

Abstract
Presentation of antigen with immune stimulating “signal” has been a cornerstone of vaccine design for decades. Here, the antigen plus immune “signal” of vaccines is modified to produce antigen-specific immunotherapies (antigen-SITs) that can potentially reprogram the immune response toward tolerance of an autoantigen. The codelivery of antigen with a cell adhesion inhibitor using Soluble Antigen Arrays (SAgAs) was previously shown to slow or halt experimental autoimmune encephalomyelitis (EAE), a murine form of multiple sclerosis (MS). SAgAs are comprised of a hyaluronic acid backbone with cografted intercellular cell adhesion molecule-1 ligand derived from αL-integrin (CD11a237–246, “LABL”) and an encephalitogenic epitope peptide of proteolipid protein (PLP139–151, “PLP”). Here, the physical characteristics of the carrier were investigated to evaluate how structure, size, and solubility drive the immune response when treating EAE. A bifunctional peptide (small, soluble), SAgAs (large, soluble), and PLGA nanoparticles (large, insoluble) all displaying PLP and LABL in equimolar ratios were compared. Maximum EAE suppression was achieved with coincident display of both peptides on a soluble construct.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-014-9654-z) contains supplementary material, which is available to authorized users.
KEY WORDS: experimental autoimmune encephalomyelitis, multivalency, proteolipid peptide, scaffold, soluble antigen array
INTRODUCTION
Investigations into the molecular processes involved in antigen recognition have led to new approaches in the design and administration of vaccines (1, 2). Traditionally, vaccines generate a robust immune response to a particular antigen by adsorbing the antigen to an alum particle surface (3–5). Though individual alum particles are quite small (10–50 nm), in solution they form insoluble agglomerates that have a typical size range of 1–10 μm to which the antigen of interest is presented on the surface (6). Protection is achieved when these insoluble alum particulates initiate an inflammatory response against the adsorbed antigen (7, 8). Efforts to optimize alum vaccines investigated the influence of agglomerate particle size on the physical and thermal stability of the vaccine as well as the ability to adsorb antigen (9, 10). Newer vaccine approaches have considered directing specific molecular responses involved in inflammation through the use of lipids, toll-like receptor mimics, and nano- and microparticle approaches (7, 11–14). The physical characteristics of vaccines is known to play a major role in defining the protective immune response that is generated, suggesting physical properties may also be important when attempting to induce tolerance to autoantigen.
While seemingly very different, vaccines and autoimmune therapeutics often share similarities based on the fundamentals of how the body recognizes and responds to a given antigen. Vaccines prepare the body for exposure to a particular antigen through innate and adaptive immune responses. “Antigen-specific immunotherapies” (antigen-SITs) also use antigen, most notably in the form of subcutaneous allergy shots. The antigen-SIT approach has also been investigated to reverse the root cause of autoimmune disease by repeatedly injecting low doses of autoantigen (15–18).
Defining underlying events during antigen recognition has led to advancements in autoimmune disease treatment (19, 20). In the autoimmune disease multiple sclerosis (MS), the disease state results from antigen-specific activation of immune cells leading to neural degeneration and attack on the central nervous system (CNS) (21, 22). The majority of MS therapeutics broadly suppress the inflammatory response despite severe side effects (23). Antigen-SITs have been developed to reprogram the immune response to the offending antigen, which is the root cause of many autoimmune diseases (15). Antigen-SITs may be improved by adding a signal that disrupts the immune signaling pathways during antigen presentation. This has been shown in previous studies where the codelivering antigen plus a molecule directing the immune response can provide protection or tolerance (24–26).
Most autoimmune therapies are soluble proteins or antigen-based polymers. Natalizumab marketed as TYSABRI®, is a soluble, ~150 kDa, anti-α4 integrin antibody that can prevent activation of CD4+ T cells and lymphocyte recruitment (27–29). Another approved therapy, Copaxone®, is a soluble polymer (~5–9 kDa) that contains repeats of autoantigen derived from myelin basic protein. It is hypothesized that polymeric presentation of myelin-specific antigen in Copaxone® helps to promote tolerance by inducing antigen-specific regulatory T-cells (18, 30, 31). Allergy shots provide frequent low doses of soluble antigen to induce antigen-specific tolerance in patients (32–35). Thus, approved immunotherapies tend toward soluble proteins or peptides. In addition to solubility, the physical size of the therapy or carrier is important (24, 25).
We investigated the structure, size, and solubility of carriers codelivering autoantigen derived from proteolipid protein (PLP139–151, “PLP”) and an intercellular cell adhesion molecule-1 ligand peptide derived from αL-integrin (CD11a237–246, “LABL”). Different constructs, codelivering PLP and LABL were synthesized to exhibit discrete properties. A bifunctional peptide inhibitor (BPI) is small and soluble (~3 kDa), achieved by covalently linking PLP and LABL with a short peptide spacer. Soluble Antigen Arrays (SAgAs) are large and soluble constructs (45 and 65 kDa) with PLP and LABL grafted to hyaluronic acid (HA). PLGA nanoparticles are much larger (500 nm) and insoluble with PLP and LABL conjugated to the surface. Each of these structures were compared to assess which scaffold suppressed experimental autoimmune encephalomyelitis (EAE).
MATERIALS AND METHODS
Materials
Hyaluronic acid, with an average molecular weight of 16 and 31 kDa was purchased from Lifecore. Analytical-grade acetonitrile and synthesis grade trifluoro acetic acid (TFA) were purchased from Fisher Scientific. Research-grade sodium acetate, acetic acid, and D2O were purchased from Sigma. Water was provided by a Labconco Water PRO PS ultrapure water purification unit. Poly(dl-lactic-coglycolic acid) (50:50) (PLGA; inherent viscosity of 1.05 dL/g, Mw ~101 kDa) was purchased from LACTEL Absorbable Polymers International (Pelham, AL, USA). Pluronic® F68 (Mw ~8.4 kDa) and Pluronic® F108 (Mw ~14.6 kDa) were obtained from BASF Corporation. Acetone, diethyl ether and 1× Tris/EDTA buffer solution (pH 8) were obtained from Fisher Scientific. d-mannitol, Dess-Martin periodinane, tert-butyl carbazate (TBC), trinitrobenzenesulfonic acid (TNBS), dichloromethane anhydrous (DCM) and Triton X-100 were purchased from Sigma-Aldrich.
Mice
SJL/J (H-2s) female mice, 4–6 weeks old, were purchased from The Jackson Laboratory, and they were housed under specific pathogen-free conditions at the University of Kansas. All protocols involving live mice were approved by the Institutional Animal Care and Use Committee.
Peptide Synthesis
Aminooxy peptides were synthesized using 9-fluorenylmethyloxycarbonyl- protected amino acid chemistry on polyethylene glycol-polystyrene resins. The peptides synthesized were aminooxy-LABL (aminooxy-ITDGEATDSG, Ao-LABL), a cell adhesion ligand antagonist, and aminooxy-PLP (aminooxy–HSLGKWLGHPDKF, Ao-PLP), an antigen derived from proteolipid protein amino acids 139–151. Peptides were deprotected, cleaved from resin, and isolated by precipitation in ether. Purification was completed using preparatory high-performance liquid chromatography (HPLC) followed by lyophilization. Peptide identity was verified and purity/content was assessed using analytical HPLC and mass spectroscopy.
Reaction of Aminooxy Peptides to Polymers
Hyaluronic acid polymer (~16,000 or 31,000 MW) was dissolved into 20 mM acetate buffer (pH 5.5 ± 0.1 pH units) and aminooxy reactive peptide(s) added. When LABL and PLP peptides were to be used, each was weighed separately, the lyophilized peptide powders mixed, and then added simultaneously. After addition of the peptide(s), the pH of the reaction solution was adjusted to pH 5.5 ± 0.1 pH units. Solutions were stirred at 500 RPM using magnetic stir bars for ~16 h. Once complete, the soluble antigen array (SAgAPLP:LABL) or PLGA nanoparticle products were purified by extensive dialysis to remove any unreacted peptide, and then purified product solutions were lyophilized.
Conversion of Terminal Hydroxyl Groups to Terminal Aldehyde Groups on Pluronic® F108
To conjugate peptide to Pluronic® on PLGA nanoparticles, the terminal hydroxyl groups were converted to aldehyde groups 33. Briefly, the Dess-Martin periodinane, an oxidizing reagent, was used to convert hydroxyl groups on Pluronic®F108 (Pluronic®F108-OH) into aldehyde groups (Pluronic®F108-CHO). One gram Pluronic®F108-OH was dissolved in 30 mL DCM. Subsequently, 58.1 mg Dess-Martin periodinane was added and reacted for 24 h at room temperature. The product was purified by precipitation in cold diethyl ether, followed by filtration. The obtained Pluronic®F108-CHO was verified by nuclear magnetic resonance spectroscopy (1H-NMR). Deuterated chloroform (CDCl3) was used to dissolve the samples. The conversion percentage was determined using TBC/TNBS. An excess amount of TBC was added to the Pluronic®F108-CHO solution as described and the amount of unreacted TBC was measured using TNBS solution. A UV/VIS Spectrophotometer (SpectraMax) operating at 334 nm was employed to quantify the colored mixture of TBC and TNBS (33–35).
Preparation of PLGA Nanoparticles
A solvent displacement method was employed to prepare PLGA nanoparticles (NPs) (36, 37). Briefly, PLGA (inherent viscosity 1.05 dL/g) was dissolved in acetone (15 mg/mL). A mixture of 1,425 μL of PLGA solution and 75 μL 1× Tris/EDTA buffer solution was injected into a 15 mL water phase containing 0.1% w/v Pluronic® (Pluronic® F108- CHO:Pluronic® F68-OH (25:75 and 0:100)) using a syringe pump (10 mL/h) while stirring (1,000 rpm). Stirring was continued for 1.5 h and then excess surfactant was removed by centrifugation (15,000 rpm, 15 min, 4°C) for 3 cycles, resuspending in water between cycles. During the wash and concentrating steps, sonication was performed to disperse NPs using sonication bath (Branson 2510 ultrasonic cleaner). The 25:75 Pluronic® ratio was used for fabrication of NPs with conjugated PLP (NP-ArrayPLP), LABL (NP-ArrayLABL), or PLP/LABL (NP-ArrayLABL-PLP). The 0:100 Pluronic® ratio was used as the control (NP-Blank) without any peptide conjugation.
Conjugation of Peptides to PLGA Nanoparticles
Stock solutions of 2 mg/mL of PLP and LABL peptides were separately prepared. To prepare NP-ArrayPLP, 3.99 mL of PLP stock was added to 102.27 mg NPs in 3.52 mL of water. For NP-ArrayLABL preparation, 2.6 mL of LABL stock was added to 112.5 mg NPs in 3.2 mL of water. Finally, to prepare the NP-ArrayLABL-PLP, 3.6 mL of PLP stock and 2.34 mL of LABL stock were added to 227.84 mg NPs in 2.844 mL water. The volume of each sample was increased to 50 mL using ddH2O. The volume of NP- Blank sample was also increased up to 50 mL as well (227.84 mg NPs). The samples reacted overnight and unreacted peptide was removed by centrifugation (15,000 rpm, 15 min, 4°C) for 3 cycles, resuspending in water between cycles.
High-Performance Liquid Chromatography
Quantification of free peptide post reaction was accomplished by gradient reversed phase HPLC (SHIMADZU) using a Vydac HPLC protein and peptide C18 column. HPLC system was composed of an SCL-20A SHIMADZU system controller, LC-10AT VP SHIMADZU liquid chromatograph, SIL-10A XL SHIMADZU auto-injector set at 75 μL injection volume, DGU-14A SHIMADZU degasser, sample cooler, and SPD-10A SHIMADZU UV/vis detector (220 nm). A personal computer equipped with SHIMADZU class VP software controlled the HPLC-UV system. Gradient elution was conducted at constant flow of 1 mL/min, from 100% A to 35% A (corresponding to 0% B to 65% B) over 50 min, followed by an isocratic elution at 75% B for 3 min. Mobile phase compositions were (A) acetonitrile-water (5:95) with 0.1% TFA and (B) acetonitrile-water (90:10, v/v) with 0.1% TFA. At the completion of each analysis, the cartridge was equilibrated at initial conditions at 1 mL/min flow rate for 5 min with A.
Gel Permeation Chromatography
The relative molecular weight of the HA and of the HA-Arrays was estimated using a Viscotek GPC max VE 2001 GPC solvent/sample module, VE 3580 refractive index detector, and 270 Dual Detector with right angle light scattering. To achieve sample separation a tandem column setup of two Viscogel, GMPWxl grade, columns (Viscotek) was used, at a flow rate of 1 ml/min with isocratic elution in water for 30 min.
Calculation of Peptide Density on the Surface of NPs
Peptide surface density was calculated by subtracting the amount of recovered peptide after conjugation from the amount of peptide added to the NP suspension. This value was then divided by the total surface area assuming a normal Gaussian particle size distribution. NP-Blank suspension was used as a negative control. PLP and LABL at molar ratios of 100:0, 50:50, and 0:100 were added to the negative controls at the same concentration as samples in which reactive sites were available as an additional control.
Induction of EAE and Therapeutic Study
Five- to 7-week-old SJL/J female mice were immunized subcutaneously (s.c). with 200 mg of PLP139–151 in a 0.2 ml emulsion composed of equal volumes of phosphate-buffered saline (PBS) and complete Freund’s adjuvant (CFA) containing killed Mycobacterium tuberculosis strain H37RA (final concentration of 4 mg/ml; Difco). The PLP139–151/CFA was administered to regions above the shoulders and the flanks (total of four sites; 50 μL at each injection site). In addition, 200 ng/100 μL of pertussis toxin (List Biological Laboratories Inc.) was injected intraperitoneally (i.p.) on the day of immunization (day 0) and 2 days post-immunization. The mice received s.c. injections of each sample, equivalent to 100 nmol PLP/100 μL, on days 4, 7, 10. All NP samples were sonicated to disperse NPs before injection. For HA samples and controls, 100 μL of each vehicle was injected. For NP vehicles, 400 μL solution was used to assure suspension stability. Disease progression was evaluated blindly by the same observer using clinical scoring as follows: 0, no clinical signs of the disease; 1, tail weakness or limp tail; 2, paraparesis (weakness or incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with forelimb weakness or paralysis; and 5, moribund (mice were euthanized if they were found to be moribund). Body weight was also measured daily.
Statistical Analysis
Statistical difference was determined by comparing treated groups to the negative control (PBS) for clinical disease score and body weight. Data was analyzed with a one- or two-way analysis of variance (ANOVA) as experimentally appropriate followed by Fisher’s least significant difference post hoc test. A p < 0.05 was considered the threshold of statistical significance. All analyses were performed using GraphPad Software (GraphPad Software Inc.).
RESULTS
Characterization of Soluble Antigen Arrays
SAgAs were analyzed for relative molecular weight (MW) and peptide concentration as previously reported (26). The concentration of conjugated peptide was determined by HPLC (Table I). To evaluate the change in relative MW of the SAgAs compared to the starting materials, gel permeation chromatography (GPC) was employed and products were compared to starting HA material and pullulan standards. The SAgAs showed the appropriate shift in retention time indicating an increase in MW equal to that of the number of grafted peptides determined by HPLC. The combination of the HPLC and GPC data also showed the desired 1:1 ratio of PLP and LABL peptides was achieved MW relative to pulluan standards was calculated as previously reported (Table I) (26, 38).
Table I.
Sample identification, peptide concentration, and calculated number of peptides per HA (16.9 kDa) chain as determined by HPLC. Peptide concentration was calculated based on HPLC analysis of 1 mg SAgA complex
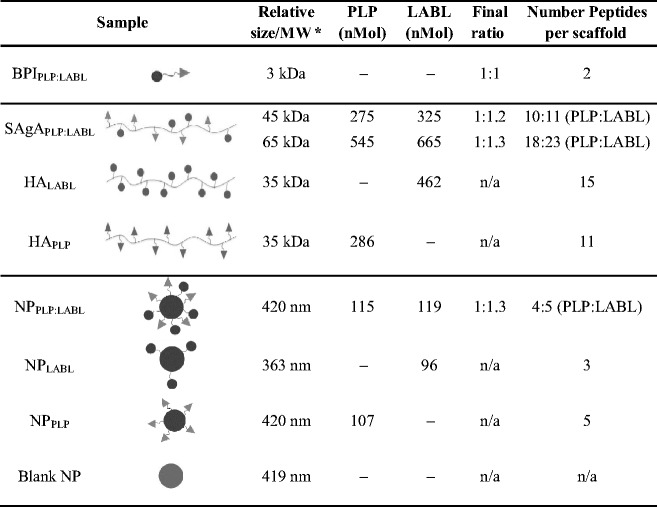
MW of arrays was determined using SEC as reported in the “MATERIALS AND METHODS” section
aSize (diameter) of NP was determined using light scattering
Conversion of Terminal Hydroxyl Groups to Terminal Aldehyde Groups on Pluronic® F108
The hydroxyl groups of Pluronic® were converted to aldehyde groups in order to utilize Pluronic® for conjugation to the terminal aminooxy of the PLP and LABL peptides. Pluronic® F108-CHO with aldehyde groups were prepared by the Dess-Martin oxidation reaction. To confirm conversion, 1H NMR spectra were collected before and after the reaction. The signal corresponding to the aldehyde group (δ = 9.75) appeared which indicated the conversion of hydroxyl groups to aldehyde groups (Supplementary Figure 1) (39). The yield of the conversion was also determined via a colormetric TBC/TNBS assay (74.0%) (40, 41).
Characterization of Nanoparticle Arrays
Reversed phase HPLC was used to quantify the amount of peptide conjugated to NPs and values (Table I). NPs were centrifuged from solution and the amount of unreacted peptide was quantified from the supernatant. Blank NPs and empty vials were used as controls to ensure that peptide was not being adsorbed to surfaces non-specifically. All NP groups targeted a dose of ~100 nMol per 400 μL (Fig. 1a). Additionally, the peptide density on the surface of NPs was calculated, as previously reported, based on the total NP surface area, assuming a normal Gaussian particle size distribution (Fig. 1b) (42, 43). The NPPLP:LABL had a 1:1 ratio of both peptides on the surface. For the nanoparticles displaying only one of the peptides, the grafting density of both the NPPLP and the NPLABL were statistically similar (Table I).
Fig. 1.
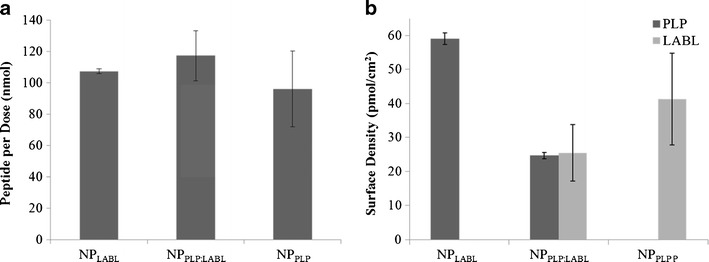
a Amount of peptide per 400 μL dose of PLGA NPs (nMol). b Calculated surface density of peptide for each NP formulation. All data represent the mean ± SD (n = 3)
Backbone Scaffold Size and Structure Affect Degree of EAE Suppression In Vivo
The therapeutic efficacy of BPI, SAgAs, and NP-Arrays were evaluated in the EAE murine model of MS. A summary of the test articles is shown in Table I. Disease onset typically begins after day 8 and progresses to remission after day 20 in the EAE model; therefore, the weights are normalized to the day 8 value for all mice. Upon disease onset, the mice show disease signs such as weakness, paralysis of their tail and limbs, and loss of body weight. Measurements grading the extent of paralysis are used to evaluate clinical score and accompany the weight measurements.
Previous studies showed that subcutaneous injection of SAgAs (HA grafted with PLP and LABL peptides) suppressed EAE in vivo (26). Here, the effect of the size of HA backbone was explored. A 16.9 kDa HA and a 32.0 kDa HA were used to produce the 45-kDa SAgAPLP:LABL and 65-kDa SAgAPLP:LABL, respectively. The relative MW was determined using GPC. Additionally, the much smaller fusion peptide, BPIPLP:LABL, was evaluated. All BPI and SAgA treatments showed significant suppression of EAE disease score (p < 0.05) for days 11–17 (Fig. 2a). The smaller 45 kDa SAgAPLP:LABL maintained body weight longer (days 11–17) than the larger 65 kDa SAgAPLP:LABL (days 11–15) and longer than the much smaller BPIPLP:LABL (days 11–15) as shown in Fig. 2b. Additionally, the 45-kDa SAgAPLP:LABL delayed EAE disease onset for 2 days longer than other treatments and had a lower overall incidence of disease (Supplementary Figure 2). Furthermore, free peptides alone (PLP and LABL), a mixture of these free peptides with HA, and PLP or LABL individually grafted to HA were all previously shown to have no effect when treating EAE (26, 44).
Fig. 2.
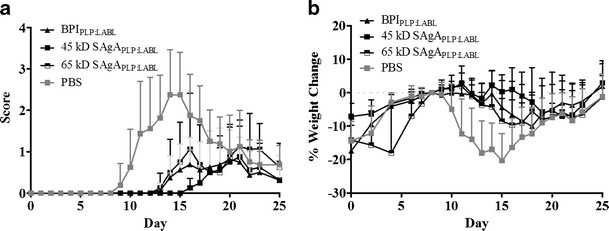
SAgAs composed of a smaller HA Backbone potently suppress EAE. EAE was induced in SJL mice (day 0) and were subsequently treated with a 45-kDa SAgAPLP:LABL, a 65-kDa SAgAPLP:LABL, and the smaller BPIPLP:LABL, as well as PBS control on days 4, 7, and 10. Significant difference from PBS was seen in a daily clinical scores for the all samples (p <0.05, days 11–17) and b percent weight change was for BPI and the 65-kDa SAgAPLP:LABL (Daily a clinical scores (p < 0.05, days 11–17) and b percent weight change was determined (p < 0.05, days 11–17)) and 45 kDa SAgAPLP:LABL (Daily a clinical scores (p < 0.05, days 11–17) and b percent weight change was determined (p < 0.05, days 11–17)). The 45-kDa SAgAPLP:LABL had greater effect in maintaining weight and showed delay of disease onset by 2 days over BPI and 65 kDa SAgAPLP:LABL. Data is expressed as mean ± SD, n = 6 animals per group
To further probe the importance of structure, PLGA NPs were tested as the carrier. The large, insoluble nanoparticle offers a scaffold more similar to colloidal adjuvants such as alum. NPs with only PLP or LABL grafted to the surface were examined. Surprisingly, NPs with only PLP or only LABL had little to no effect on clinical score or weight change when compared to PBS and to the soluble HA scaffold (Fig. 3). The NPPLP:LABL array was then compared to the soluble 45 kDa SAgAPLP:LABL and to much smaller BPIPLP:LABL, which had already demonstrated efficacy. The 45-kDa SAgAPLP:LABL and BPIPLP:LABL suppressed EAE scores for days 12–14 of the study, but the NPPLP:LABL only suppressed the disease on day 12 (Fig. 4a). The percent weight change data showed NPPLP:LABL arrays maintained body weight (Days 12–16) for a similar duration as the 45-kDa SAgAPLP:LABL and BPIPLP:LABL (Days 12–15) when compared to PBS (Fig. 4b). NPs prepared without peptides were also tested and showed no statistical difference in clinical score or percent weight change when compared to control (Supplementary Figure 3).
Fig. 3.
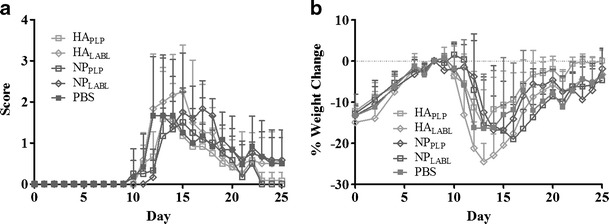
Both HA and NP carriers displaying only one peptide were not effective in EAE. EAE was induced in SJL mice (day 0) and were subsequently treated with arrays displaying only PLP or LABL along HA and NP carriers as well as a PBS control on days 4, 7, and 10. All samples showed no statistical difference when compared to PBS in clinical score or % weight change. Data is expressed as mean ± SD, n = 6 animals per group
Fig. 4.
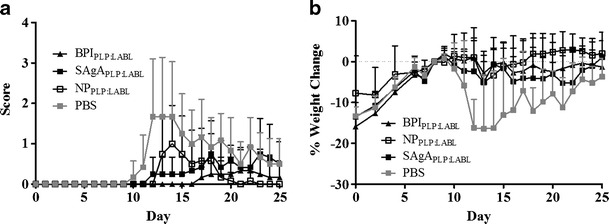
Arrays displaying both PLP and LABL suppressed EAE regardless of carrier, but small, soluble constructs enhanced suppression. EAE was induced in SJL mice (day 0) and were subsequently treated with a 65-kDa SAgAPLP:LABL, NPPLP:LABL, and BPIPLP:LABL, as well as PBS control on days 4, 7, and 10. Significant difference from PBS was seen in a daily clinical scores for the all samples NPPLP:LABL (p < 0.05, day 12) and 65 kDa SAgAPLP:LABL and BPIPLP:LABL (p < 0.05, days 12–14) and b percent weight change for 65 kDa SAgAPLP:LABL and BPIPLP:LABL (p < 0.05, days 12–15) and NPPLP:LABL (p < 0.05, day 12–16). Data is expressed as mean ± SD, n = 6 animals per group
DISCUSSION
The induction of immune response by presentation of antigen on or with immune stimulating “signal” is a primary design metric of vaccines. We rationalized that a similar strategy could also be used to design antigen-SITs to induce tolerance to autoantigen. Both approaches focus on the recognition, attack, and removal of a “foreign material” through stimulation and specific activation of subpopulations of immune cells by presentation of specific antigen. In autoimmune diseases, the antigen is autologous. For example, myelin surrounding the nerves in the CNS and destruction leads to paralysis and eventually death (17). Similarities between “vaccine induced” and “autoimmune disease induced” antigen attack, suggest we may be able to learn from emerging vaccine strategies in our design of tolerogenic therapies.
Current vaccine development focuses on directing specific molecular responses involved in inflammation and the effect of altering the physical characteristics of adjuvants that define the protective immune response (7, 11–14). Optimal immune protection is often achieved with larger (500 nm–5 μm), insoluble constructs that can entrap or adsorb antigen (7). Similarly, antigen-SITs utilize antigen or autoantigen to hyposensitize immune response. Vaccines tend to have similar physical characteristics, existing as insoluble colloids or emulsions that deliver antigen and an adjuvant to stimulate an inflammatory immune response. The most effective antigen-SITs also tend to share physical similarities of small size (10–200 kDa) and solubility in water (31, 35, 45).
One currently approved therapy for multiple sclerosis, Copaxone®, a synthetic copolymer glatiramer acetate, focuses on disrupting autoantigen recognition events. Alternatively, another therapy, TYSABRI®, an anti-α4 integrin antibody, aims to disrupt the recruitment and translocation of immune cells. Both therapies show clinical success in halting the activation of CD4+ T cells and lymphocyte recruitment, but target very different pathways (e.g. “signal” recognition or stimulation) (28, 29). TYSABRI® is non-specific and leads to unwanted immunosuppressive effects and the effectiveness of Copaxone® has been questioned (17, 19, 27). Enhancing the specificity of these types of therapies (e.g., by combining them) could reduce side effects and improve potency.
Current strategies target either antigen-specific hyposensitization or disruption of costimulation “signal”. Antigen-specific strategies attempt to generate Th2 immunity and/or induce tolerance (Treg) by altering recognition of modified antigens (46, 47). The use of altered peptide ligands (APL), splenocytes coupled with myelin basic protein (MBP)- or PLP-derived peptides, or soluble MHC-PLP139–151 constructs have all produced positive results (48–54). Conversely, strategies that target costimulation often aim to disrupt cell adhesion or molecular signaling events through the use of monoclonal antibodies such as anti-CD28 fAb (55) and anti-CD40L antibody (27, 56). Both the antigen-SIT and disruption of costimulation provide some level of therapeutic benefit and most of these therapies share physical attributes (soluble polymers, proteins, or constructs less than 200 kDa).
Combining these strategies may offer an opportunity for antigen-specific disruption of costimulation to hyposensitize patients to autoantigens or even induce tolerance. Previously, we showed the codelivery of antigen plus an inhibitor of immune cell adhesion cografted to a polymer backbone induced tolerance to the antigen (26, 38, 44). We suspected that efficacy would be linked to the physical properties of these “antigen arrays”, thus prompting exploration of size and solubility of carriers that codeliver PLP and LABL. Treatment with covalently linked PLP antigen and LABL peptide via a bifunctional peptide (BPI) suppressed EAE (29, 57, 58). Both the BPIPLP:LABL and the graft copolymer SAgAPLP:LABL were effective, yet their size and structure were quite different. Next, two sizes of SAgAPLP:LABL, 45 and 65 kDa were synthesized, to determine if there was an effect on EAE suppression. The smaller 45 kDa SAgAPLP:LABL did not extend suppression of EAE when compared to other treatments, it did however delay onset of disease by 2 days and extended the maintenance of weight in the animals. This difference could be due to the ability of the smaller SAgAPLP:LABL to move away from the injection site more rapidly than the larger SAgAPLP:LABL. Polymers of this size may also drain with interstitial fluid through regional lymph nodes instead of entering the systemic circulation from the injection site, whereas BPIPLP:LABL (~3 kDa) may directly enter the circulation due to its much smaller size.
Size and solubility of “antigen arrays” was further evaluated by using 400 nm PLGA particles. These particles are much larger than the SAgAPLP:LABL and BPIPLP:LABL and they are insoluble. NP displaying only the PLP antigen or only LABL cell adhesion inhibitor peptide did not significantly suppress EAE. The NPPLP:LABL showed very slight disease suppression, however, this was clearly not as effective as the BPIPLP:LABL or SAgAPLP:LABL treatments. Similar to vaccine strategies, the presence of both the PLP “antigen” and LABL “adjuvant” was not sufficient to reduce EAE symptoms; the physical characteristics of the carrier play an essential role in efficacy. Finally, NP treatments typically produced nodules at the injection site (clinical observation), suggesting these materials did not transport, at least during the disease observation period. Our studies suggested small, soluble carriers displaying both peptides provided the greatest activity; however, further studies are needed to determine the biodistribution and molecular mechanisms of these arrays post-injection.
CONCLUSIONS
Antigen-SITs that combined delivery of antigen with delivery of an immune cell adhesion inhibitor peptide were synthesized using carriers that varied in structure, size, and solubility. These antigen-SITs were tested in EAE, a murine model of MS. Co-delivery of both antigenic peptide (PLP) and cell adhesion inhibitor (LABL) significantly suppressed clinical scores compared to no treatment regardless of carrier structure and size. When peptides were conjugated to a small, soluble carrier, disease suppression was improved. In vivo data also showed that disease suppression required the presence of both PLP and LABL. These results support previous studies where codelivery of conjugated antigen and a secondary “context” signal were necessary to ameliorate EAE. Unlike vaccines which often use an insoluble adjuvant to induce a protective immune response, solubility may play an important role when attempting to hyposensitize or induce tolerance to autoantigen. The effect of structure, size, and solubility of “antigen arrays” on the drainage rate away from the injection site and into adjacent compartments such as lymphatic or systemic circulation should be further investigated.
Electronic supplementary material
(DOCX 1.29 kb)
(DOCX 162 kb)
(DOCX 104 kb)
ACKNOWLEDGMENTS
This work was supported by the NIH (1R56AI091996-01A1), KINBRE (P20 RR016475/P20 GM103418), Madison and Lila Self Graduate Fellowship (University of Kansas), and The Institute for Advancing Medical Innovation Graduate Fellowship (University of Kansas).
REFERENCES
- 1.Xiang SD, Scholzen A, Minigo G, David C, Apostolopoulos V, Mottram PL, et al. Pathogen recognition and development of particulate vaccines: does size matter? Methods. 2006;40(1):1–9. doi: 10.1016/j.ymeth.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 2.Phanse Y, Carrillo-Conde BR, Ramer-Tait AE, Broderick S, Kong CS, Rajan K, et al. A systems approach to designing next generation vaccines: combining α-galactose modified antigens with nanoparticle platforms. Sci Rep. 2014;4. doi: 10.1038/srep03775http://www.nature.com/srep/2014/140120/srep03775/abs/srep03775.html#supplementary-information. [DOI] [PMC free article] [PubMed]
- 3.Kool M, Pétrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181(6):3755–9. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed N, Gottschalk S. How to design effective vaccines: lessons from an old success story. Expert Rev Vaccines. 2009;8(5):543–6. doi: 10.1586/erv.09.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rolland JM, Gardner LM, O'Hehir RE. Allergen-related approaches to immunotherapy. Pharmacol Ther. 2009;121(3):273–84. doi: 10.1016/j.pharmthera.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Clapp T, Siebert P, Chen D, Jones BL. Vaccines with aluminum-containing adjuvants: optimizing vaccine efficacy and thermal stability. J Pharm Sci. 2011;100(2):388–401. doi: 10.1002/jps.22284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alving CR, Peachman KK, Rao M, Reed SG. Adjuvants for human vaccines. Curr Opin Immunol. 2012;24(3):310–5. doi: 10.1016/j.coi.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox JC, Coulter AR. Adjuvants–a classification and review of their modes of action. Vaccine. 1997;15(3):248–56. doi: 10.1016/S0264-410X(96)00183-1. [DOI] [PubMed] [Google Scholar]
- 9.Clausi A, Cummiskey J, Merkley S, Carpenter JF, Braun LJ, Randolph TW. Influence of particle size and antigen binding on effectiveness of aluminum salt adjuvants in a model lysozyme vaccine. J Pharm Sci. 2008;97(12):5252–62. doi: 10.1002/jps.21390. [DOI] [PubMed] [Google Scholar]
- 10.Brandau DT, Jones LS, Wiethoff CM, Rexroad J, Middaugh CR. Thermal stability of vaccines. J Pharm Sci. 2003;92(2):218–31. doi: 10.1002/jps.10296. [DOI] [PubMed] [Google Scholar]
- 11.Ireton GC, Reed SG. Adjuvants containing natural and synthetic Toll-like receptor 4 ligands. Expert Rev Vaccines. 2013;12(7):793–807. doi: 10.1586/14760584.2013.811204. [DOI] [PubMed] [Google Scholar]
- 12.Kumar S, Tummala H. Development of soluble inulin microparticles as a potent and safe vaccine adjuvant and delivery system. Mol Pharm. 2013;10(5):1845–53. doi: 10.1021/mp3006374. [DOI] [PubMed] [Google Scholar]
- 13.Moingeon P, Lombardi V, Saint-Lu N, Tourdot S, Bodo V, Mascarell L. Adjuvants and vector systems for allergy vaccines. Immunol Allergy Clin N Am. 2011;31(2):407–19. doi: 10.1016/j.iac.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Reed SG, Bertholet S, Coler RN, Friede M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009;30(1):23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 15.Dolgin E. The inverse of immunity. Nat Med. 2010;16(7):740–3. doi: 10.1038/nm0710-740. [DOI] [PubMed] [Google Scholar]
- 16.Derfuss T, Meinl E. Identifying autoantigens in demyelinating diseases: valuable clues to diagnosis and treatment? Curr Opin Neurol. 2012;25(3):231–8. doi: 10.1097/WCO.0b013e3283533a64. [DOI] [PubMed] [Google Scholar]
- 17.Zenichiro K, Kataoka H, Shimano K, Seki N, Sugahara K, Sugita T, et al. Multiple sclerosis (PP-038) Int Immunol Meet Abstr. 2010;22(Suppl 1 Pt 1):i148–55. [Google Scholar]
- 18.Aharoni R, Teitelbaum D, Leitner O, Meshorer A, Sela M, Arnon R. Specific Th2 cells accumulate in the central nervous system of mice protected against experimental autoimmune encephalomyelitis by copolymer 1. Proc Natl Acad Sci. 2000;97(21):11472–7. doi: 10.1073/pnas.97.21.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brück W, Gold R, Lund BT, et al. Therapeutic decisions in multiple sclerosis: moving beyond efficacy. JAMA Neurol. 2013;70(10):1315–24. doi: 10.1001/jamaneurol.2013.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucchinetti C, Bruck W, Lassmann H. Evidence for pathogenic heterogeneity in multiple sclerosis. Ann Neurol. 2004;56:308. doi: 10.1002/ana.20182. [DOI] [PubMed] [Google Scholar]
- 21.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 22.Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain. 2006;129:606–16. doi: 10.1093/brain/awl007. [DOI] [PubMed] [Google Scholar]
- 23.Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol. 2007;7(9):665–77. doi: 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- 24.Dustin ML. The cellular context of T cell signaling. Immunity. 2009;30(4):482–92. doi: 10.1016/j.immuni.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sant AJ, Chaves FA, Jenks SA, Richards KA, Menges P, Weaver JM, et al. The relationship between immunodominance, DM editing, and the kinetic stability of MHC class II:peptide complexes. Immunol Rev. 2005;207(1):261–78. doi: 10.1111/j.0105-2896.2005.00307.x. [DOI] [PubMed] [Google Scholar]
- 26.Sestak J, Mullins M, Northrup L, Thati S, Laird Forrest M, Siahaan TJ, et al. Single-step grafting of aminooxy-peptides to hyaluronan: a simple approach to multifunctional therapeutics for experimental autoimmune encephalomyelitis. J Control Release. 2013;168(3):334–40. doi: 10.1016/j.jconrel.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353(4):375–81. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- 28.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310(5751):1191–3. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- 29.Ridwan R, Kiptoo P, Kobayashi N, Weir S, Hughes M, Williams T, et al. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor: structure optimization and pharmacokinetics. J Pharmacol Exp Ther. 2010;332(3):1136–45. doi: 10.1124/jpet.109.161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Senti G, Prinz Vavricka BM, Erdmann I, Diaz MI, Markus R, McCormack SJ, et al. Intralymphatic allergen administration renders specific immunotherapy faster and safer: a randomized controlled trial. Proc Natl Acad Sci. 2008;105(46):17908–12. doi: 10.1073/pnas.0803725105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinman L, Conlon P. Antigen specific immunotherapy of multiple sclerosis. J Clin Immunol. 2001;21(2):93–8. doi: 10.1023/A:1011020225433. [DOI] [PubMed] [Google Scholar]
- 32.Vickery BP, Burks AW. Immunotherapy in the treatment of food allergy: focus on oral tolerance. Curr Opin Allergy Clin Immunol. 2009;9(4):364–70. doi: 10.1097/ACI.0b013e32832d9add. [DOI] [PubMed] [Google Scholar]
- 33.Vickery BP, Scurlock AM, Jones SM, Burks AW. Mechanisms of immune tolerance relevant to food allergy. J Allergy Clin Immunol. 2011;127(3):576–84. doi: 10.1016/j.jaci.2010.12.1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Senti G, Crameri R, Kuster D, Johansen P, Martinez-Gomez JM, Graf N, et al. Intralymphatic immunotherapy for cat allergy induces tolerance after only 3 injections. J Allergy Clin Immunol. 2012;129(5):1290–6. doi: 10.1016/j.jaci.2012.02.026. [DOI] [PubMed] [Google Scholar]
- 35.Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr Opin Immunol. 2010;22(5):609–15. doi: 10.1016/j.coi.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang N, Chittasupho C, Duangrat C, Siahaan TJ, Berkland C. PLGA nanoparticle–peptide conjugate effectively targets intercellular cell-adhesion molecule-1. Bioconjug Chem. 2007;19(1):145–52. doi: 10.1021/bc700227z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chittasupho C, Xie S-X, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. ICAM-1 targeting of doxorubicin-loaded PLGA nanoparticles to lung epithelial cells. Eur J Pharm Sci. 2009;37(2):141–50. doi: 10.1016/j.ejps.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chittasupho C, Sestak J, Shannon L, Siahaan TJ, Vines CM, Berkland C. Hyaluronic acid graft polymers displaying peptide antigen modulate dendritic cell response in vitro. Mol Pharm. 2013;11(1):367–73. doi: 10.1021/mp4003909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang T-F, Chen C-N, Chen M-C, Lai C-H, Liang H-F, Sung H-W. Shell-crosslinked Pluronic L121 micelles as a drug delivery vehicle. Biomaterials. 2007;28(4):725–34. doi: 10.1016/j.biomaterials.2006.09.035. [DOI] [PubMed] [Google Scholar]
- 40.Bouhadir KH, Hausman DS, Mooney DJ. Synthesis of cross-linked poly(aldehyde guluronate) hydrogels. Polymer. 1999;40(12):3575–84. doi: 10.1016/S0032-3861(98)00550-3. [DOI] [Google Scholar]
- 41.Lee KY, Bouhadir KH, Mooney DJ. Degradation behavior of covalently cross-linked poly(aldehyde guluronate) hydrogels. Macromolecules. 1999;33(1):97–101. doi: 10.1021/ma991286z. [DOI] [Google Scholar]
- 42.Chittasupho C, Shannon L, Siahaan TJ, Vines CM, Berkland C. Nanoparticles targeting dendritic cell surface molecules effectively block T cell conjugation and shift response. ACS Nano. 2011;5(3):1693–702. doi: 10.1021/nn102159g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fakhari A, Baoum A, Siahaan TJ, Le KB, Berkland C. Controlling ligand surface density optimizes nanoparticle binding to ICAM-1. J Pharm Sci. 2011;100(3):1045–56. doi: 10.1002/jps.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sestak JO, Sullivan BP, Thati S, Northrup L, Hartwell B, Antunez L, et al. Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Molecular Therapy—Methods & Clinical Development. 2014;1. doi: 10.1038/mtm.2014.8http://www.nature.com/articles/mtm20148#supplementary-information. [DOI] [PMC free article] [PubMed]
- 45.Tisch R, McDevitt HO. Antigen-specific immunotherapy: is it a real possibility to combat T-cell-mediated autoimmunity? Proc Natl Acad Sci. 1994;91(2):437–8. doi: 10.1073/pnas.91.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing‐remitting multiple sclerosis: results of a phase III multicenter, double‐blind, placebo‐controlled trial. Neurology. 1995;45(7):1268–76. doi: 10.1212/WNL.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 47.Neuhaus O, Farina C, Wekerle H, Hohlfeld R. Mechanisms of action of glatiramer acetate in multiple sclerosis. Neurology. 2001;56(6):702–8. doi: 10.1212/WNL.56.6.702. [DOI] [PubMed] [Google Scholar]
- 48.Kuchroo VK, Greer JM, Kaul D, Ishioka G, Franco A, Sette A, et al. A single TCR antagonist peptide inhibits experimental allergic encephalomyelitis mediated by a diverse T cell repertoire. J Immunol. 1994;153(7):3326–36. [PubMed] [Google Scholar]
- 49.Stern JNH, Illés Z, Reddy J, Keskin DB, Fridkis-Hareli M, Kuchroo VK, et al. Peptide 15-mers of defined sequence that substitute for random amino acid copolymers in amelioration of experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2005;102(5):1620–5. doi: 10.1073/pnas.0409022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Margot CD, Ford ML, Evavold BD. Amelioration of established experimental autoimmune encephalomyelitis by an MHC anchor-substituted variant of proteolipid protein 139–151. J Immunol. 2005;174(6):3352–8. doi: 10.4049/jimmunol.174.6.3352. [DOI] [PubMed] [Google Scholar]
- 51.Samson MF, Smilek DE. Reversal of acute experimental autoimmune encephalomyelitis and prevention of relapses by treatment with a myelin basic protein peptide analogue modified to form long-lived peptide-MHC complexes. J Immunol. 1995;155(5):2737–46. [PubMed] [Google Scholar]
- 52.Vanderlugt CL, Neville KL, Nikcevich KM, Eagar TN, Bluestone JA, Miller SD. Pathologic role and temporal appearance of newly emerging autoepitopes in relapsing experimental autoimmune encephalomyelitis. J Immunol. 2000;164(2):670–8. doi: 10.4049/jimmunol.164.2.670. [DOI] [PubMed] [Google Scholar]
- 53.Smith CE, Eagar TN, Strominger JL, Miller SD. Differential induction of IgE-mediated anaphylaxis after soluble vs. cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2005;102(27):9595–600. doi: 10.1073/pnas.0504131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang C, Gold BG, Kaler LJ, Yu X, Afentoulis ME, Burrows GG, et al. Antigen-specific therapy promotes repair of myelin and axonal damage in established EAE. J Neurochem. 2006;98(6):1817–27. doi: 10.1111/j.1471-4159.2006.04081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perrin PJ, June CH, Maldonado JH, Ratts RB, Racke MK. Blockade of CD28 during in vitro activation of encephalitogenic t cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J Immunol. 1999;163(3):1704–10. [PubMed] [Google Scholar]
- 56.Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, et al. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci. 1996;93(6):2499–504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murray JS, Oney S, Page JE, Kratochvil-Stava A, Hu Y, Makagiansar IT, et al. Suppression of Type 1 diabetes in NOD mice by bifunctional peptide inhibitor: modulation of the immunological synapse formation. Chem Biol Drug Des. 2007;70(3):227–36. doi: 10.1111/j.1747-0285.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. J Pharmacol Exp Ther. 2007;322(2):879–86. doi: 10.1124/jpet.107.123257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 1.29 kb)
(DOCX 162 kb)
(DOCX 104 kb)