

Abstract
Hsp70, Hsp32, and Hsp27 were induced by celastrol in rat cerebral cortical cultures at dosages that did not affect cell viability. Pronounced differences were observed in the cellular localization of these heat shock proteins in cell types of cerebral cortical cultures. Celastrol-induced Hsp70 localized to the cell body and cellular processes of neurons that were identified by neuron-specific βIII-tubulin. Hsp70 was not detected in adjacent GFAP-positive glial cells that demonstrated a strong signal for Hsp27 and Hsp32 in both glial cell bodies and cellular processes. Cells in the cerebral cortex region of the brain are selectively impacted during the progression of Alzheimer’s disease which is a “protein misfolding disorder.” Heat shock proteins provide a line of defense against misfolded, aggregation-prone proteins. Celastrol is a potential agent to counter this neurodegenerative disorder as recent evidence indicates that in vivo administration of celastrol in a transgenic model of Alzheimer’s reduces an important neuropathological hallmark of this disease, namely, amyloid beta pathology that involves protein aggregation.
Keywords: Hsp70 (HSPA1A), Hsp32, Hsp27 (HSPB1), Cerebral cortical cultures, Celastrol, Alzheimer’s disease
Introduction
As average life expectancy lengthens, the prevalence of neurodegenerative diseases is increasing; hence, the search for effective treatment and preventive measures is imperative. Neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, and amyotropic lateral sclerosis (ALS) have been termed “protein misfolding disorders” that are characterized by the accumulation of misfolded, aggregation-prone proteins in neural cells (Muchowski and Wacker 2005; Asea and Brown 2008; Brown 2008). Manipulation of the cellular stress response involving the induction of heat shock proteins offers a potential approach to counter misfolded proteins that trigger pathogenic cascades resulting in neurodegenerative diseases (Asea and Brown 2008). Heat shock proteins are protein repair agents that provide a line of defense against misfolded, aggregation-prone proteins (Muchowski and Wacker 2005; Lanneau et al. 2010; Stetler et al. 2010).
A collaborative drug screen aimed at identifying therapeutic agents against neurodegenerative diseases from a panel of existing drugs and natural products has identified celastrol as a potential neuroprotective candidate based on an assay for ability to suppress protein aggregation (Abbott 2002; Heemskerk et al. 2002). In vivo administration of celastrol in a transgenic model of Alzheimer’s disease has been shown to reduce an important neuropathological aspect of this “protein misfolding” disorder, namely, amyloid beta pathology that involves protein aggregation (Paris et al. 2010). Cerebral cortical cultures have been utilized as a model system to investigate aging of the nervous system (Lesuisse and Martin 2002). Cells in the cerebral cortex of the brain are selectively impacted during the progression of Alzheimer’s disease (Martin 1999; Duyckaerts et al. 2009; Stranahan and Mattson 2010). Recently, we have demonstrated that celastrol, but not classical heat shock treatment, induces a set of heat shock proteins at 24 h in cerebral cortical cultures (Chow et al. 2013). In the present investigation, we examine the localization of celastrol-induced Hsp70, Hsp32, and Hsp27 in cell types of the cerebral cortical cultures following celastrol dosages that do not affect cell viability. Hsp27 is the product of the HSPB1 gene, and Hsp70 includes the product of the HSPA1A gene.
Material and methods
Cerebral cortical cultures
Primary cortical cultures were prepared from cerebral cortices of 19 days embryonic rats (E19). Timed pregnant Sprague-Dawley rats were obtained from Charles River Laboratories (Saint-Constant, Quebec, Canada). E1 was considered as the day on which a vaginal plug was observed. Cerebral cortices were gently dissociated with a plastic pipette after digestion with trypsin (TripLE, Invitrogen, Burlington, ON, Canada) at 37 °C. Dissociated cells were resuspended in minimal essential medium (Gibco, Burlington, ON, Canada) supplemented with 10 % fetal calf serum, 5.56 mM glucose, and 2 mM glutamine. Cell viability was assessed by trypan blue exclusion, and live cells were counted, and plated at a final density of 1.8 × 105 cells/cm2 on poly-ornithine (Sigma, Mississauga, ON, Canada) coated culture dishes (Sarstedt, Montreal, QC, Canada), or laminin/poly-l-lysine-coated culture glass slides (BD, Mississauga, ON, Canada). Cultures were incubated at 37 °C in an atmosphere of 5 % CO2 in Neurobasal media with B27 supplement (Gibco) for 11 days. Extensive neuronal and glial cellular processes were observed as shown in Figs. 4, 5, and 6. All procedures using animals were approved by the Animal Care Committee of the University of Toronto and were in accordance with the guidelines established by the Canadian Council on Animal Care.
Fig. 4.

Localization of Hsp70 in cerebral cortical cultures following induction by celastrol. a Celastrol-induced Hsp70 (red) localized to neurons identified by neuronal-specific βIII-tubulin (blue, upper panels) and was not detectable in GFAP-positive glial cells (green, lower panels). Scale bar = 20 μM. b Higher magnification indicated that Hsp70 (red) was detected in neuronal cell bodies (arrow) and neuronal cellular processes (arrowheads) that were positive for neuron-specific βIII-tubulin (blue). Scale bar = 20 μM
Fig. 5.

Cellular localization of celastrol-induced Hsp27. a Celastrol-induced Hsp27 (red) co-localized with GFAP-positive (green) glial cell bodies (arrow) and glial cellular processes (arrowheads, upper panels) and was not detected in βIII-tubulin-positive neurons (blue, lower panels). Scale bar = 20 μM. b Higher magnification demonstrated a punctate Hsp27 signal (red, arrowheads) in GFAP-positive glial cellular processes (green). Scale bar = 20 μM
Fig. 6.

Localization of Hsp32 in cerebral cortical cultures following induction by celastrol. a Celastrol-induced Hsp32 (red) co-localized with GFAP-positive (green) glial cell bodies (arrow) and glial cellular processes (arrowheads, upper panels) and not with βIII-tubulin positive neurons (blue). Scale bar = 20 μM. b Higher magnification showed a punctate Hsp32 signal (red, arrowheads) in GFAP-positive glial cellular processes (green). Scale bar = 20 μM
Celastrol treatment and cell viability assays
The cerebral cortical cultures were incubated in the presence of celastrol at the specified concentrations for 24 h prior to analysis by immunoblotting. Celastrol was obtained from Gaia Chemical Corporation (Gaylordsville, CT, USA) and dissolved in DMSO as the vehicle. Cell viability was assayed using a combination of three stains. CellTrace calcein violet AM (Invitrogen, Burlington, ON, Canada) was used for identifying live cells. LIVE/DEAD fixable blue dead cell stain (Invitrogen) is taken up selectively by dead cells. Anti-MAP2 antibody (ab5392, Abcam) was used to visualize neuronal cellular processes.
Western blotting analysis
Cerebral cortical cultures were solubilized in Laemmli buffer and boiled for 20 min. Lowry assays with an additional deoxcholate-trichloroacetic procedure for removal of interfering substances were performed for protein quantification (Peterson 1977). Equal loadings of 30 μg of protein per lane were separated by 12 % SDS-PAGE using the Mini-PROTEAN 3 Electrophoresis Module Assembly (Bio-Rad Laboratories, Hercules, CA) with a stacking gel of 4 %, using the standard buffer system of Laemmli before transfer to nitrocellulose membranes. Western blotting was performed with antibodies specific to Hsp70 (ADI-SPA-810, Enzo Life Sciences, Farmingdale, NY), Hsp32 (ADI-OSA-111, Enzo), and Hsp27 (ADI-SPA-801, Enzo) (Chow and Brown 2007; Chow et al. 2010). Anti-β-tubulin antibody (MAB3408, Chemicon, Temecula, CA, USA) was employed to detect tubulin as loading control. Horseradish peroxidase-conjugated secondary antibodies (Sigma) were detected by enhanced chemiluminescence assay (Amersham, Piscataway, NJ, USA). Western blots representative of three experimental repeats are shown.
Immunocytochemistry
Cerebral cortical culture cells treated with celastrol for 24 h were fixed with 4 % paraformaldehyde in phosphate buffer solution (PBS) at room temperature for 30 min. Cells were then permeabilized with 0.1 % Triton X-100 in PBS containing 100 mM glycine for 30 min, washed, blocked with 5 % fetal bovine serum (FBS) in PBS overnight, and incubated with the indicated primary antibodies in 1 % FBS in PBS overnight. Primary antibodies against microtubule-associated protein 2 (MAP2) (ab5392, Abcam), βIII-tubulin (T8660, Sigma Aldrich), doublecortin (Ab18723, Abcam), Hsp70 (ADI-SPA-810, Enzo Life Sciences, Farmingdale, NY), Hsp32 (ADI-OSA-111, Enzo), and Hsp27 (ADI-SPA-801, Enzo) were employed. Cells were then washed and incubated with fluorescent secondary antibodies at 1:1,000 dilution (DyLight, Jackson ImmunoResearch Labs; Alexa, Invitrogen). Culture slides were mounted and subjected to fluorescence microscopy imaging using AxioCam HRm camera with ApoTome module on Axiovert 200M microscope (Carl Zeiss).
Results
Induction of heat shock proteins in cerebral cortical cultures by celastrol
The effect of celastrol at concentrations ranging from 0.25 to 1.5 μM on the induction of heat shock proteins in rat cerebral cortical cultures is shown in Fig. 1. Celastrol dosages of 1 μM, and above, induced Hsp70, Hsp32, and Hsp27. Basal expression of Hsp32 and Hsp27 was noted in the cerebral cortical cultures; however, Hsp70 was only detected after 1–1.5 μM celastrol.
Fig. 1.

Heat shock protein induction by celastrol in cerebral cortical cultures. Celastrol induced Hsp70, Hsp32, and Hsp27 in cultures derived from the rat cerebral cortex. Basal expression of Hsp32 and Hsp27 was noted; however, Hsp70 was detected only after celastrol addition at 1–1.5 μM. NT no treatment control, DMSO vehicle control
Cell viability is not affected by celastrol concentrations of up to 1.5 μM
In the cerebral cortical cultures, cellular viability was assayed by CellTrace calcein (green), identifying live cells, MAP2 (red) a marker for neuronal cellular processes and dead cell stain (blue). As shown in Fig. 2, celastrol concentrations from 0.75 to 1.5 μM did not affect cellular viability (green) or the morphology of cellular processes (red). As a positive control, increasing the celastrol to 3 μM triggered cell death in the cerebral cortical cultures as indicated by cells binding dead cell stain (blue). Cellular processes were retracted at 3 μM celastrol but were not affected by celastrol concentrations of up to 1.5 μM. In the subsequent immunocytochemical analysis, a celastrol dosage of 1 μM was employed.
Fig. 2.

Effect of celastrol concentration on cell viability in cerebral cortical cultures. Cell viability effects were not detected at celastrol concentrations of up to 1.5 μM. Cell viability was assayed by CellTrace calcein (green) identifying live cells, MAP2, a marker for neuronal cellular processes (red) and dead cell stain (blue). Celastrol treatment at 3 μM was included as a positive control that resulted in the retraction of cellular processes (red), a decrease in the live cell signal (green), and an increase in dead cell signal (blue). Scale bar = 20 μM
Identification of neuronal cells in cerebral cortical cultures
Three neuronal marker proteins were investigated for their ability to visually identify neuronal cells in cerebral cortical cultures by immunocytochemical analysis (Fig. 3). βIII-tubulin (green) is a cytoskeletal protein that is specific for differentiated neurons and their cellular processes and is not present in glia cells (Katsetos et al. 2003). MAP2 (red) is a microtubule-associated protein found in neuronal cellular processes (Farah and Leclerc 2008). Doublecortin (blue) is a neuronal marker protein that is exclusively expressed in post-mitotic neurons with preferential localization at the ends of neuronal cellular processes (Friocourt et al. 2003). As shown in Fig. 3, βIII-tubulin (green) resulted in superior visualization of neuronal cells in the cerebral cortical cultures, identifying both neuronal cell bodies (arrow) and their cellular processes (arrowheads); hence, it was employed in the subsequent immunocytochemical analysis.
Fig. 3.
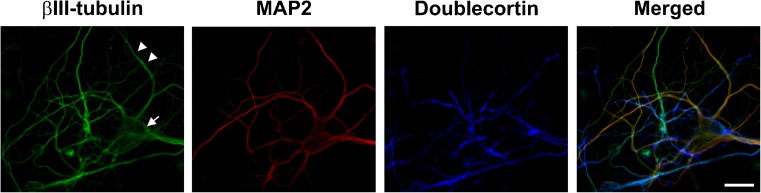
Marker proteins for the identification of neuronal cells in cortical cultures. βIII-tubulin (green), MAP2 (red), and doublecortin (blue) were examined. βIII-tubulin resulted in superior visualization of neuronal cells in the cerebral cortical cultures, identifying both neuronal cell bodies (arrow) and neuronal cellular processes (arrowheads). Scale bar = 20 μM
Celastrol-induced Hsp70 localizes to neuronal cells in cerebral cortical cultures
Immunocytochemistry revealed that the Hsp70 protein (red) that was induced in cerebral cortical cultures by celastrol, co-localized with the neuron-specific βIII-tubulin marker protein (blue) as shown in Fig. 4a, upper panels. In contrast, celastrol-induced Hsp70 (red) did not co-localize with glial fibrillary acidic protein (GFAP)-positive glial cells and their long cellular processes (green) that were present in the cerebral cortical cultures (Fig. 4a, lower panels). As previously shown in Fig. 1 by Western blot analysis, basal expression of Hsp70 was not detected in the cerebral cortical cultures, and Hsp70 induction required a celastrol dosage of 1 μM. Higher magnification (Fig. 4b) showed that celastrol-induced Hsp70 (red) was detected in neuronal cell bodies (indicated by arrow) and neuronal cellular processes (arrowheads) that were positive for neuron-specific βIII-tubulin (blue).
Hsp27 is induced in glial cells by celastrol
As shown in Fig. 5a (upper panels), celastrol-induced Hsp27 (red) co-localized with GFAP-positive (green) glial cell bodies (arrow) and glial cellular processes (arrowheads) and was not detected in βIII-tubulin-positive neurons (blue, Fig. 5a, lower panels) in the cerebral cortical cultures. At higher magnification (Fig. 5b), a punctate Hsp27 signal (red), indicated by arrowheads, was apparent in GFAP-positive glial cellular processes (green).
Celastrol-induced Hsp32 localizes to glial cell bodies and glial cellular processes
Celastrol-induced Hsp32 (red) demonstrated a striking co-localization with GFAP-positive (green) glial cell bodies (arrow) and glial cellular processes (arrowheads) in the cerebral cortical cultures (Fig. 6a, upper panels). At higher magnification, the Hsp32 signal (red), in GFAP-positive glial cellular processes (green) demonstrated a punctate appearance (Fig. 6b, arrowheads). Co-localization of Hsp32 (red) with βIII-tubulin-positive neuronal cells (blue) was not apparent in the cerebral cortical cultures (Fig. 6a, lower panels). Basal expression of Hsp32 and Hsp27 was noted in the cerebral cortical cultures by Western blotting (Fig. 1). Immunocytochemistry indicated that this basal expression was localized to GFAP-positive glial cells (data not shown).
Discussion
The present results indicate that celastrol induces Hsp70, Hsp32, and Hsp27, in rat cerebral cortical cultures at dosages that do not affect cell viability. The inducibility of a set of neuroprotective Hsps (Brown 2007, 2008; Sharp et al. 2013) in cerebral cortical cultures suggests that celastrol is a potential agent to counter Alzheimer’s disease, a neurodegenerative “protein misfolding” disorder that targets neural cells in the cerebral cortex. Hsps are “protein repair agents” that provide a line of defense against misfolded, aggregation-prone proteins (Muchowski and Wacker 2005; Lanneau et al. 2010; Stetler et al. 2010). Differing levels of constitutively expressed Hsps in different populations of neural cells could confer a variable buffering capacity against “protein misfolding disorders” that correlates with the relative frequencies of different neurodegenerative diseases (Chen and Brown 2007). The high frequency of Alzheimer’s disease relative to Parkinson’s disease and ALS may be due to the low level of constitutively expressed Hsc70 and Hsp27 in impacted cells in the cerebral cortex; hence, upregulation of Hsps could be a beneficial therapeutic strategy (Chen and Brown 2007; Brown 2008). Interestingly, in vivo administration of celastrol in a transgenic model of Alzheimer's disease has recently been shown to reduce an important neuropathological hallmark of this “protein misfolding” disorder, namely, amyloid beta pathology that involves protein aggregation (Paris et al. 2010). We have previously reported that celastrol, but not classical heat shock treatment, induces Hsps in cultures derived from the cerebral cortex (Chow et al. 2013). We have now investigated the pattern of that induction at the cellular level.
Striking cell type differences were observed in the pattern of celastrol-induced Hsps that was noted in neurons and glial cells that are present in cerebral cortical cultures. Celastrol induced Hsp70 in neurons with signal detected in both the neuronal cell body and neuronal cellular processes. Hsp70 induction was not detected in glial cells that were present in the cerebral cortical cultures and situated adjacent to neurons that exhibited Hsp70 induction in response to celastrol. In contrast, celastrol induced Hsp27 and Hsp32 in glial cells in the cerebral cortical cultures and not in adjacent neurons. Prominent Hsp27 and Hsp32 signals were noted in both glial cell bodies and also in glial cellular processes, where a punctate pattern of Hsp27 and Hsp32 was apparent. Oligodendrocytes have been reported in the cerebral cortical cultures (Ramakers et al. 1991). However, they do not appear to induce Hsps following celastrol as we did not observe any Hsp-positive cells in the culture that were not positive for either the GFAP or the neuronal marker.
During the progression of neurodegenerative diseases, disruption of synaptic function is an early event that precedes cell death (Shankar and Walsh 2009; Marcello et al. 2012; Sheng et al. 2012). Upregulation of Hsps and their transport to synapses could mitigate that synaptic dysfunction (Brown 2008). Our previous studies involving both the biochemical isolation of synaptic compounds and electron microscopy have shown that Hsp70 is associated with synaptic elements (Bechtold et al. 2000) and that Hsp27 and Hsp32 are transported into perisynaptic glial processes and subsequently associated with synaptic elements (Bechtold and Brown 2000). In addition, we have reported functional protection of synapses is conferred by prior stress that is sufficient to induce heat shock proteins (Karunanithi et al. 1999) and that transgenic overexpression of Hsp70 enhances synaptic protection (Karunanithi et al. 2002). Synapses are tri-partite structures composed of pre- and post-synaptic neuronal elements plus surrounding perisynaptic glial processes (Perea et al. 2009). Our present results indicate that celastrol induces Hsp70 in neurons and Hsp27 and Hsp32 in glial cells at dosages that do not affect cell viability in cerebral cortical cultures that are derived from a brain region that is impacted during Alzheimer’s disease. Celastrol has been reported to induce Hsps by acting on HSF1 (Westerheide et al. 2004; Klaic et al. 2012). Given that Hsp27 and Hsp32 are transported into perisynaptic glial processes and subsequently associated with synaptic elements (Bechtold and Brown 2000) that are Hsp70-positive (Bechtold et al. 2000), these heat shock proteins could be involved in mechanisms that mitigate against synaptic dysfunction that occurs during Alzheimer’s disease (Brown 2008). In vivo administration of celastrol in a transgenic model of Alzheimer’s disease reduces an important hallmark of the disease, namely, amyloid beta pathology that involves protein aggregation (Paris et al. 2010). Amyloid beta aggregation correlates with synaptic failure through the targeting of synaptic elements such as NMDA receptors (Parihar and Brewer 2010; Sheng et al. 2012).
Acknowledgments
This study was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) to I.R.B who also holds a Canada Research Chair (Tier I) in Neuroscience.
References
- Abbott A. Neurologists strike gold in drug screen effort. Nature. 2002;417:109. doi: 10.1038/417109a. [DOI] [PubMed] [Google Scholar]
- Asea AA, Brown IR, editors. Heat shock proteins and the brain: implications for neurodegenerative diseases and neuroprotection. New York: Springer Science Publishers; 2008. [Google Scholar]
- Bechtold DA, Brown IR. Heat shock proteins Hsp27 and Hsp32 localize to synaptic sites in the rat cerebellum following hyperthermia. Brain Res Mol Brain Res. 2000;75:309–320. doi: 10.1016/S0169-328X(99)00323-X. [DOI] [PubMed] [Google Scholar]
- Bechtold DA, Rush SJ, Brown IR. Localization of the heat-shock protein Hsp70 to the synapse following hyperthermic stress in the brain. J Neurochem. 2000;74:641–646. doi: 10.1046/j.1471-4159.2000.740641.x. [DOI] [PubMed] [Google Scholar]
- Brown IR. Heat shock proteins and protection of the nervous system. Ann N Y Acad Sci. 2007;1113:147–158. doi: 10.1196/annals.1391.032. [DOI] [PubMed] [Google Scholar]
- Brown IR. Heat shock proteins at the synapse: implications for functional protection of the nervous system. In: Asea AAA, Brown IR, editors. Heat shock proteins and the brain: implications for neurodegenerative diseases and neuroprotection. New York: Springer Science Publishers; 2008. pp. 239–254. [Google Scholar]
- Chen S, Brown IR. Neuronal expression of constitutive heat shock proteins: implications for neurodegenerative diseases. Cell Stress Chaperones. 2007;12:51–58. doi: 10.1379/CSC-236R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow AM, Brown IR. Induction of heat shock proteins in differentiated human and rodent neurons by celastrol. Cell Stress Chaperones. 2007;12:237–244. doi: 10.1379/CSC-269.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow AM, Mok P, Xiao D, Khalouei S, Brown IR. Heteromeric complexes of heat shock protein 70 (HSP70) family members, including Hsp70B′, in differentiated human neuronal cells. Cell Stress Chaperones. 2010;15:545–553. doi: 10.1007/s12192-009-0167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow AM, Tang DW, Hanif A, Brown IR. Induction of heat shock proteins in cerebral cortical cultures by celastrol. Cell Stress Chaperones. 2013;18:155–160. doi: 10.1007/s12192-012-0364-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- Farah CA, Leclerc N. HMWMAP2: new perspectives on a pathway to dendritic identity. Cell Motil Cytoskeleton. 2008;65:515–527. doi: 10.1002/cm.20284. [DOI] [PubMed] [Google Scholar]
- Friocourt G, Koulakoff A, Chafey P, Boucher D, Fauchereau F, Chelly J, Francis F. Doublecortin functions at the extremities of growing neuronal processes. Cereb Cortex. 2003;13:620–626. doi: 10.1093/cercor/13.6.620. [DOI] [PubMed] [Google Scholar]
- Heemskerk J, Tobin AJ, Bain LJ. Teaching old drugs new tricks. Meeting of the neurodegeneration drug screening consortium, 7–8 April 2002, Washington, DC, USA. Trends Neurosci. 2002;25:494–496. doi: 10.1016/S0166-2236(02)02236-1. [DOI] [PubMed] [Google Scholar]
- Karunanithi S, Barclay JW, Robertson RM, Brown IR, Atwood HL. Neuroprotection at Drosophila synapses conferred by prior heat shock. J Neurosci. 1999;19:4360–4369. doi: 10.1523/JNEUROSCI.19-11-04360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunanithi S, Barclay JW, Brown IR, Robertson RM, Atwood HL. Enhancement of presynaptic performance in transgenic Drosophila overexpressing heat shock protein Hsp70. Synapse. 2002;44:8–14. doi: 10.1002/syn.10048. [DOI] [PubMed] [Google Scholar]
- Katsetos CD, Legido A, Perentes E, Mork SJ. Class III beta-tubulin isotype: a key cytoskeletal protein at the crossroads of developmental neurobiology and tumor neuropathology. J Child Neurol. 2003;18:851–866. doi: 10.1177/088307380301801205. [DOI] [PubMed] [Google Scholar]
- Klaic L, Morimoto RI, Silverman RB. Celastrol analogues as inducers of the heat shock response. Design and synthesis of affinity probes for the identification of protein targets. ACS Chem Biol. 2012;7:928–937. doi: 10.1021/cb200539u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanneau D, Wettstein G, Bonniaud P, Garrido C. Heat shock proteins: cell protection through protein triage. Sci World J. 2010;10:1543–1552. doi: 10.1100/tsw.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesuisse C, Martin LJ. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol. 2002;51:9–23. doi: 10.1002/neu.10037. [DOI] [PubMed] [Google Scholar]
- Marcello E, Epis R, Saraceno C, Di Luca M. Synaptic dysfunction in Alzheimer’s disease. Adv Exp Med Biol. 2012;970:573–601. doi: 10.1007/978-3-7091-0932-8_25. [DOI] [PubMed] [Google Scholar]
- Martin JB. Molecular basis of the neurodegenerative disorders. N Engl J Med. 1999;340:1970–1980. doi: 10.1056/NEJM199906243402507. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Parihar MS, Brewer GJ. Amyloid-beta as a modulator of synaptic plasticity. J Alzheimers Dis. 2010;22:741–763. doi: 10.3233/JAD-2010-101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris D, Ganey NJ, Laporte V, Patel NS, Beaulieu-Abdelahad D, Bachmeier C, March A, Ait-Ghezala G, Mullan MJ. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2010;7:17. doi: 10.1186/1742-2094-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ, Raadsheer FC, Corner MA, Ramaekers FC, Van Leeuwen FW. Development of neurons and glial cells in cerebral cortex, cultured in the presence or absence of bioelectric activity: morphological observations. Eur J Neurosci. 1991;3:140–153. doi: 10.1111/j.1460-9568.1991.tb00074.x. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR, Zhan X, Liu DZ. Heat shock proteins in the brain: role of Hsp70, Hsp 27, and HO-1 (Hsp32) and their therapeutic potential. Transl Stroke Res. 2013;4:685–692. doi: 10.1007/s12975-013-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, Sudhof TC (2012) Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol 4 [DOI] [PMC free article] [PubMed]
- Stetler RA, Gan Y, Zhang W, Liou AK, Gao Y, Cao G, Chen J. Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog Neurobiol. 2010;92:184–211. doi: 10.1016/j.pneurobio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP. Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer’s disease. Neural Plast. 2010;2010:108190. doi: 10.1155/2010/108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]