ovarian, P = 0.005), marginally predictive in two analyses (Dhanaskearan_Prostate, Welsh_Prostate, P < 0.15), and not predictive in one analysis (Garber_Lung, P = not applicable). Taken together, it appears that this meta-signature is common to undifferentiated breast cancer, lung cancer, ovarian cancer, bladder cancer, and medulloblastoma, and may be marginally associated with undifferentiated prostate cancer.
Independent Data Set Validation of Meta-Signatures. To confirm the validity and biological relevance of the meta-signatures, we tested their discriminative power on 12 independent data sets that became recently available and were collected after the initial discovery of the metasignatures (28, 39–49) (see supporting information). To validate the universal cancer meta-signature, we analyzed nine independent data sets representing nine distinct cancer types, three of which were not represented in the original analysis [adrenocortical carcinoma (40), pilocytic astrocytoma (41), meningioma (39)]. Table 3, which is published as supporting information on the PNAS web site, shows that in seven of the nine data sets, including all three data sets representing new cancer types, the metasignature significantly discriminated between cancer and respective normal tissue (Fisher's Exact Test, P < 0.05). In the two other data sets, LaTulippe_Prostate (45) and Rosenwald_Lymphoma (28), the metasignature made many more correct than incorrect predictions; however, the discrimination did not reach statistical significance (P = 0.085 and 0.115, respectively).
To assess the discriminative power of the undifferentiated metasignature, we identified five independent data sets that included low- and high-grade cancer samples (Table 4, which is published as supporting information on the PNAS web site). In three of five data sets [Katua_Astrocytoma (44), Schaner_Ovarian (48), and Sotiriou_Breast (49)] the meta-signature significantly discriminated between low- and high-grade cancer samples (Table 1, all P < 0.01), whereas in the remaining two data sets the signature was not predictive (P > 0.5). In these two data sets [Mutter_Endometrium (46), Powell_Lung (47)], no genes were found to be significantly differentially expressed between high- and low-grade cancers (Q < 0.10) and only a small number of cases defined each class (low grade: n = 4, high grade: n = 3). Similar to the results in the training set, this signature seems to perform well in data sets that identified significant gene expression differences, but poorly in those that do not. It is unclear whether this represents cancer types for which the undifferentiated meta-signature is not present, or if it was not detected because of technical issues in particular data sets. Regardless, the signature appears to define a wide variety of undifferentiated cancer types both in the training and test sets and likely points to common transcriptional mechanisms by which cancer cell avoid differentiation. In summary, this validation on independent microarray data sets confirms that the meta-signatures represent common gene expression programs that may be important to the processes of neoplastic transformation and progression.
In conclusion, the systematic collection of public microarray data (see www.oncomine.org/meta) combined with the comparative meta-profiling framework generated a useful platform for drawing conclusions that span multiple microarray data sets and importantly, multiple cancer types. By integrating microarray data and analysis from a number of cancer types, we characterized a metasignature of neoplastic transformation, defining a transcriptional program that is almost always activated in cancer, regardless of cell of origin. This universal activation suggests that these genes may be essential to carcinogenesis, and likely represent the convergence of a number of transforming mechanisms in a variety of cellular contexts. Furthermore, universal overexpression suggests that these genes may serve as attractive therapeutic targets. Interestingly, topoisomerase II and the proteasome complex, both members of the meta-signature, have been targeted therapeutically with some degree of success. We also identified a meta-signature of cancer progression, demonstrating that various types of high-grade cancer share common transcriptional features, including the overexpression of specific chromatin remodeling and transcriptional memory genes that may play a role in the cancer cells' ability to avoid differentiation. Finally, this work provides a simple, scalable framework for comparing and assessing the intersection of multiple gene expression signatures from disparate data sets. This approach will be increasingly useful as the mass of published transcriptome data continues to grow.
Supplementary Material
Acknowledgments
We thank Douglas Gibbs for hardware support. This work was funded by pilot funds from the Dean's Office, Department of Pathology, the American Cancer Society RSG-02-179-01, Cancer Center Support Grant 5P30 CA46592, the Specialized Program of Research Excellence in Prostate Cancer (P50 CA69568), and the Bioinformatics Program. D.R.R. is a fellow of the Medical Scientist Training Program, and A.M.C. is a Pew Biomedical Scholar.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Alon, U., Barkai, N., Notterman, D. A., Gish, K., Ybarra, S., Mack, D. & Levine, A. J. (1999) Proc. Natl. Acad. Sci. USA 96, 6745-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dhanasekaran, S. M., Barrette, T. R., Ghosh, D., Shah, R., Varambally, S., Kurachi, K., Pienta, K. J., Rubin, M. A. & Chinnaiyan, A. M. (2001) Nature 412, 822-826. [DOI] [PubMed] [Google Scholar]
- 3.Luo, J., Duggan, D. J., Chen, Y., Sauvageot, J., Ewing, C. M., Bittner, M. L., Trent, J. M. & Isaacs, W. B. (2001) Cancer Res. 61, 4683-4688. [PubMed] [Google Scholar]
- 4.Luo, J. H., Yu, Y. P., Cieply, K., Lin, F., Deflavia, P., Dhir, R., Finkelstein, S., Michalopoulos, G. & Becich, M. (2002) Mol. Carcinog. 33, 25-35. [DOI] [PubMed] [Google Scholar]
- 5.Magee, J. A., Araki, T., Patil, S., Ehrig, T., True, L., Humphrey, P. A., Catalona, W. J., Watson, M. A. & Milbrandt, J. (2001) Cancer Res. 61, 5692-5696. [PubMed] [Google Scholar]
- 6.Notterman, D. A., Alon, U., Sierk, A. J. & Levine, A. J. (2001) Cancer Res. 61, 3124-3130. [PubMed] [Google Scholar]
- 7.Welsh, J. B., Zarrinkar, P. P., Sapinoso, L. M., Kern, S. G., Behling, C. A., Monk, B. J., Lockhart, D. J., Burger, R. A. & Hampton, G. M. (2001) Proc. Natl. Acad. Sci. USA 98, 1176-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Welsh, J. B., Sapinoso, L. M., Su, A. I., Kern, S. G., Wang-Rodriguez, J., Moskaluk, C. A., Frierson, H. F., Jr., & Hampton, G. M. (2001) Cancer Res. 61, 5974-5978. [PubMed] [Google Scholar]
- 9.Frierson, H. F., Jr., El-Naggar, A. K., Welsh, J. B., Sapinoso, L. M., Su, A. I., Cheng, J., Saku, T., Moskaluk, C. A. & Hampton, G. M. (2002) Am. J. Pathol. 161, 1315-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garber, M. E., Troyanskaya, O. G., Schluens, K., Petersen, S., Thaesler, Z., Pacyna-Gengelbach, M., van de Rijn, M., Rosen, G. D., Perou, C. M., Whyte, R. I., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 13784-13789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh, D., Febbo, P. G., Ross, K., Jackson, D. G., Manola, J., Ladd, C., Tamayo, P., Renshaw, A. A., D'Amico, A. V., Richie, J. P., et al. (2002) Cancer Cell 1, 203-209. [DOI] [PubMed] [Google Scholar]
- 12.Ye, Q. H., Qin, L. X., Forgues, M., He, P., Kim, J. W., Peng, A. C., Simon, R., Li, Y., Robles, A. I., Chen, Y., et al. (2003) Nat. Med. 9, 416-423. [DOI] [PubMed] [Google Scholar]
- 13.Varambally, S., Dhanasekaran, S. M., Zhou, M., Barrette, T. R., Kumar-Sinha, C., Sanda, M. G., Ghosh, D., Pienta, K. J., Sewalt, R. G., Otte, A. P., et al. (2002) Nature 419, 624-629. [DOI] [PubMed] [Google Scholar]
- 14.van't Veer, L. J., Dai, H., van de Vijver, M. J., He, Y. D., Hart, A. A., Mao, M., Peterse, H. L., van der Kooy, K., Marton, M. J., Witteveen, A. T., et al. (2002) Nature 415, 530-536. [DOI] [PubMed] [Google Scholar]
- 15.Sørlie, T., Perou, C. M., Tibshirani, R., Aas, T., Geisler, S., Johnsen, H., Hastie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 10869-10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shipp, M. A., Ross, K. N., Tamayo, P., Weng, A. P., Kutok, J. L., Aguiar, R. C., Gaasenbeek, M., Angelo, M., Reich, M., Pinkus, G. S., et al. (2002) Nat. Med. 8, 68-74. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz, D. R., Kardia, S. L., Shedden, K. A., Kuick, R., Michailidis, G., Taylor, J. M., Misek, D. E., Wu, R., Zhai, Y., Darrah, D. M., et al. (2002) Cancer Res. 62, 4722-4729. [PubMed] [Google Scholar]
- 18.Rickman, D. S., Bobek, M. P., Misek, D. E., Kuick, R., Blaivas, M., Kurnit, D. M., Taylor, J. & Hanash, S. M. (2001) Cancer Res. 61, 6885-6891. [PubMed] [Google Scholar]
- 19.Ramaswamy, S., Ross, K. N., Lander, E. S. & Golub, T. R. (2003) Nat. Genet. 33, 49-54. [DOI] [PubMed] [Google Scholar]
- 20.Dyrskjot, L., Thykjaer, T., Kruhoffer, M., Jensen, J. L., Marcussen, N., Hamilton-Dutoit, S., Wolf, H. & Orntoft, T. F. (2003) Nat. Genet. 33, 90-96. [DOI] [PubMed] [Google Scholar]
- 21.Chen, X., Cheung, S. T., So, S., Fan, S. T., Barry, C., Higgins, J., Lai, K. M., Ji, J., Dudoit, S., Ng, I. O., et al. (2002) Mol. Biol. Cell 13, 1929-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharjee, A., Richards, W. G., Staunton, J., Li, C., Monti, S., Vasa, P., Ladd, C., Beheshti, J., Bueno, R., Gillette, M., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 13790-13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas, J. W., Touchman, J. W., Blakesley, R. W., Bouffard, G. G., Beckstrom-Sternberg, S. M., Margulies, E. H., Blanchette, M., Siepel, A. C., Thomas, P. J., McDowell, J. C., et al. (2003) Nature 424, 788-793. [DOI] [PubMed] [Google Scholar]
- 24.Rubin, G. M., Yandell, M. D., Wortman, J. R., Gabor Miklos, G. L., Nelson, C. R., Hariharan, I. K., Fortini, M. E., Li, P. W., Apweiler, R., Fleischmann, W., et al. (2000) Science 287, 2204-2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhodes, D. R., Barrette, T. R., Rubin, M. A., Ghosh, D. & Chinnaiyan, A. M. (2002) Cancer Res. 62, 4427-4433. [PubMed] [Google Scholar]
- 26.Storey, J. D. & Tibshirani, R. (2003) Proc. Natl. Acad. Sci. USA 100, 9440-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95, 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenwald, A., Wright, G., Chan, W. C., Connors, J. M., Campo, E., Fisher, R. I., Gascoyne, R. D., Muller-Hermelink, H. K., Smeland, E. B., Giltnane, J. M., et al. (2002) N. Engl. J. Med. 346, 1937-1947. [DOI] [PubMed] [Google Scholar]
- 29.Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., et al. (2000) Nat. Genet. 25, 25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramaswamy, S., Tamayo, P., Rifkin, R., Mukherjee, S., Yeang, C. H., Angelo, M., Ladd, C., Reich, M., Latulippe, E., Mesirov, J. P., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 15149-15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hande, K. R. (1998) Biochim. Biophys. Acta 1400, 173-184. [DOI] [PubMed] [Google Scholar]
- 32.Adams, J. (2002) Curr. Opin. Oncol. 14, 628-634. [DOI] [PubMed] [Google Scholar]
- 33.Cao, R., Wang, L., Wang, H., Xia, L., Erdjument-Bromage, H., Tempst, P., Jones, R. S. & Zhang, Y. (2002) Science 298, 1039-1043. [DOI] [PubMed] [Google Scholar]
- 34.Meneghini, M. D., Wu, M. & Madhani, H. D. (2003) Cell 112, 725-736. [DOI] [PubMed] [Google Scholar]
- 35.Kleer, C. G., Cao, Q., Varambally, S., Shen, R., Ota, I., Tomlins, S. A., Ghosh, D., Sewalt, R. G., Otte, A. P., Hayes, D. F., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 11606-11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bracken, A. P., Pasini, D., Capra, M., Prosperini, E., Colli, E. & Helin, K. (2003) EMBO J. 22, 5323-5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heyer, B. S., Kochanowski, H. & Solter, D. (1999) Dev. Dyn. 215, 344-351. [DOI] [PubMed] [Google Scholar]
- 38.Li, F., Ambrosini, G., Chu, E. Y., Plescia, J., Tognin, S., Marchisio, P. C. & Altieri, D. C. (1998) Nature 396, 580-584. [DOI] [PubMed] [Google Scholar]
- 39.Watson, M. A., Gutmann, D. H., Peterson, K., Chicoine, M. R., Kleinschmidt-DeMasters, B. K., Brown, H. G. & Perry, A. (2002) Am. J. Pathol. 161, 665-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giordano, T. J., Thomas, D. G., Kuick, R., Lizyness, M., Misek, D. E., Smith, A. L., Sanders, D., Aljundi, R. T., Gauger, P. G., Thompson, N. W., et al. (2003) Am. J. Pathol. 162, 521-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gutmann, D. H., Hedrick, N. M., Li, J., Nagarajan, R., Perry, A. & Watson, M. A. (2002) Cancer Res. 62, 2085-2091. [PubMed] [Google Scholar]
- 42.Higgins, J. P., Shinghal, R., Gill, H., Reese, J. H., Terris, M., Cohen, R. J., Fero, M., Pollack, J. R., van de Rijn, M. & Brooks, J. D. (2003) Am. J. Pathol. 162, 925-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iacobuzio-Donahue, C. A., Maitra, A., Olsen, M., Lowe, A. W., van Heek, N. T., Rosty, C., Walter, K., Sato, N., Parker, A., Ashfaq, R., et al. (2003) Am. J. Pathol. 162, 1151-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khatua, S., Peterson, K. M., Brown, K. M., Lawlor, C., Santi, M. R., LaFleur, B., Dressman, D., Stephan, D. A. & MacDonald, T. J. (2003) Cancer Res. 63, 1865-1870. [PubMed] [Google Scholar]
- 45.LaTulippe, E., Satagopan, J., Smith, A., Scher, H., Scardino, P., Reuter, V. & Gerald, W. L. (2002) Cancer Res. 62, 4499-4506. [PubMed] [Google Scholar]
- 46.Mutter, G. L., Baak, J. P., Fitzgerald, J. T., Gray, R., Neuberg, D., Kust, G. A., Gentleman, R., Gullans, S. R., Wei, L. J. & Wilcox, M. (2001) Gynecol. Oncol. 83, 177-185. [DOI] [PubMed] [Google Scholar]
- 47.Powell, C. A., Spira, A., Derti, A., DeLisi, C., Liu, G., Borczuk, A., Busch, S., Sahasrabudhe, S., Chen, Y., Sugarbaker, D., et al. (2003) Am. J. Respir. Cell Mol. Biol. 29, 157-162. [DOI] [PubMed] [Google Scholar]
- 48.Schaner, M. E., Ross, D. T., Ciaravino, G., Sorlie, T., Troyanskaya, O., Diehn, M., Wang, Y. C., Duran, G. E., Sikic, T. L., Caldeira, S., et al. (2003) Mol. Biol. Cell 14, 4376-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sotiriou, C., Powles, T. J., Dowsett, M., Jazaeri, A. A., Feldman, A. L., Assersohn, L., Gadisetti, C., Libutti, S. K. & Liu, E. T. (2002) Breast Cancer Res. 4, R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.