

Abstract
Although arctigenin (AR) has attracted substantial research interests due to its promising and diverse therapeutic effects, studies regarding its biotransformation were limited. The current study aims to provide information regarding the pharmacokinetic properties of AR via various in vitro and in vivo experiments as well as semi-mechanistic pharmacokinetic modeling. Our in vitro rat microsome incubation studies revealed that glucuronidation was the main intestinal and liver metabolic pathway of AR, which occurred with Vmax, Km, and Clint of 47.5 ± 3.4 nmol/min/mg, 204 ± 22 μM, and 233 ± 9 μl/min/mg with intestinal microsomes and 2.92 ± 0.07 nmol/min/mg, 22.7 ± 1.2 μM, and 129 ± 4 μl/min/mg with liver microsomes, respectively. In addition, demethylation and hydrolysis of AR occurred with liver microsomes but not with intestinal microsomes. In vitro incubation of AR and its metabolites in intestinal content demonstrated that glucuronides of AR excreted in bile could be further hydrolyzed back to the parent compound, suggesting its potential enterohepatic circulation. Furthermore, rapid formation followed by fast elimination of arctigenic acid (AA) and arctigenin-4′-O-glucuronide (AG) was observed after both intravenous (IV) and oral administrations of AR in rats. Linear pharmacokinetics was observed at three different doses for AR, AA, and AG after IV administration of AR (0.48–2.4 mg/kg, r2 > 0.99). Finally, an integrated semi-mechanistic pharmacokinetic model using in vitro enzyme kinetic and in vivo pharmacokinetic parameters was successfully developed to describe plasma concentrations of AR, AA, and AG after both IV and oral administration of AR at all tested doses.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-014-9664-x) contains supplementary material, which is available to authorized users.
KEY WORDS: arctigenic acid, arctigenin, arctigenin-4′-O-glucuronide, pharmacokinetics, semi-mechanistic pharmacokinetic modeling
INTRODUCTION
Arctigenin (AR), a key bioactive phenylpropanoid dibenzyl butyrolactone lignan presented in medicinal plants such as Arctium lappa, Torreya nucifera, and Saussurea medusa, has received substantial attention in the medicinal and pharmaceutical research literature in the past two decades. It has been suggested that AR possesses anti-viral (1–3), anti-inflammation (4–6), and immunomodulation effects (7) in a variety of in vitro and in vivo models. It was also demonstrated, in a diseased mice model, that AR had potential as a drug candidate for the treatment of Alzheimer’s disease through targeting β-amyloid formation and clearance, with the effect of ameliorating memory impairment (8). However, the most extensively studied therapeutic aspect of AR is its anti-cancer activity, resulting in the launch of its phase I and II clinical trials, in which AR was used as the second-line therapeutic agent for the treatment of pancreatic cancer (9). Furthermore, mechanistic studies of the anti-cancer effect of AR have associated it with various pathways, including chemo-preventative (10), induction of apoptosis (11,12), cytotoxicity (13,14), inhibition of angiogenesis (15), and anti-oxidation (16). Recent publications also indicated that it could enhance the chemo-sensitivity of cisplatin through inhibition of the STAT3 signaling pathway (17) and downregulation of survivin expression (18) in human carcinoma cells.
Although previous studies have discovered many promising pharmacological activities of AR, pharmacokinetic investigation of it is rather scanty. Demethylation mediated by gastrointestinal (GI) bacteria was once believed to be the major biotransformation pathway of AR, leading to enterolactone formation in in vitro incubation settings (19–21). However, our recent study revealed that arctigenic acid (AA) and arctigenin-4′-O-glucuronide (AG), rather than parent compound AR or 4-O-demethyl-arctigenin (DA) (Fig. 1), were the main compounds found in plasma after oral administration of AR in rats (22,23). Although our previous rat single-pass intestinal perfusion study demonstrated that extensive hydrolysis and glucuronidation of AR occurred when it passed through the intestinal epithelium (22), all its available in vivo pharmacokinetic studies by far monitored only the parent compound (24–26). Therefore, pharmacokinetic studies providing concrete plasma concentration profiles of both AR and its metabolites are essential for the understanding of its in vivo biotransformation.
Fig. 1.
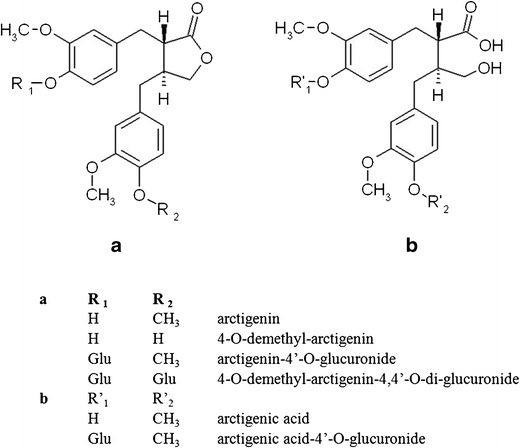
a, b Chemical structures of arctigenin and its metabolites identified in plasma and bile after oral and intravenous administration of arctigenin in rats
In the current report, in vitro enzyme kinetic studies of both hepatic and intestinal metabolism of AR followed by qualitative and quantitative determinations of its bile secretion and intestinal lumen metabolism were carried out for mechanistic mapping of AR biotransformation pathways in vitro. Additionally, plasma concentration profiles of AR, AA, and AG after both IV and oral administration of several doses were obtained in rats. Finally, the observed results from in vitro enzyme kinetics and the estimated in vivo pharmacokinetic parameters of all the analytes were incorporated into a semi-mechanistic model to describe the biotransformation of AR after its oral and IV administration in rats.
MATERIAL AND METHODS
Materials
AR was purchased from Standhill Technology Limited (Hong Kong, HKSAR). AA was synthesized with a purity >98.0% as described in our previous report (23). Diclofenac sodium (IS) was purchased from Wing Hing Chemical Co., Ltd (Hong Kong, HKSAR). Acetonitrile and methanol (HPLC grade) were purchased from RCI Labscan (Bangkok, Thailand). Other reagents were of at least analytical grade and were used without further purification. Distilled and deionized water was used for the preparation of solutions. Uridine 5′diphosphoglucuronic acid (UDPGA) and alamethicin were obtained from Sigma-Aldrich Chem. Co. (Milwaukee, WI, USA). Pooled male rat liver microsome (RLM) was purchased from BD Biosciences (Woburn, MA, USA). Pooled male rat intestinal microsome (RIM) and RapidStart nicotinamide adenine dinucleotide phosphate (NADPH) Regenerating System (RNRS) were obtained from XenoTech, LLC (Lenexa, KS, USA).
Metabolic Profiles of AR in Rat Tissues and Organs
Hepatic and Intestinal Metabolism of AR
A series of concentrations of AR was prepared by dilution of 2 mg/ml DMSO stock solution with 50% methanol (final methanol concentration of 0.5%, v/v). To evaluate its intestinal and hepatic glucuronidation, incubations of AR in RIM/RLM with glucuronidation cofactors were carried out as described previously (23). Briefly, AR (0.3–67.2 μM) was pre-incubated with 0.2 mg/ml RIM/RLM in 50 mM Tris–HCl buffer (pH 7.4) containing 8 mM MgCl2 and 25 μg/ml of alamethicin for 5 min at 37°C. The reaction was initiated by adding 2 mM UDPGA. Demethylation and hydrolysis reactions of AR were carried out with 5 min pre-incubation of AR (0.3–67.2 μM) in RIM/RLM followed by addition of 2 mM NADPH (equivalent amount generated from RNRS). To explore the possibility of its secondary metabolism, AA was incubated in RIM/RLM with 5 mM of either UDPGA or NADPH (equivalent amount generated from RNRS) for 30 min or 2 h, respectively.
All the above reactions were carried out at 37°C and terminated by the addition of an equal volume of ice-cold methanol containing 20% ascorbic acid and 80 μg/ml diclofenac acid (IS) followed by content analyses of AR and its metabolites using our previously developed ultra-performance liquid chromatography tandem mass spectrometry (UPLC/MS/MS) method (22). The calibration samples were prepared in microsomal matrices similar to those of the reaction samples except for the absence of corresponding enzyme reaction cofactors. Optimizations of protein concentration and incubation time were performed to ensure linear metabolite formation for each of the above metabolic reactions. All the experiments were conducted in triplicate.
Biliary Secretion of AR
To investigate the biliary secretion of AR after IV administration, the bile duct and right jugular vein of Sprague Dawley (SD) rats (n = 2) were cannulated in advance as previously described (27). On the day of experiment, 0.96 mg/kg AR (dissolved in saline containing 20% polyethylene glycol 400 (PEG400) and 0.5% Tween 80, w/w) was administered to rats via the cannulated right jugular vein. Bile was collected in vials containing 5 μM ethylenediaminetetraacetic acid (EDTA) for consecutive 90 min with a 15-min time interval followed by further treatment as described before (14). Briefly, the collected bile samples were diluted with an equal volume of 35% methanol in 25 mM NaH2PO4 buffer (pH 2.5) containing 1% ascorbic acid. After 5-min centrifugation at 16,000×g, 20 μl of supernatant was injected into UPLC/MS/MS for content analyses of AR and its metabolites by our previously developed assay (22). For qualitative detection of AR and its metabolites in bile, multiple reaction mode (MRM) transitions corresponding with DA (m/z 357 → 83) and potential secondary metabolites were also monitored in addition to the parent compound and the two major metabolites identified in plasma (AR 371 → 83, AA 389 → 330, and AG 547 → 371). Secondary metabolites screened in the collected bile included arctigenic acid-4′-O-glucuronide (AAG, m/z 565 → 389), 4-O-demethyl-arctigenin-4 or 4′-O-glucuronide (DAG, m/z 533 → 357), and 4-O-demethyl-arctigenin-4, 4′-O-di-glucuronide (DA-diG, m/z 709 → 533) (Fig. 1).
Metabolism of AR in GI Contents
Biotransformation of drug molecules in the GI tract has played an important role in the metabolic evaluation of some xenobiotics. Besides the parent compound, metabolites formed after absorption could also occur in the GI tract via either efflux from enterocyte or bile secretion. As demonstrated in our previous in situ single-pass rat intestinal perfusion study, over 90% of the AA and AG formed during AR intestinal absorption was found in the perfusate (22), which were in turn susceptible to further biotransformation in the intestinal lumen. To comprehend the potential biotransformation of AR in the GI tract, in vitro incubations of AR, AA, and collected bile (after IV administration of AR) with both simulated GI fluid and rat GI content solution were conducted based on their potential site of appearance in vivo, followed by qualitative and semi-quantitative determinations of relevant metabolites. Simulated gastric fluid and simulated intestinal fluid were prepared as described in the United States Pharmacopoeia (USP) (28). Rat GI content solution was prepared as described previously (29). Briefly, five SD rats were sacrificed by CO2 inhalation after overnight fasting followed by immediate harvesting of their GI tract. Rat gastric and small/large intestinal contents were then collected and suspended in fivefold ice-cold simulated gastric fluid or simulated intestinal fluid, respectively. All suspensions were mixed extensively by vortex followed by centrifugation at 8,000×g for 5 min. The supernatants were kept on ice for subsequent experiments.
To investigate its potential metabolism in the GI tract, AR (0.8 μg/ml) was incubated with simulated gastric/intestinal fluid and rat GI content solution. In addition, AA (0.8 μg/ml) and the collected bile (1:50, v/v) were incubated separately with simulated intestinal fluid or rat small/large intestinal content solution to estimate the further biotransformation of these in vivo metabolites of AR in intestinal lumen after either efflux of enterocyte or bile secretion. All the above incubations were carried out at 37°C followed by sampling at 0, 5, 15, 30, 60, 120, and 240 min. The collected samples were mixed with equal volume of methanol containing 80 μg/ml of IS and 20% ascorbic acid. After centrifugation at 16,000×g for 5 min, 20 μl of the supernatant was injected into UPLC/MS/MS for analysis. Due to the unavailability of authentic standards of the metabolites identified in bile, it was not possible to accurately quantify them. Thus, a semi-quantitative approach was adopted, in which the relative intensity of the metabolites compared with that from time zero was used to estimate the percentage remaining in intestinal content for all the metabolites identified in bile.
Further Estimation of Formation Pathways for Secondary Metabolites of AR
Three additional in vitro incubation studies with either RLM or rat plasma were carried out to explore the possible formation pathways of secondary metabolites of AR identified above:
To investigate the possibility of further hydrolysis of AG to AAG, 2 μl of bile containing approximately 2.67 μM AG (calculated based on calibration curve of AR) was obtained as previously described in the section on bile secretion of AR metabolites and incubated in freshly harvested rat plasma in the presence or absence of 5 mM EDTA for 30 min at 37°C.
To test the possibility of further glucuronidation of DA, incubation of 0.8 μg/ml AR in 0.4 mg/ml RLM was carried out with 2 mM NADPH for 2 h at 37°C to generate DA, then 0.4 mg/ml fresh RLM with 2 mM UDPGA was added and allowed to react for another 15 min.
To explore the potential further metabolism of AG, similar incubation of AR in RLM with UDPGA added prior to NADPH was carried out.
All collected samples were immediately mixed with an equal volume of methanol containing 80 μg/ml of IS and 20% ascorbic acid. After centrifugation of the mixture at 16,000×g for 5 min, 20 μl of the supernatant was injected into UPLC/MS/MS for analysis.
Enzyme Kinetic Data Analysis
GraphPad Prism® was employed to analyze enzyme kinetic data using the Michaelis-Menten equation by fitting the result of a single experiment with three replicates into non-linear regression. The reported Km and Vmax as well as their standard deviations were the best-fit values predicted by the software based on simulation.
In Vivo Pharmacokinetics in Rats
Male SD rats (weighting 250–260 g) used in the experiment were provided by Laboratory Animal Services Center of The Chinese University of Hong Kong (Hong Kong SAR, People’s Republic of China). The study was approved by both the Department of Health of Hong Kong SAR and the Animal Ethics Committee at the Chinese University of Hong Kong. For the jugular vein cannulation surgery, the rats were anesthetized (intramuscular injection of a mixture of 80 mg/kg ketamine and 8 mg/kg xylazine) followed by inserting a polyethylene tube (0.5 mm ID, 1 mm OD, Portex Ltd., England) into the right jugular vein. The cannulated rats were allowed to recover overnight while fasting with free access to water.
For IV administrations, AR (dissolved in saline containing 20% PEG400 + 0.5% Tween 80, w/w) was given through the jugular vein catheter at 0.48, 0.96, and 2.4 mg/kg (IV low (L), medium (M), high (H) dose group, n = 6 for each group), followed by rinsing the catheter with 1 ml blank blood and 1 ml saline. During the preliminary studies, it was found that IV injection of 4.8 mg/kg AR triggered an immediate toxic reaction in SD rats (one out of two rats tested for 4.8 mg/kg IV died within 5 min), while IV injection of 2.4 mg/kg of AR did not cause any obvious irritation reaction in SD rats. Thus, 2.4 mg/kg was set as the H dose. The IV L dose of 0.48 mg/kg was estimated based on analytical detection limits of AR, AA, and AG. IV M dose of 0.96 mg/kg was set at the approximate mean of the L and H doses. For oral administration, AR (dissolved with 50% PEG400, 5% Tween 80 in H2O, w/w) was given to rats by oral gavage at 2.4, 4.8, and 12 mg/kg (n = 6 for each group), representing oral L, M, and H doses, respectively. Oral L dose was set to be the same as IV H dose to allow direct calculation of absolute bioavailability. In addition, the fold changes among oral L, M, and H doses were similar to those for the IV administration. At pre-determined time points post-dosing (2, 5, 10, 15, 20, 30, 45, 60, 90, and 120 min for IV administration groups; 15, 30, 45, 60, 90, 120, 240, 480, 720, and 1,440 min for oral administration groups), approximately 300 μl blood was collected into a heparinized centrifuge tube containing 5 μM EDTA followed by centrifugation at 16,000×g for 5 min to obtain the plasma. The rats were allowed free access to food 4 h after drug administrations with 300 μl saline containing 50 IU heparin injected through the cannulated jugular vein after each blood sampling.
Analyses of all the obtained plasma samples followed a previously validated UPLC/MS/MS assay method, which demonstrated satisfactory sensitivity, selectivity, accuracy, and precision of quantitative measurement of AR, AA, and AG in rat plasma, as well as recovery, matrix effect, and sample stability (22,30). Briefly, an aliquot of 5 μl IS was added to a 100-μl plasma sample, and 500 μl acetonitrile (ACN) was added into the mixture followed by vortex mixing for 1 min. The mixture was then centrifuged for 5 min at 16,000×g, and the supernatant was collected. After evaporation to dryness by vacuum evaporator at 35°C, the residue was reconstituted in 100 μl 50% ACN. After 5 min centrifugation at 16,000×g, 20 μl of the supernatant was then injected to UPLC/MS/MS for analysis of AR and metabolites. Analyte concentrations were considered as 0 ng/ml if they were lower than the lowest level of quantification (LLOQ). Samples with analyte concentrations higher than the upper limit of quantification were diluted with blank plasma before sample preparation.
Pharmacokinetic parameters including maximum concentration (Cmax), volume of distribution (V), and elimination rate constant (Ke) were calculated by WinNonlin using non-compartmental approach (NCA). Area under the curve for monitored time period (area under the concentration curve (AUC)0→∞) was calculated using linear/log trapezoidal method. In addition, metabolic ratio (MR) for each biotransformation pathway was calculated as below to estimate the related extent of metabolisms.
Based on the molar concentrations of both parent and metabolites, the absorbed fraction (F) was calculated as follows:
Comparisons among different dose groups were performed with ANOVA independent test using SPSS (version 22.0.0.0) with p < 0.05 considered as significantly different.
Integrated Semi-mechanistic PK Model for AR and Its Metabolites
The plasma concentration versus time profiles of AR and its two metabolites AG and AA after both IV and oral administrations were fitted into one integrated model as demonstrated in Fig. 2 (left). This model consists of five compartments: AR in intestinal lumen (Compt. 1), AR in enterocytes (Compt. 5), AR in central compartment (Compt. 2), AG in central compartment (Compt. 3), and AA in central compartment (Compt. 4).
Fig. 2.
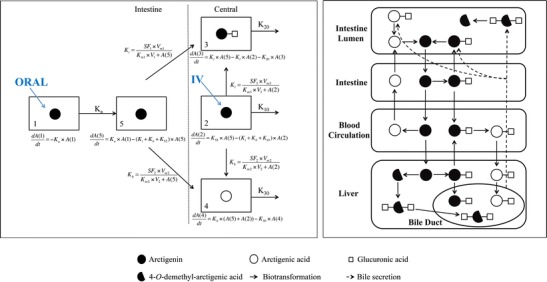
Proposed biotransformation diagram of arctigenin after various routes of administrations in rats. Dotted double line represents quantitative model used for fitting of the plasma concentrations of arctigenin and its metabolites (Oral oral administration; IV intravenous administration; K i, K b, K l metabolic constant in intestinal cells, blood, and liver, respectively; K 10, K 20, K 30 elimination constant of AR, AA, and AG from central compartment, respectively. Solid double line represents qualitative description of biotransformation of arctigenin based on in vitro and in vivo observations
After oral administration, AR was considered to be absorbed into Compt. 5 first and then metabolized into AA and AG with Kb (metabolic rate constant in blood) and Ki (metabolic rate constant in intestine), respectively, before entering the central compartment. For IV administrations, AR was introduced into Compt. 2 directly, followed by metabolism into AA in plasma (Compt. 3) and AG in liver, which was assumed to be transferred back into central compartment (Compt. 4) immediately. Compt. 3 and 4 were connected with Compt. 2 via the formation of AG in liver and AA in plasma at a rate of Kl (metabolic rate constant in liver) and Kb, respectively. Since glucuronidation and hydrolysis were found to be the dominating metabolic pathways in RLM/RIM and plasma, respectively, no other metabolite was included in the integrated model. In addition, the volumes of Compt. 2, 3, and 4 were assumed to be the same.
The formation of AA and AG was characterized by the Michaelis-Menten equation with Vm and Km determined from our current in vitro experiments and previous studies (23). Since it is difficult to accurately measure the anatomical parameters of each experimental animal, further adjustments on Vm and Km were carried out based on anatomical parameters obtained from literatures: the in vivo Km of glucuronidation in intestine or liver was calculated as in vitro Km multiplied by organ weight per rat and content of microsomal protein per gram of relevant organ. The liver weight and the content of microsomal protein in liver of a 250-g SD rat were suggested to be 11.021 g/rat and 38 mg/g, respectively (GastroPlus™). The rat intestine weight and content of microsomal protein were set to be 5.58 g/rat (31) and 47 mg/g (32), respectively. Moreover, since only the free form of drug molecules could be metabolized, Km values generated from in vitro studies were divided by the percentage of free drug before introduction into the in vivo model. As suggested by GastroPlus™, the protein binding of AR was set to be 90%. Furthermore, since plasma accounts for approximately half the volume of blood, the in vivo Km of plasma hydrolysis was calculated as twice the in vitro Km. The plasma volume of a rat was set to be 7.75 ml (33), and the rat plasma protein concentration was measured to be 30 mg/ml by Bio-Rad® protein assay according to manufacturer’s manual. Thus, the in vivo Vmax of plasma hydrolysis was calculated as in vitro Vmax multiplied by plasma protein concentration and plasma volume per rat. Moreover, considering the potential differences in anatomical parameters between the literature and the actual value of each experimental animal, scale factors were introduced into the model, which is a common practice employed in both literatures (34) and simulation software such as GastroPlus™.
Our model was implemented using NONMEM7.2, and the corresponding control file was provided in the Supplementary material. The parameters of this model were estimated using first-order conditional estimation (FOCE), and the variance–covariance matrix was calculated using the default method to estimate the RSE of each parameter. The subroutine of ADVAN6, which could support the user-defined models, was adopted to define the semi-mechanistic model. The residue model of Y = F + EPS (1)* SQRT(THETA(17)**2*IPRED**2 + THETA(18)**2) was employed with only additive errors considered, where THETA(17) stands for the proportional residual error and THETA(18) stands for the additive residual error. Since all the data were acquired from experimental rats, and the inter-animal variation was considered small, the OMEGA values of all pharmacokinetic (PK) parameters were fixed to be zero. The criteria for model selection included (a) objective function value (OFV): a reduction of more than 5.02 is considered as significant; (b) estimated PK parameters are physiologically reasonable and comparable with those calculated by NCA; (c) graphical fitting of predicted versus experimental concentrations; and (d) the covariance matrix could be successfully calculated. The differential equations used in the model were incorporated in Fig. 2. Non-parametric (case resampling) bootstrap and visual predictive check (VPC) plots were used as internal validation tools to ensure the ruggedness and predictability of the integrated PK model.
RESULTS
Metabolic Profiles of AR
Extensive glucuronidation of AR was observed in both RLM and RIM. Demethylation and hydrolysis of AR were observed in RLM but not in RIM. No further glucuronidation or demethylation of AA was demonstrated in RLM or RIM under the tested conditions. As shown in Fig. 3, the enzyme kinetic profile of AR metabolism in RLM and RIM was obtained with Vmax, Km, and Clint calculated to be 47.5 ± 3.4 nmol/min/mg, 204 ± 22 μM, and 233 ± 9 μl/min/mg for glucuronidation in RIM; 2.92 ± 0.07 nmol/min/mg, 22.7 ± 1.2 μM, and 129 ± 4 μl/min/mg for glucuronidation in RLM; 1.75 ± 0.19 nmol/min/mg, 52.2 ± 10.4 μM, and 33.4 ± 3.2 μl/min/mg for demethylation in RLM; and 0.21 ± 0.19 nmol/min/mg, 363 ± 239 μM, and 0.57 ± 0.25 μl/min/mg for hydrolysis in RLM, respectively.
Fig. 3.
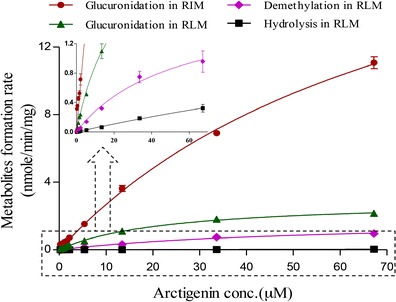
Metabolic kinetics profiles of arctigenin (0.269–67.2 μM) in rat intestinal (RIM) and liver microsome (RLM) (n = 3) Glucuronidation in RIM: 50 mM Tris–HCl buffer (pH 7.4), 0.05 mg/ml, 2 mM UDPGA 37°C, 10 min; glucuronidation in RLM: 50 mM Tris–HCl buffer (pH 7.4), 0.2 mg/ml, 2 mM UDPGA, 37°C, 15 min; hydrolysis and demethylation in RLM: 20 mM potassium phosphate buffer (pH 7.4), 0.2 mg/ml, 2 mM NADPH, 37°C, 2 h
As shown in Fig. 4, in addition to a large amount of AG, which was also observed in plasma after AR administrations (IV and oral), two novel glucuronides, DA-diG and AAG, as well as trace amount of AA were identified in bile after IV administration of 0.96 mg/kg AR, whereas no AR was detected in bile.
Fig. 4.
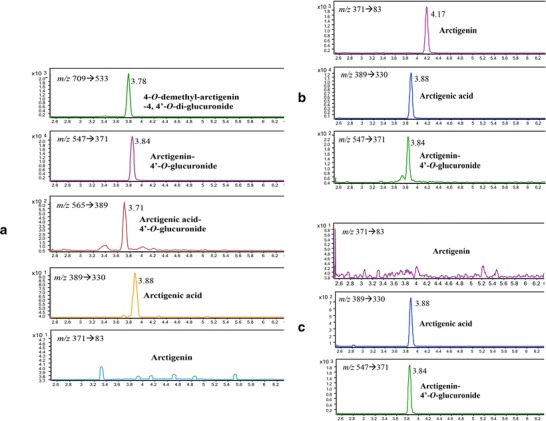
Representative extracted ion chromatographs (multiple reaction monitoring) of a metabolites identified in bile (0–15 min) after intravenous administration of 0.96 mg/kg arctigenin in rats; b arctigenin and metabolites identified in plasma (2 min) after intravenous administration of 0.96 mg/kg arctigenin; and c arctigenin and metabolites identified in plasma (15 min) after oral administration of 4.8 mg/kg arctigenin
AAG was identified in the matrices after 30 min plasma incubation of bile at 37°C, accounting for around 20% of AG added (calculated based on parent loss). Such hydrolysis could be efficiently suppressed by the addition of 5 mM EDTA, indicating the involvement of calcium-dependent plasma esterase, probably paraoxonase I (35,36), which is also responsible for AR hydrolysis in plasma (23). Incubation of AR in RLM with NADPH and UDPGA added in either order revealed that DA-diG could be formed by further metabolism of either DA or AG. The extent of formation of DA-diG from AR was found to be 11.6 ± 6.5% when UDPGA was added first and 23.5 ± 2.3% when NADPH was introduced prior to UDPGA, suggesting possibilities of both pathways for the formation of DA-diG.
As demonstrated in Fig. 5a, AR and all its tested metabolites were found to be stable in both simulated gastric and intestinal fluids. Stabilities of all the analytes were also demonstrated after 4 h incubation in rat gastric/small intestinal content solutions, except for AAG the content of which was tripled after the incubation (Fig. 5b). However, considering the low concentration of AAG presented in the incubation solution (∼1% of AG, suggested by relative intensity shown in Fig. 5a), such a change would not be significant for the overall biotransformation of AR. Regarding metabolism in the rat large intestinal content solution, AR was rather stable, while the amount of AA gradually declined during the 4-h incubation (Fig. 5c). AA was found to be converted back to AR at as early as time zero, indicating instant transformation of AA to AR when incubated with rat large intestinal content (Fig. 5d). Furthermore, as shown in Fig. 5c, the amount of all three glucuronides, AG, DA-diG, and AAG, dropped dramatically when incubated with rat large intestinal content, while the amount of corresponding parent compounds, AR, DA, and AA gradually increased (Fig. 5e). These findings suggested that the large amount of glucuronides identified in bile would be hydrolyzed after being excreted into rat intestinal lumen. Based on these results, we therefore composed a qualitative description of AR metabolism in rats, as shown in Fig. 2 (right).
Fig. 5.
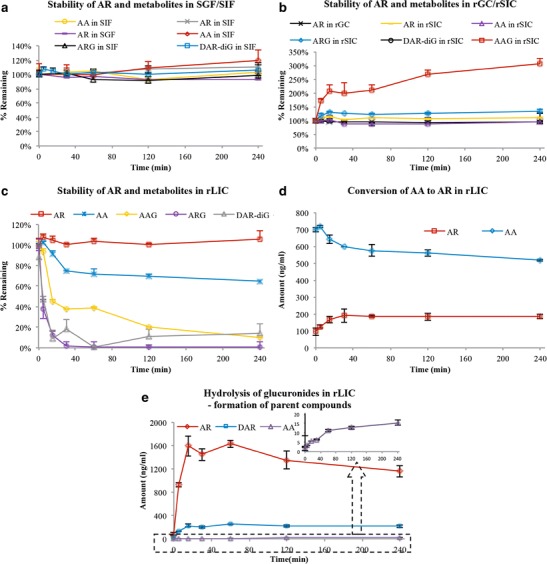
Stability of arctigenin (AR, 0.8 μg/ml), arctigenic acid (AA, 0.8 μg/ml), and bile (1:50, v/v, 15 min collection after intravenous administration of 0.96 mg/kg AR in SD rats) in a simulated gastric and intestinal fluid (SGF/SIF), b rat gastric and small intestine content solution (rGC/rSIC), c rat large intestine content solution (rLIC), d the conversion of AA to AR in rLIC, and e hydrolysis of glucuronic acid conjugated metabolites in rLIC. All experiments were carried out at 37°C. Metabolites monitored in bile were arctigenin-4′-O-glucuronide (AG), 4-O-demethyl-arctigenin-4,4′-O-di-glucuronide (DA-diG), and arctigenic acid-4′-O-glucuronide (AAG)
In Vivo Pharmacokinetics of AR in Rats
As shown in Fig. 6 (upper panels), the plasma concentration versus time profiles of AR and its two major metabolites AA and AG followed similar patterns after IV administrations of 0.48, 0.96, and 2.4 mg/kg of AR in rats. Pharmacokinetic parameters determined based on NCA in Table I indicated rapid formations of both AA and AG with Tmax of approximately 5 and 2 min, respectively, with no significant difference observed among the three dose groups. In addition, T1/2 of both AR and its metabolites were found to be shorter than 20 min, with no significant differences among different dose groups, suggesting a rather fast elimination process for all analytes. Based on the linear correlations between Cmax and AUC0→∞ of each analyte and dose (r2 > 0.99), linear kinetics of AR, AA, and AG were demonstrated after IV administrations of AR. In addition, consistent MR for both hydrolysis (39.1 ± 19.9%, 41.5 ± 7.5, and 41.6 ± 6.2 at 0.48, 0.96, and 2.4 mg/kg, respectively) and glucuronidation (9.4 ± 2.9, 10.8 ± 2.1, and 8.6 ± 0.0 at 0.48, 0.96, and 2.4 mg/kg, respectively) were observed, indicating neither glucuronidation nor hydrolysis of AR was saturated under the dose range tested.
Fig. 6.
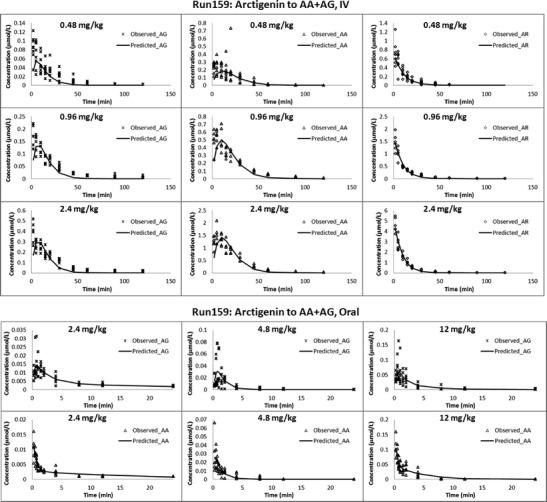
Observation (dots) and typical model prediction (line) of plasma concentration versus time profiles of arctigenin and metabolites in rats after intravenous administration (upper); oral administration (lower) of arctigenin at various doses (n = 6 per dose group)
Table I.
Non-compartmental Model-Based Pharmacokinetic Parameters of AR and Its Metabolites After Intravenous (IV) and Oral AR Administrations in Rats (n = 6 per Treatment Group, Results Were Presented as Mean ± SD)
| Route | Analyte | Dose (μmol) | K e (1/min) | T 1/2 (min) | T max (min) | C max (μmol/L) | AUC0–∞ (min μmol/L) | V/V_F (L) | Cl/Cl_F (L/min) | AUMC (min min μmol/L) | MRT (min) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IV | AR | 0.32 | 0.08 ± 0.02 | 9.35 ± 2.15 | 2.38 ± 1.07 | 0.74 ± 0.23 | 9.32 ± 2.80 | 0.46 ± 0.11 | 0.04 ± 0.02 | 98.5 ± 39.4 | 10.3 ± 1.8 |
| 0.64 | 0.05 ± 0.01 | 13.4 ± 2.2 | 2.00 ± 0.00 | 1.45 ± 0.33 | 19.4 ± 2.9 | 0.65 ± 0.09 | 0.03 ± 0.00 | 278 ± 33 | 14.4 ± 1.0 | ||
| 1.61 | 0.05 ± 0.01 | 13.0 ± 1.8 | 2.00 ± 0.01 | 4.57 ± 0.69 | 54.1 ± 8.1 | 0.57 ± 0.13 | 0.03 ± 0.01 | 660 ± 126 | 12.1 ± 1.2 | ||
| AG | – | 0.05 ± 0.01 | 14.9 ± 2.1 | 2.38 ± 1.07 | 0.09 ± 0.03 | 1.69 ± 0.52 | 29.7 ± 8.3 | 18.6 ± 6.3 | |||
| – | 0.04 ± 0.00 | 15.6 ± 1.0 | 2.00 ± 0.00 | 0.18 ± 0.05 | 4.38 ± 0.88 | 113 ± 33 | 25.6 ± 2.3 | ||||
| – | 0.05 ± 0.01 | 15.0 ± 2.2 | 4.17 ± 5.30 | 0.39 ± 0.10 | 9.32 ± 1.21 | 235 ± 49 | 25.0 ± 3.0 | ||||
| AA | – | 0.04 ± 0.01 | 17.9 ± 4.1 | 6.13 ± 5.76 | 0.30 ± 0.19 | 7.06 ± 3.60 | 127 ± 75 | 17.6 ± 3.0 | |||
| – | 0.04 ± 0.00 | 19.8 ± 2.2 | 6.17 ± 3.21 | 0.56 ± 0.11 | 16.9 ± 3.0 | 409 ± 65 | 24.4 ± 1.7 | ||||
| – | 0.04 ± 0.01 | 19.5 ± 3.3 | 5.33 ± 2.56 | 1.51 ± 0.35 | 45.3 ± 6.8 | 1,082 ± 162 | 24.0 ± 1.4 | ||||
| Oral | AR | 1.61 | – | – | – | – | – | – | – | – | – |
| 3.22 | – | – | – | – | – | – | – | – | – | ||
| 8.06 | – | – | – | – | – | – | – | – | – | ||
| AA | – | 0.004 ± 0.001 | 196 ± 49 | 15.0 ± 0.0 | 0.01 ± 0.00 | 1.81 ± 0.58 | – | – | 14.4 ± 8.2 | 385 ± 158 | |
| – | 0.004 ± 0.001 | 176 ± 40 | 30.0 ± 16.5 | 0.03 ± 0.02 | 3.63 ± 1.27 | – | – | 10.8 ± 6.2 | 177 ± 51 | ||
| – | 0.004 ± 0.001 | 182 ± 27 | 17.5 ± 6.1 | 0.10 ± 0.03 | 12.3 ± 4.2 | – | – | 32.4 ± 16.2 | 154 ± 49 | ||
| AG | – | 0.001 ± 0.001 | 547 ± 175 | 57.5 ± 25.9 | 0.02 ± 0.01 | 5.42 ± 1.4 | – | – | 42.6 ± 16.6 | 437 ± 105 | |
| – | 0.003 ± 0.001 | 250 ± 70 | 67.5 ± 43.2 | 0.04 ± 0.04 | 5.39 ± 2.8 | – | – | 21.4 ± 9.6 | 268 ± 115 | ||
| – | 0.002 ± 0.000 | 307 ± 61 | 40.2 ± 12.5 | 0.08 ± 0.04 | 13.2 ± 8.2 | – | – | 59.4 ± 45.7 | 259 ± 52 |
K e elimination rate constant, T 1/2 half time, T max time of maximum concentration, C max maximum concentration, AUC 0–∞ area under the concentration curve from time zero to infinity, V/V_F volume of distribution, Cl/Cl_F clearance, AUMC area under the first moment curve, MRT mean residence time, “–” not available
Unlike the results observed after IV administration, only trace amounts of AR were identified in rat plasma after its oral administration at 2.4, 4.8 and 12 mg/kg. Since the concentrations of AR observed were lower than LLOQ at most time points, only the plasma concentration versus time profiles of AA and AG could be presented (Fig. 6, lower panels). As shown in Table I, the relatively short Tmax (around 0.5 and 1 h, respectively) and T1/2 (1–2 and 2–3 h, respectively) observed indicated rapid formation and elimination of both AA and AG. MR for the hydrolysis of AR was found to be increasing, from 25.0 ± 8.0% to 40.0 ± 14.0% to 47.6 ± 16.2%, while MR for glucuronidation was found to be decreasing from 75.0 ± 19.5% to 60.0 ± 31.2% to 52.4 ± 32.5%, at the administered doses of 2.4, 4.8, and 12 mg/kg, respectively. Linear kinetics of AA was observed after oral administrations of AR, evidenced by linear correlations between Cmax and AUC0–∞ of the analyte and AR dose (r2 > 0.99). However, Cmax and AUC0–∞ of AG were not dose proportional at the highest dose. These results suggested that glucuronidation had a higher tendency to be saturated than hydrolysis after oral administration of AR. The absorbed fraction F of AR calculated by NCA was 0.0583 ± 0.0226 (mean ± standard deviation), indicating its rather low oral bioavailability even with AA and AG taken into account.
Integration of In Vitro and In Vivo Data by Compartmental Analysis and Modeling
As shown in Fig. 6, the integrated model can describe the elimination of AR, AG, and AA from the central compartment simultaneously with reasonable pharmacokinetic parameters after both IV and oral administrations at all dose levels tested. The data estimated by the integrated model was presented as estimate ± standard error. The absorption rate constant (Ka) and V of AR in central compartment were estimated to be 0.0101 ± 0.0020 min−1 and 0.283 ± 0.011 L, respectively. Ke of AR was estimated to be near zero, suggesting that all the absorbed AR was converted to AA or AG in vivo prior to its elimination. In the meantime, Ke of AA and AG were estimated to be 0.0768 ± 0.0061 and 0.215 ± 0.019 min−1, respectively, indicating rather rapid elimination of both major metabolites. The scale factors accounting for individual differences of anatomical parameters were all between 0.8 and 1.5 (Table II), indicating a reasonable deviation of actual anatomical parameters from the literature values. The low F of AR calculated (0.0862 ± 0.0147) could be explained by the extensive metabolism followed by efflux of the resulting metabolites during the absorption process, as demonstrated in our previous single-pass rat intestinal perfusion study (22). Furthermore, the objective function value and residual were calculated to be −2,145.12 and 0.0199 ± 0.0060, respectively, which were considered reasonable for such a small population size (37,38).
Table II.
Parameter Estimates of the Integrated PK Model and Results of the Bootstrap Internal Validation Procedure
| Parameters | Original dataset | Bootstrap datasets (n = 2,000) | ||
|---|---|---|---|---|
| Estimate ± SE | Mean ± SD | Median | 2.5th–97.5th percentiles | |
| Fixed effect parameters | ||||
| K a (AR) (1/min) | 0.0101 ± 0.0020 | 0.0105 ± 0.0305 | 0.00977 | 0.007–0.016 |
| K e (AR) (1/min) | 0 (fixed) | 0 (fixed) | 0 (fixed) | – |
| K e (AG) (1/min) | 0.213 ± 0.019 | 0.215 ± 0.019 | 0.214 | 0.183–0.249 |
| K e (AA) (1/min) | 0.0768 ± 0.0061 | 0.0819 ± 0.0090 | 0.0783 | 0.071–0.095 |
| V (L) | 0.283 ± 0.011 | 0.279 ± 0.011 | 0.278 | 0.258–0.302 |
| K m1 (μmol/L) | 204 (fixed) | 204 (fixed) | 204 (fixed) | – |
| V m1 (μmol/min) | 12.4 (fixed) | 12.4 (fixed) | 12.4 (fixed) | – |
| K m2 (μmol/L) | 13.5 (fixed) | 13.5 (fixed) | 13.5 (fixed) | – |
| V m2 (μmol/min) | 0.292 (fixed) | 0.292 (fixed) | 0.292 (fixed) | – |
| K m3 (μmol/L) | 230 (fixed) | 230 (fixed) | 230 (fixed) | – |
| V m3 (μmol/min) | 1.23 (fixed) | 1.23 (fixed) | 1.23 (fixed) | – |
| SF1 | 0.907 ± 0.272 | 0.885 ± 0.283 | 0.836 | 0.528–1.418 |
| SF2 | 0.874 ± 0.043 | 0.881 ± 0.053 | 0.881 | 0.795–0.967 |
| SF3 | 1.43 ± 0.10 | 1.41 ± 0.10 | 1.41 | 1.249–1.577 |
| F1 | 0.0862 ± 0.0147 | 0.0852 ± 0.0162 | 0.0853 | 0.059–0.112 |
| Prop.RE | 4.37 ± 0.61 | 4.20 ± 0.55 | 4.31 | 3.302–5.057 |
| Add.RE | 0.0849 ± 0.0187 | 0.0906 ± 0.0181 | 0.0859 | 0.067–0.123 |
| Residual | 0.0199 ± 0.0060 | 0.0155 ± 0.0020 | 0.0153 | 0.012–0.019 |
SE standard error, K a absorption rate constant, K e elimination rate constant, V volume of distribution, V m1 , V m2, and V m3 maximum rate of metabolism in intestine, plasma, and liver of arctigenin, respectively, K m1 , K m2 , and K m3 Michaelis constant of metabolism in intestine, plasma, and liver of arctigenin, respectively, SF 1 , SF 2 , and SF 3 scale factor of metabolism in intestine, plasma, and liver of arctigenin, respectively, “–” not available, F1 fraction absorbed, Prop.RE proportional residual error, Add.RE additive residual error
To ensure the validity of the developed model, parameter estimates for each of the re-sampled bootstrap datasets were obtained by fitting them to the integrated PK model determined based on the original dataset, with 95% confidence intervals (95% CIs) of all model parameters calculated. As demonstrated in Table II, the medians/means of the bootstrap estimates were found to be comparable to those obtained from the original dataset. In addition, for the VPC plots, a total of 1,000 datasets were simulated from the integrated PK model developed based on the original dataset. As shown in Fig. 7, the 2.5th, 50th, and 97.5th percentiles of simulated datasets matched the distribution of the original datasets well, indicating good predictability of our integrated PK model.
Fig. 7.
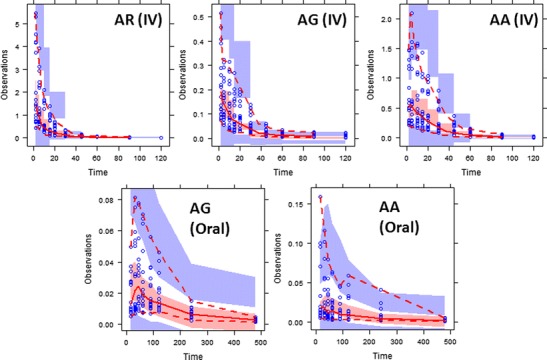
Visual predictive check (VPC) plots of the integrated PK model for arctigenin (AR) and its metabolites (AG and AA) after intravenous (IV) or oral administration. The gray-shaded areas represent the 95% confidence intervals of the corresponding 2.5th, 50th, and 97.5th percentiles of the simulated data; the red-dashed areas represent the 2.5th and 97.5th percentiles of the observed data; and red solid line represents median of the observed data
DISCUSSION
In Vitro and In Vivo Characterization of AR Pharmacokinetics
As demonstrated in the current study, AR underwent rapid hydrolysis to form AA in rat plasma, which could not be further metabolized via either glucuronidation or demethylation. Hydrolysis was further confirmed to be the dominating biotransformation pathway after IV injection of AR. On the other hand, the larger MR for glucuronidation observed after oral administration could be explained by the higher susceptibility of AR to intestinal and hepatic first-pass metabolism. As demonstrated in the rat intestinal single-pass perfusion model (22) and the current rat intestinal microsomal incubation studies, enterocytes play an important role in AR glucuronidation with much greater Clint in intestine (60.8 L/min) than in liver (5.21 L/min) and hydrolysis in plasma (22.2 L/min) (calculated based on parameters listed in Table II).
Enterohepatic circulation of glucuronides has been suggested to play an essential role in the absorption and metabolism of plant polyphenols, especially isoflavones (39–41). Glits et al. reported the high recovery of conjugated plant lignans in the ileum of pigs after a rye-bread diet, which could serve as an indirect evidence for enterohepatic circulation of plant lignans (42). The current report provides concrete evidence of enterohepatic circulation of plant lignans in rats by direct identification of conjugated metabolites of plant lignans in bile and demonstration of conversion of conjugated metabolites to parent compounds through hydrolysis in intestinal content.
Furthermore, identification of the secondary metabolite DA-diG in bile solved the discrepancy between our findings and previous studies regarding biotransformation of AR. It was believed that demethylation was the predominating metabolic pathway of AR as demonstrated in previous in vitro studies (19–21). Our in vitro rat liver microsomal incubation studies also revealed that AR could be efficiently demethylated in RLM. However, the amount of DA identified in rat plasma after either IV or oral administration was found to be trivial. Identification of DA-diG in bile suggested that DA formed either in intestine lumen or liver could be further conjugated to this secondary metabolite followed by direct secretion back into intestine lumen through bile without necessarily entering systemic blood circulation, which would further explain the absence of demethylated metabolites in plasma.
Establishment of Integrated Semi-mechanistic PK Model
Since the current model aims to describe simultaneously both AR and its in vivo metabolites, pharmacokinetic parameters for both parent and metabolites are essential. However, related parameters for metabolites are generally limited due to lack of authentic standards and the follow-up pharmacokinetic studies on the metabolites themselves. Thus, simplification and estimation of the parameters regarding metabolites are necessary for the establishment of our current model.
Estimation of Volume of Distributions for AR Metabolites
Since AA was formed by esterase hydrolysis of AR in blood (virtual representation of central compartment), it is reasonable to deduce that AA was distributed in the central compartment after formation. On the other hand, since AG is formed in either enterocytes or liver, separate formation compartments of AG (AG in enterocytes and AG in liver) were placed outside the central compartment during the early development of this model. Fitting results showed that the transfer rate of AG from either enterocytes or liver to the central compartment is rather high (data not shown), which may be due to the highly perfused nature of both intestine and liver. For this reason, the transfer between central compartment and either enterocytes or liver was not included in the final model. AG was considered to be immediately distributed into the central compartment after formation. Regarding the distribution of AA and AG into peripheral compartments, since only the concentrations of AA and AG in the central compartment were available, it was impossible to determine whether the disappearance of metabolites was due to direct elimination from the central compartment or irreversible transfer to a peripheral site. Furthermore, considering the high formation rate and elimination rate of AG and AA, it is reasonable to assume that their distribution to peripheral compartments is rather limited. Therefore, no peripheral compartment was included in the final model. Finally, since insufficient data was available for estimation of the relationship between V2, V3, and V4, equal volumes of distribution were assumed for AR, AA, and AG in order to make the model identifiable. The reasonableness of such assumption could be supported by the fact that V generated from the integrated model was close to the V of AR calculated from NCA.
Estimation of Ke of AR
Ke of AR was found to be extremely low, suggesting no direct elimination of AR from the central compartment, which could be buttressed by the fact that no AR was found in the urine sample collected during the in vivo pharmacokinetic study (data not shown). Thus, Ke of AR was fixed to be zero in the current model. On the other hand, the fact that Ke of AR itself was already found to be close to zero with only AA and AG taken into consideration could serve as indirect support for the decision to include only AA and AG in the PK model as major metabolites of AR.
Modeling of Intestinal Absorption and Metabolism Processes of AR
Since only AA and AG could be detected in the blood after oral administration, it was suggested that all the AR absorbed was metabolized to either AA or AG before entering the systemic circulation. Considering the much higher efficiency of glucuronidation in intestine than that in liver, it was deduced that the AG found in blood after oral administration was mainly from intestinal metabolism during AR absorption. Furthermore, it was demonstrated in the previous single-pass intestinal perfusion study that AG and AA formed during absorption would be transported to both intestinal lumen and mesenteric blood (22). Since the concentrations of AR, AA, and AG in intestine cells are unavailable, it is impossible to evaluate the metabolism during absorption based only on the AG and AA concentrations in the systemic circulation. Thus, to simplify the model, absorption and metabolism of AR were considered separately. AR was regarded as being absorbed into the compartment of AR in enterocytes (Compt. 5) first. Then, Ki and Kb were used to represent the formation rate constants of AG in enterocytes and AA in plasma after oral administration, respectively.
This developed semi-mechanistic model combined a traditional compartmental model with in vitro observations of drug biotransformation, providing more bio-relevant descriptions of ADME processes of AR. For example, Ke of AR itself was found to be quite high by NCA, as listed in Table I. However, once the biotransformation of AR to its metabolites was considered simultaneously as in the newly developed model, Ke of AR was found to be close to zero, suggesting that direct elimination of AR was insignificant. Since the pharmacokinetic profiles of the parent compound and metabolites were evaluated separately, the traditional compartmental model analysis could not clearly distinguish whether the disappearance of the parent compound was due to direct elimination of itself or biotransformation of the parent compound into metabolites. In this circumstance, the elimination of AR could be overestimated by the traditional compartmental analysis due to its extensive metabolism to either AG or AA. Furthermore, due to the involvement of specific mechanistic considerations of biotransformation, the current developed model managed to explain the dose range tested. In addition, the calculation of F by NCA was completely based on AUC, with the elimination rates of the analytes arbitrarily assumed to be equal. While for the integrated PK model, F was calculated as the fraction being absorbed into enterocytes with no assumption regarding elimination, which would appear to be more bio-relevant.
CONCLUSION
This report provides a systematic investigation of in vitro and in vivo pharmacokinetic profiles of AR and its major metabolites. Extensive hepatic and intestinal as well as GI content metabolism of AR and its metabolites were demonstrated in vitro. Rapid formation and elimination of AA and AG were also observed after both oral and IV administration of AR. A semi-mechanistic PK model integrating in vitro and in vivo parameters was successfully developed to describe the biotransformation process of AR at various doses via both oral and IV administration routes.
Electronic supplementary material
(DOCX 23.4 kb)
ACKNOWLEDGMENTS
This work is supported by Direct Grant 3800005 from the Chinese University of Hong Kong and research fund 7010213 from the Health Authority of Hong Kong.
References
- 1.Kim Y, Hollenbaugh JA, Kim DH, Kim B. Novel PI3K/Akt inhibitors screened by the cytoprotective function of human immunodeficiency virus type 1 Tat. PLOS One. 2011;6. doi 10.1371/journal.pone.0021781. [DOI] [PMC free article] [PubMed]
- 2.Yang Z, Liu N, Huang B, Wang Y, Hu Y, Zhu Y. Effect of anti-influenza virus of arctigenin in vivo. Zhongyaocai. 2005;28:1012–1014. [PubMed] [Google Scholar]
- 3.Hayashi K, Narutaki K, Nagaoka Y, Hayashi T, Uesato S. Therapeutic effect of arctiin and arctigenin in immunocompetent and immunocompromised mice infected with influenza A virus. Biol Pharm Bull. 2010;33:1199–1205. doi: 10.1248/bpb.33.1199. [DOI] [PubMed] [Google Scholar]
- 4.Kou XJ, Qi SM, Dai WX, Luo L, Yin ZM. Arctigenin inhibits lipopolysaccharide-induced iNOS expression in RAW264.7 cells through suppressing JAK-STAT signal pathway. Int Immunopharmacol. 2011;11:1095–1102. doi: 10.1016/j.intimp.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Tsai WJ, Chang CT, Wang GJ, Lee TH, Chang SF, Lu SC, et al. Arctigenin from Arctium lappa inhibits interleukin-2 and interferon gene expression in primary human T lymphocytes. Chin Med. 2011;6. doi 10.1186/1749-8546-6-12. [DOI] [PMC free article] [PubMed]
- 6.Hyam SR, Lee IA, Gu W, Kim KA, Jeong JJ, Jang SE, et al. Arctigenin ameliorates inflammation in vitro and in vivo by inhibiting the PI3K/AKT pathway and polarizing M1 macrophages to M2-like macrophages. Eur J Pharmacol. 2013;708:21–29. doi: 10.1016/j.ejphar.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Cho MK, Jang YP, Kim YC, Kim SG. Arctigenin, a phenylpropanoid dibenzylbutyrolactone lignan, inhibits MAP kinases and AP-1 activation via potent MKK inhibition: the role in TNF-alpha inhibition. Int Immunopharmacol. 2004;4:1419–1429. doi: 10.1016/j.intimp.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Z, Yan J, Jiang W, Yao X-g, Chen J, Chen L, et al. Arctigenin effectively ameliorates memory impairment in Alzheimer’s disease model mice targeting both beta-amyloid production and clearance. J Neurosci. 2013;33:13138–13149. doi: 10.1523/JNEUROSCI.4790-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strimpakos AS, Saif MW. Update on phase I studies in advanced pancreatic adenocarcinoma. Hunting in darkness. JOP. 2013;14:354–358. doi: 10.6092/1590-8577/1664. [DOI] [PubMed] [Google Scholar]
- 10.Kang K, Lee HJ, Kim CY, Lee SB, Tunsag J, Batsuren D, et al. The chemopreventive effects of Saussurea salicifolia through induction of apoptosis and phase II detoxification enzyme. Biol Pharm Bull. 2007;30:2352–2359. doi: 10.1248/bpb.30.2352. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Zhao F, Liu K. Induction of apoptosis of the human leukemia cells by arctigenin and its mechanism of action. Chin J Chem. 2008;43:542–547. [PubMed] [Google Scholar]
- 12.Hsieh CJ, Kuo PL, Hsu YC, Huang YF, Tsai EM, Hsu YL. Arctigenin, a dietary phytoestrogen, induces apoptosis of estrogen receptor-negative breast cancer cells through the ROS/p38 MAPK pathway and epigenetic regulation. Free Radic Biol Med. 2013;67C:159–170. doi: 10.1016/j.freeradbiomed.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Awale S, Lu J, Kalauni SK, Kurashima Y, Tezuka Y, Kadota S, et al. Identification of arctigenin as an antitumor agent having the ability to eliminate the tolerance of cancer cells to nutrient starvation. Cancer Res. 2006;66:1751–1757. doi: 10.1158/0008-5472.CAN-05-3143. [DOI] [PubMed] [Google Scholar]
- 14.Susanti S, Iwasaki H, Inafuku M, Taira N, Oku H. Mechanism of arctigenin-mediated specific cytotoxicity against human lung adenocarcinoma cell lines. Phytomedicine. 2013;21:39–46. doi: 10.1016/j.phymed.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Gu Y, Scheuer C, Feng D, Menger MD, Laschke MW. Inhibition of angiogenesis: a novel antitumor mechanism of the herbal compound arctigenin. Anticancer Drugs. 2013;24:781–791. doi: 10.1097/CAD.0b013e328362fb84. [DOI] [PubMed] [Google Scholar]
- 16.Shoeb M, Celik S, Jaspars M, Kumarasamy Y, MacManus SM, Nahar L, et al. Isolation, structure elucidation and bioactivity of schischkiniin, a unique indole alkaloid from the seeds of Centaurea schischkinii. Tetrahedron. 2005;61:9001–9006. doi: 10.1016/j.tet.2005.07.047. [DOI] [Google Scholar]
- 17.Yao X, Zhu F, Zhao Z, Liu C, Luo L, Yin Z. Arctigenin enhances chemosensitivity of cancer cells to cisplatin through inhibition of the STAT3 signaling pathway. J Cell Biochem. 2011;112:2837–2849. doi: 10.1002/jcb.23198. [DOI] [PubMed] [Google Scholar]
- 18.Hq W, Jj J, Wang J. Arctigenin enhances chemosensitivity to cisplatin in human nonsmall lung cancer H460 cells through downregulation of survivin expression. J Biochem Mol Toxicol. 2013;28:39–45. doi: 10.1002/jbt.21533. [DOI] [PubMed] [Google Scholar]
- 19.Kasper R, Gansser D, Doehmer J. Biotransformation of the naturally-occurring lignan (−)-arctigenin in mammalian-cell lines genetically-engineered for expression of single cytochrome-P450 isoforms. Planta Med. 1994;60:441–444. doi: 10.1055/s-2006-959528. [DOI] [PubMed] [Google Scholar]
- 20.Zhao YF, Song FR, Zhao LP, Liu SY. Studies on the biotransformation of arctigenin using electrospray ionization mass spectrometry. Chin J Chem. 2009;67:1123–1126. [Google Scholar]
- 21.Heinonen S, Nurmi T, Liukkonen K, Poutanen K, Wahala K, Deyama T, et al. In vitro metabolism of plant lignans: new precursors of mammalian lignans enterolactone and enterodiol. J Agric Food Chem. 2001;49:3178–3186. doi: 10.1021/jf010038a. [DOI] [PubMed] [Google Scholar]
- 22.Gao Q, Zhang Y, Wo S, Zuo Z. Extensive intestinal first-pass metabolism of arctigenin: evidenced by simultaneous monitoring of both parent drug and its major metabolites. J Pharm Biomed Anal. 2014;91:60–67. doi: 10.1016/j.jpba.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 23.Gao Q, Zhang Y, Wo S, Zuo Z. Hydrolysis is the dominating in vivo metabolism pathway for arctigenin: identification of novel metabolites of arctigenin by LC/MS/MS after oral administration in rats. Planta Med. 2013;79:471–479. doi: 10.1055/s-0033-1336555. [DOI] [PubMed] [Google Scholar]
- 24.Zhao D, Zhang Y, Xu C, Dong C, Lin H, Zhang L, et al. Pharmacokinetics, tissue distribution, and plasma protein binding study of platinum originating from dicycloplatin, a novel antitumor supramolecule, in rats and dogs by ICP-MS. Biol Trace Elem Res. 2012;148:203–208. doi: 10.1007/s12011-012-9364-2. [DOI] [PubMed] [Google Scholar]
- 25.Zou Q, Gu Y, Lu R, Zhang T, Zhao GR, Liu C, et al. Development of an LC/MS/MS method in order to determine arctigenin in rat plasma: its application to a pharmacokinetic study. Biomed Chromatogr. 2013;27:1123–1128. doi: 10.1002/bmc.2916. [DOI] [PubMed] [Google Scholar]
- 26.He F, Dou DQ, Hou Q, Sun Y, Kang TG. Pharmacokinetic study of arctigenin in rat plasma and organ tissue by RP-HPLC method. Nat Prod Res. 2013;27:903–906. doi: 10.1080/14786419.2012.666745. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, Li C, Lin G, Krajcsi P, Zuo Z. Hepatic metabolism and disposition of baicalein via the coupling of conjugation enzymes and transporters—in vitro and in vivo evidences. AAPS J. 2011;13:378–389. doi: 10.1208/s12248-011-9277-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagert KB. In: United States Pharmacopeia and National Formulary (USP-NF) Zhang Y, editor. Thousand Oaks: SAGE Publications; 2008. [Google Scholar]
- 29.Akao T, Kawabata K, Yanagisawa E, Ishihara K, Mizuhara Y, Wakui Y, et al. Baicalin, the predominant flavone glucuronide of scutellariae radix, is absorbed from the rat gastrointestinal tract as the aglycone and restored to its original form. J Pharm Pharmacol. 2000;52:1563–1568. doi: 10.1211/0022357001777621. [DOI] [PubMed] [Google Scholar]
- 30.FDA. Guidance for industry, bioanalytical method validation. US Food and Drug Administration, Centre for Drug Evaluation and Research, Rockville2001.
- 31.Martignoni M, Groothuis G, de Kanter R. Comparison of mouse and rat cytochrome P450-mediated metabolism in liver and intestine. Drug Metab Dispos. 2006;34:1047–1054. doi: 10.1124/dmd.105.009035. [DOI] [PubMed] [Google Scholar]
- 32.Miller DL. Rat small intestine—development, composition and effects of perfusion. Am J Dig Dis. 1971;16:247. doi: 10.1007/BF02235247. [DOI] [PubMed] [Google Scholar]
- 33.Kwon Y. Handbook of essential pharmacokinetics, pharmacodynamics and drug metabolism for industrial scientists: Springer; 2002. 277 p.
- 34.Gardiner P, Paine SW. The impact of hepatic uptake on the pharmacokinetics of organic anions. Drug Metab Dispos. 2011;39:1930–1938. doi: 10.1124/dmd.111.039842. [DOI] [PubMed] [Google Scholar]
- 35.Teiber JF, Billecke SS, La Du BN, Draganov DI. Estrogen esters as substrates for human paraoxonases. Arch Biochem Biophys. 2007;461:24–29. doi: 10.1016/j.abb.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 36.Teiber JF, Draganov DI, La Du BN. Lactonase and lactonizing activities of human serum paraoxonase (PON1) and rabbit serum PON3. Biochem Pharmacol. 2003;66:887–896. doi: 10.1016/S0006-2952(03)00401-5. [DOI] [PubMed] [Google Scholar]
- 37.Syvanen S, Schenke M, van den Berg DJ, Voskuyl RA, de Lange EC. Alteration in P-glycoprotein functionality affects intrabrain distribution of quinidine more than brain entry—a study in rats subjected to status epilepticus by Kainate. AAPS J. 2012;14:87–96. doi: 10.1208/s12248-011-9318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johansson CC, Gennemark P, Artursson P, Abelo A, Ashton M, Jansson-Lofmark R. Population pharmacokinetic modeling and deconvolution of enantioselective absorption of eflornithine in the rat. J Pharmacokinet Pharmacodyn. 2013;40:117–128. doi: 10.1007/s10928-012-9293-x. [DOI] [PubMed] [Google Scholar]
- 39.Setchell KD. Phytoestrogens: the biochemistry, physiology, and implications for human health of soy isoflavones. Am J Clin Nutr. 1998;68:1333–1346. doi: 10.1093/ajcn/68.6.1333S. [DOI] [PubMed] [Google Scholar]
- 40.Moon YJ, Sagawa K, Frederick K, Zhang S, Morris ME. Pharmacokinetics and bioavailability of the isoflavone biochanin A in rats. AAPS J. 2006;8:433–442. doi: 10.1208/aapsj080351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, Lin H, Hu M. Metabolism of flavonoids via enteric recycling: role of intestinal disposition. J Pharmacol Exp Ther. 2003;304:1228–1235. doi: 10.1124/jpet.102.046409. [DOI] [PubMed] [Google Scholar]
- 42.Glitso LV, Mazur WM, Adlercreutz H, Wahala K, Makela T, Sandstrom B, et al. Intestinal metabolism of rye lignans in pigs. Br J Nutr. 2000;84:429–437. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 23.4 kb)