

Abstract
SNCA/α-synuclein and its rare mutations are considered as the culprit proteins in Parkinson disease (PD). Wild-type (WT) SNCA has been shown to impair macroautophagy in mammalian cells and in transgenic mice. In this study, we monitored the dynamic changes in autophagy process and confirmed that overexpression of both WT and SNCAA53T inhibits autophagy in PC12 cells in a time-dependent manner. Furthermore, we showed that SNCA binds to both cytosolic and nuclear high mobility group box 1 (HMGB1), impairs the cytosolic translocation of HMGB1, blocks HMGB1-BECN1 binding, and strengthens BECN1-BCL2 binding. Deregulation of these molecular events by SNCA overexpression leads to autophagy inhibition. Overexpression of BECN1 restores autophagy and promotes the clearance of SNCA. siRNA knockdown of Hmgb1 inhibits basal autophagy and abolishes the inhibitory effect of SNCA on autophagy while overexpression of HMGB1 restores autophagy. Corynoxine B, a natural autophagy inducer, restores the deficient cytosolic translocation of HMGB1 and autophagy in cells overexpressing SNCA, which may be attributed to its ability to block SNCA-HMGB1 interaction. Based on these findings, we propose that SNCA-induced impairment of autophagy occurs, in part, through HMGB1, which may provide a potential therapeutic target for PD.
Keywords: Parkinson disease, SNCA, autophagy, HMGB1, corynoxine B
Introduction
Parkinson disease is characterized by the formation of SNCA-containing inclusions termed Lewy bodies and the degeneration of dopaminergic neurons in the midbrain. 1 Protein degradation pathways remove damaged or abnormally modified proteins in neurons, and thus play central roles in maintaining proper neuronal function. 1 Current literature provides evidence that the ubiquitin-proteasome system (UPS) and autophagy-lysosomal pathway (ALP) are primary mechanisms for the degradation of wild-type SNCA and its mutant variants (A53T and A30P). 1 - 3 It is generally accepted that under normal conditions unmodified soluble SNCA is recognized by the UPS and chaperone-mediated autophagy (CMA) and subsequently degraded. However, for the more insoluble oligomeric and aggregated SNCA, macroautophagy is the only mechanism for their clearance. 4 , 5
The initiation of macroautophagy (here referred to as autophagy) is regulated by multiple signaling pathways involving 2 macromolecular complexes: the MTOR -ULK1-ATG13-RB1CC1-C12orf44/ATG101 complex (the latter gene product, C12orf44, is also known as RGD1359310 in the rat and 9430023L20Rik in mice), and the BECN1-PIK3C3 (ortholog of yeast Vps34) complex. 6 BECN1 plays an essential role in autophagy initiation by interacting with various cofactors, 7 one of which is high mobility group box 1. 8 Autophagic stimuli trigger HMGB1 translocation from the nucleus into the cytosol, where it binds to BECN1 and results in dissociation of BECN1-BCL2, and subsequent induction of autophagy. 8 Autophagy is very important for preventing the accumulation of abnormal proteins, as evidenced by the neuronal accumulation of inclusion bodies in mice deficient for ATG5 or ATG7. 9 - 11 In contrast, overexpression of BECN1 (ortholog of yeast Vps30/Atg6) 12 or ATG7 13 activates autophagy, reduces accumulation of SNCA and attenuates associated neurodegeneration in SNCA-transgenic mice. Pharmacological induction of autophagy by a range of small molecules promotes the clearance of aggregate-prone proteins including SNCA species. 14 - 16
SNCA forms a reciprocal relationship with protein degradation pathways. 17 Both UPS and ALP activity decline with aging, which may result in SNCA accumulation due to impaired clearance. Conversely, increased SNCA burden and generation of aberrant species may further impair UPS and ALP functions. 17 However, conflicting results exist regarding whether SNCA inhibits or overactivates autophagy. 17 Some studies have reported that autophagic vacuoles are increased in cultured cells and transgenic mice overexpressing mutant or, in some cases, WT SNCA. 13 , 18 , 19 Conversely, other studies showed that overexpression of WT SNCA inhibited autophagy while the mutant SNCAA53T had no effect. 20 Variability in the findings might be a result of different experimental settings. 1
In this study, we set out to monitor the time-course changes in autophagy process affected by SNCA overexpression and to determine whether, and at what time points, WT and/or SNCAA53T inhibit autophagy in PC12 cells. Based on a previous report showing SNCA filaments bind to HMGB1 in brain tissues from PD patients, 21 we further determined the role of HMGB1 in SNCA overexpression-induced autophagy impairment and tested the effect of autophagy inducers during this process.
Results
Both WT and the A53T mutant SNCA inhibit autophagy in PC12 cells
In a time-course study using a stable inducible PC12 cell (iPC12) model 22 where the expression of hemagglutinin (HA)-tagged-SNCA (~20 kDa) can be induced by doxycycline (Dox), we found that the endogenous LC3-II (which correlates with autophagosome number) and BECN1 levels decreased significantly in cells overexpressing both WT and SNCAA53T at 24 h (Fig. 1A–C). The decrease in LC3-II was more significant in cells overexpressing WT than that of SNCAA53T. Notably, LC3-II levels increased at the earlier time points (1 to 6 h) and then decreased at 24 h in SNCAA53T-overexpressing cells, compared with controls (Fig. 1A–C). Since the decrease in LC3-II levels can be attributed to deficient LC3-II generation or excessive lysosomal degradation, the change in LC3-II and BECN1 levels induced by SNCA was also determined in the presence of chloroquine (CQ), which blocks lysosomal degradation. The decreased level of LC3-II, but not BECN1, could be restored by CQ treatment (Fig. 1D–F), indicating that overexpressed SNCA may negatively regulate BECN1 homeostatic levels.

Figure 1. WT and SNCAA53T overexpression inhibits basal autophagy in PC12 cells. Inducible PC12 cells (iPC12) were treated with 2 μg/ml doxycycline (Dox) in a 24 h time course (A) or for 24 h in the presence of chloroquine (CQ) (B). The expressions of induced hemagglutinin (HA)-SNCA, endogenous LC3-II and BECN1 were determined by western blotting. Relative intensity is normalized to that of ACTB/β-actin. Data are presented as the mean ± SD from 3 independent experiments. # P < 0.05 vs. uninduced control; *P < 0.05 vs. Dox treatment.
After confirming that SNCA overexpression inhibits basal autophagy at 24 h, we extended the time course and determined whether SNCA inhibits starvation-activated autophagy. iPC12 cells were treated with Dox for 24, 48, and 72 h respectively to induce SNCA expression and then starved by treatment with Earle's balanced salt solution (EBSS) for another 2 h. Overexpression of WT and SNCAA53T also inhibited starvation-activated autophagy at 24 h, evidenced by the decrease in LC3-II levels and the increase in SQSTM1 (formerly known as p62, a specific autophagy substrate) by Dox treatment for 24 h (Fig. 2A–C). Interestingly, no significant changes in the levels of LC3-II or SQSTM1 were observed after 48 and 72 h induction of SNCA, in comparison with the uninduced control groups at the corresponding time points (Fig. 2A–C). The results suggest that SNCA overexpression inhibits autophagy in a time-course-dependent manner. To exclude the possibility that Dox itself may affect the expression of LC3-II, BECN1, and SQSTM1, normal PC12 cells were treated with the same dosage of Dox for 24, 48, and 72 h respectively. No significant change in the levels of LC3-II, BECN1, and SQSTM1 was observed in normal PC12 cells treated with Dox (Fig. 2D), suggesting that autophagy inhibition is caused by induced SNCA, rather than Dox itself. To further confirm our finding, a PC12 cell line stably transfected with GFP-SNCA was established. We found that LC3-II and BECN1 levels also decreased in cells overexpressing both WT and SNCAA53T (Fig. 2E and F). The decreased level of LC3-II, but not BECN1, could be restored by CQ treatment, which is consistent with the results from the iPC12 cells (Fig. 1D).

Figure 2. WT and SNCAA53T overexpression inhibits starvation-activated autophagy in a time-course-dependent manner. (A) iPC12 cells were treated with 2 μg/ml Dox for 24, 48, and 72 h respectively and then starved by Earle's balanced salt solution (EBSS) treatment for 2 h. The expressions of LC3-II and SQSTM1 (p62) were determined by western blotting. (B and C) Relative intensity is normalized to that of ACTB. Data are presented as the mean ± SD from 3 independent experiments. *P < 0.05 vs. uninduced control at the corresponding time points. (D) Normal PC12 cells were treated with 2 μg/ml Dox for 24 h. The expressions of LC3-II, SQSTM1 and BECN1 were determined by western blotting. Experiments were performed 3 times with similar results and the representative blots were shown. (E) The expressions of LC3-II and BECN1 in PC12 cells constitutively expressing GFP-SNCA were determined by western blotting. Relative intensity is normalized to that of ACTB. Data are presented as the mean ± SD from 3 independent experiments. # P < 0.05 vs. untransfected control (UT); *P < 0.05 vs. SNCA transfection.
Effects of SNCA overexpression on cell viability, lysosome numbers, and proteasomal activities
In our experimental settings, we demonstrated that overexpression of both WT and SNCAA53T inhibits autophagy, and then the effect of SNCA overexpression on cell viability is evaluated by quantification of lactate dehydrogenase (LDH) release. We found that SNCA overexpression in iPC12 cells for 24 h caused mild cell injury at 48 and 72 h (Fig. S1A), which is consistent with the findings by Webb et al. 22 Considering the previous studies demonstrating the effects of SNCA on lysosomal and proteasomal system (reviewed by Xilouri et al. 17 ), we examined the lysosome numbers by LysoTracker staining and proteasomal activity by determination of polyubiquitinated proteins. As shown in Figure S1B, overexpression of WT SNCA for 24 h in iPC12 cells increased the fluorescence intensity of LysoTracker Red, indicating WT SNCA increases lysosome numbers. However, overexpression of SNCAA53T had no significant effect on lysosome at all the time points tested. SNCAA53T overexpression for 24 h caused an increase in high molecular mass (> 70 kDa) polyubiquitinated proteins in comparison with the uninduced control while WT SNCA had no obvious effect at all the time points tested (Fig. S1C). These results are consistent with the previous findings. 23 , 24
BECN1 overexpression activates basal autophagy and rescues SNCA-induced autophagy impairment
Becn1 gene transfer has been reported to activate autophagy and ameliorate neurodegeneration in SNCA models of PD. 12 In our cellular system, SNCA overexpression reduced endogenous BECN1 levels. Therefore we determined whether exogenous transfection of Becn1 could restore autophagy in our experimental settings. Becn1 transfection has no obvious effect on cell survival as evaluated by LDH assay (Fig. S2A). In uninduced iPC12 cells, overexpression of GFP-tagged BECN1 increased the level of LC3-II, compared with the untransfected or empty vector transfected controls (Fig. S2B–S2D). Dox treatment for 24 h induced significant increase in the levels of WT and SNCAA53T while significant decrease in the levels of LC3-II in iPC12 cells. Coexpression of GFP-BECN1 restored LC3-II levels and promoted the clearance of SNCA (Fig. S2B–S2D).
SNCA overexpression inhibits the cytosolic translocation of HMGB1
Autophagic stimuli trigger HMGB1 translocation from the nucleus into cytosol and subsequent induction of autophagy. 8 Therefore, we tested whether the inhibition of autophagy by overexpressing SNCA is mediated by HMGB1. In iPC12 (WT) cells treated with Dox for 24 h, the expression of HMGB1 decreased in the cytosolic fraction while it increased in the nuclear fraction (Fig. 3A and B), indicating that SNCA overexpression inhibits the cytosolic transportation of HMGB1. Autophagy inhibition by wortmannin (WM) further blocked the cytosolic translocation of HMGB1, while lysosomal inhibition by CQ had no obvious effect (Fig. 3A and B). To exclude the possibility that Dox itself may affect the cellular distribution of HMGB1, normal PC12 cells were treated with Dox (2 μg/ml) for 24 h. No obvious change in HMGB1 distribution was detected by Dox treatment (Fig. 3C). In uninduced (UI) iPC12 cells, starvation (EBSS treatment for 2 h) triggered the cytosolic translocation of HMGB1 (Fig. 3D). However, both WT and SNCAA53T overexpression (induced by Dox) blocked starvation-induced cytosolic translocation of HMGB1 (Fig. 3D and E).

Figure 3. SNCA overexpression inhibits the cytosolic translocation of HMGB1. (A) iPC12 cells (WT) were treated with 2 μg/ml Dox for 24 h in the presence of wortmannin (WM) or chloroquine (CQ). (A) The expression of HMGB1 in the cytosolic (Cyt.) and nuclear (Nuc.) fractions were determined by western blotting. ACTB and LMNB1 were used as loading controls of cytoplasmic and nuclear fraction respectively. (B) Data are presented as the mean ± SD from 3 independent experiments. # P < 0.05 vs. uninduced control; *P < 0.05 vs. Dox treatment. (C) Normal PC12 cells were treated with 2 μg/ml Dox for 24 h. The expression of HMGB1 in the cytosolic (Cyt.) and nuclear (Nuc.) fractions were determined by western blotting. Experiments were performed 3 times with similar results and the representative blots were shown. (D) iPC12 cells were treated with 2 μg/ml Dox for 24 h and then starved by Earle's balanced salt solution (EBSS) treatment for 2 h (UI: uninduced, I: induced). The distribution of HA-SNCA (green) and HMGB1 (red) were double stained. Arrows indicate cytosolic translocation of HMGB1 and *indicates nuclear retention of HMGB1. (E) Images of at least 400 cells for each treatment group were analyzed to obtain the mean nuclear and cytosolic HMGB1 intensity. Data are presented as the mean ± SD of 3 replicates in a representative experiment. # P < 0.05 vs. uninduced control; *P < 0.05 vs. uninduced starvation treatment.
SNCA binds to HMGB1 and inhibits HMGB1-BECN1 interaction
An early report showed SNCA filaments bound to HMGB1, a chromatin-associated nuclear protein in brain tissues from PD patients. 21 However, the consequence of SNCA-HMGB1 binding has not been determined. By coimmunoprecipitation experiments, we demonstrated that both endogenous (Fig. S3) and overexpressed SNCA (WT and A53T) interacted with endogenous HMGB1 in iPC12 cells (Fig. 4A and B). Since SNCA overexpression inhibited the expression of BECN1, we normalized the levels of immunoprecipitated BECN1 to the levels of BECN1 in whole cell lysates and found that the binding of endogenous BECN1 with HMGB1 decreased (Fig. 4B). Next, the endogenous BECN1 was coimmunoprecipitated with endogenous BCL2 in iPC12 cells. SNCA overexpression had no obvious effect on the expression of BCL2. However, the binding between BCL2 and BECN1 increased in SNCA-overexpressing cells (Fig. 4A and B), suggesting that SNCA-HMGB1 binding blocks HMGB1-BECN1 interaction and strengthens BECN1-BCL2 interaction. Furthermore, SNCA and endogenous BECN1 were coimmunoprecipitated with HMGB1 in the cytosolic and nuclear fraction of PC12 cells transfected with GFP-SNCA. The results demonstrated that both endogenous and overexpressed SNCA bound to HMGB1 in the cytosolic and nuclear fractions. SNCA-HMGB1 binding weakened the cytosolic HMGB1-BECN1 binding (Fig. 4C).

Figure 4. Interactions of SNCA-HMGB1, HMGB1-BECN1, and BECN1-BCL2. (A) Immunoprecipitation (IP) of overexpressed HA-SNCA and endogenous BECN1 with endogenous HMGB1 and immunoprecipitation of endogenous BECN1 with endogenous BCL2 in iPC12 cells treated with 2 μg/ml Dox for 24 h. Experiments were performed 3 times with similar results and the representative blots were shown. (B) Ratio of HMGB1-BECN1 and BCL2-BECN1 interaction. The levels of immunoprecipitated BECN1 were normalized to the corresponding levels of BECN1 in whole cell lysates. Data are presented as the mean ± SD from 3 independent experiments. *P < 0.05 vs. uninduced control. (C) Immunoprecipitation of SNCA and endogenous BECN1 with endogenous HMGB1 in cytosolic (Cyt.) and nuclear (Nuc.) fractions of PC12 cells transfected with GFP-SNCA. 0.5 mg cytosolic protein and 1 mg nuclear protein were used for immunoprecipitation, respectively. In parallel, 10 μg cytosol protein and 20 μg nuclear protein were loaded as inputs. ACTB and LMNB1 were used as loading control of cytoplasmic and nuclear fraction respectively. Experiments were performed 3 times with similar results and the representative blots were shown.
HMGB1 mediates SNCA-induced autophagy impairment
As shown above, overexpressed SNCA binds to HMGB1, blocks HMGB1 cytosolic translocation and HMGB1-BECN1 interaction. To validate the roles of HMGB1 in this process, we first knocked down Hmgb1 by siRNA prior to inducing SNCA overexpression. Using a pool of specific siRNAs targeting Hmgb1, 25 endogenous HMGB1 expression was suppressed by more than 90% (Fig. 5A). In uninduced iPC12 cells, Hmgb1 knockdown inhibited basal autophagy, as shown by the decrease in LC3-II levels (Fig. 5A). iPC12 cells treated with Dox for 24 h inhibited basal autophagy. However, Dox treatment failed to inhibit autophagy in HMGB1-depleted iPC12 cells, compared with uninduced iPC12 cells deficient in HMGB1 (Fig. 5B). The data suggest that HMGB1 mediates the autophagy inhibition induced by SNCA. Next, we determined whether overexpression of HMGB1 could restore the deficient autophagy. iPC12 cells were treated with Dox and transfected with Flag-HMGB1 plasmids or empty vector for 24 h. The results showed that Hmgb1 transfection had no obvious effect on the level of overexpressed SNCA (WT and A53T). However, the decrease in LC3-II by SNCA overexpression was restored by transfection of Hmgb1, but not by the empty vector (Fig. 5C and D), indicating that although HMGB1 overexpression could not clear SNCA, it can, however restore autophagy induction.

Figure 5. Effects of Hmgb1 knockdown and overexpression on SNCA-induced autophagy impairment. (A) iPC12 cells were transfected with Hmgb1 siRNA or control siRNA for 48 h and then induced to express SNCA for another 24 h. The expression of SNCA, HMGB1 and LC3 were determined by western blot. Experiments were performed 3 times with similar results and the representative blots were shown. (B) Relative intensity of LC3-II is normalized to the corresponding ACTB levels. Data are presented as the mean ± SD from 3 independent experiments. *P < 0.05 vs. uninduced and untransfected (UT) controls. (C) iPC12 cells were treated with 2 μg/ml Dox and transfected with Flag-HMGB1 plasmid or the relative empty vector for 24 h. The expressions of LC3-II and HA-SNCA were determined by western blotting. Experiments were performed 3 times with similar results and the representative blots were shown. (D) Relative levels of LC3-II are normalized to those of ACTB. Data are presented as the mean ± SD from 3 independent experiments. # P < 0.05 vs. uninduced and untransfected (UT) controls; *P < 0.05 vs. induced and N.T. controls.
Corynoxine B restores the deficient cytosolic translocation of HMGB1 and autophagy
Based on the above findings, we further wondered whether autophagy inducers could restore the impaired autophagy caused by SNCA overexpression. In iPC12 cells, the decrease in LC3-II and BECN1 levels was reversed by treatment with corynoxine B (Cory B, a natural autophagy inducer previously discovered by us 16 ) and rapamycin (Rap, an MTOR-dependent autophagy inducer) (Fig. 6A–C). Cory B could upregulate LC3-II and BECN1 protein levels in normal PC12 cells and inhibit the decrease in LC3-II and BECN1 in PC12 cells transfected with GFP-SNCA (data not shown). Notably, the deficient cytosolic translocation of HMGB1 induced by SNCA overexpression could be restored by treatment with Cory B, but not by Rap (Fig. 6D and E). Cory B had no obvious effect on the expression and cellular distribution of HMGB1 in normal PC12 cells (data not shown), suggesting that Cory B may act on HMGB1 under SNCA-overexpression conditions. Autophagy inhibition by WM diminished the effect of Cory B on restoring the cytosolic translocation of HMGB1 while lysosome inhibition by chloroquine (CQ) had no obvious effect (Fig. 6E). Since Cory B, but not Rap, could restore the deficient cytosolic translocation of HMGB1 induced by SNCA, we hypothesized that Cory B may interfere SNCA-HMGB1 interaction. By coimmunoprecipitation experiment, we found that the levels of SNCA binding to HMGB1 decreased significantly in iPC12 cells treated with Cory B for 12 h and 24 h, compared with that of SNCA induction alone (Fig. 6F and G). It is notable that Cory B treatment for 24 h could promote, though not significantly, the clearance of SNCA. After normalizing the level of immunoprecipitated SNCA to the level of SNCA in whole cell lysates, there is a significant decrease in SNCA-HMGB1 binding in the presence of Cory B. This result indicates that the formation of SNCA-HMGB1 complex could be inhibited by Cory B.

Figure 6. Corynoxine B restores the deficient cytosolic translocation of HMGB1 and autophagy. iPC12 cells were treated with 2 μg/ml Dox for 24 h in the presence of corynoxine B (Cory B), rapamycin (Rap), wortmannin (WM) or chloroquine (CQ). (A) The expressions of induced HA-SNCA, endogenous LC3-II and BECN1 were determined by western blotting. (B and C) Data are presented as the mean ± SD from 3 independent experiments. # P < 0.05 vs. uninduced control; *P < 0.05 vs. Dox treatment. (D) The expression of HMGB1 in the cytosolic and nuclear fractions were determined by western blotting. ACTB and LMNB1 were used as relative loading controls. Experiments were performed 3 times with similar results and the representative blots were shown. (E) Data are presented as the mean ± SD from 3 independent experiments. # P < 0.05 vs. uninduced control; *P < 0.05 vs. Dox treatment; **P < 0.05 vs. Dox + Cory B treatment. (F) iPC12 cells (WT) were treated with Dox for 12 and 24 h in the presence or absence of Cory B. Induced HA-SNCA was coimmunoprecipitated with HMGB1. The levels of SNCA binding to HMGB1 and in whole cell lysates were analyzed by western blotting. (G) Relative levels of SNCA in Dox treatment group are set to 1 and all other values are calculated relative to it. Data are presented as the mean ± SD from 3 independent experiments. *P < 0.05 vs. Dox treatment.
Discussion
In the current study, we demonstrated that overexpression of WT and SNCAA53T inhibits autophagy and revealed the possible involvement of HMGB1 in this process. In iPC12 cells, overexpressed SNCA binds to HMGB1 and inhibits its translocation from nuclei into cytosol, which may thus lead to the observed decrease in HMGB1-BECN1 interaction, increase in BCL2-BECN1 interaction. Deregulation of these events by SNCA may disturb autophagy induction (Fig. 7). SNCA-induced autophagy inhibition can be blocked by Hmgb1 knockdown. Overexpression of HMGB1 restores the deficient autophagy induced by SNCA, though it is not sufficient to clear SNCA. Cory B could restore the deficient cytosolic translocation of HMGB1 and autophagy in cells overexpressing SNCA, which may be attributed to its ability to interfere with SNCA-HMGB1 interaction (Fig. 7).
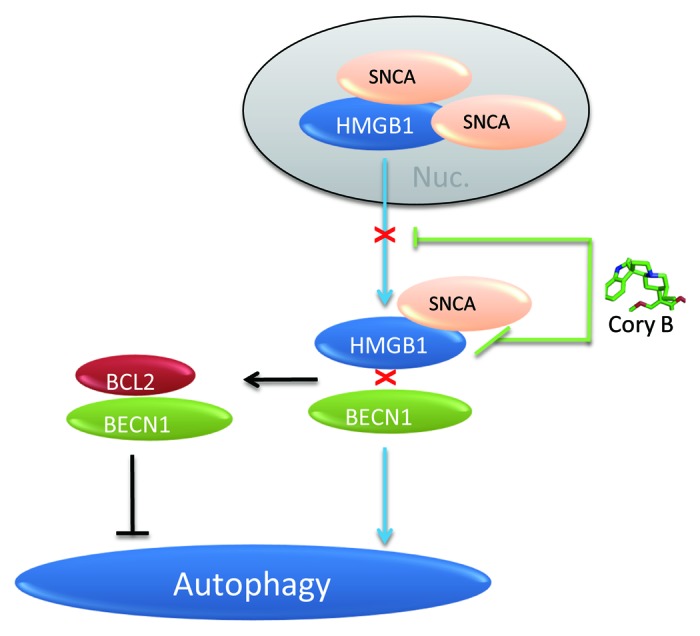
Figure 7. Diagram illustrating the involvement of HMGB1 in autophagy impairment triggered by SNCA overexpression. SNCA binds to both cytosolic and nuclear HMGB1 and inhibits HMGB1 translocation into the cytosol, which may thus lead to the decrease in HMGB1-BECN1 interaction and increase in BCL2-BECN1 interaction. Corynoxine B restores the deficient cytosolic translocation of HMGB1 and rescues the impaired autophagy, which may be attributed by its ability to disrupt SNCA-HMGB1 interaction.
In the previous studies, inconsistent results have been reported concerning the effects of SNCA overexpression on autophagy process. WT but not SNCAA53T was shown to inhibit autophagy. 20 In contrast, SNCAA53T was shown to inhibit autophagy while WT SNCA had no obvious effect in a recent study. 26 However, the time-course changes in autophagy status had not been determined in these studies. To evaluate whether SNCA overexpression inhibits or activates autophagy, a range of factors should be considered, such as different neuronal cell lines used, transfection methods, SNCA species used, relative transgenic levels, and duration of expression. The inducible SNCA cell lines generated by the Tet-On system 22 are ideal models to investigate the effect of SNCA overexpression on autophagy status since the expression of SNCA can be induced in a time-dependent manner. Therefore, the dynamic change in autophagy status by increasing dosage of SNCA and incubation time can be monitored.
In a 24 h time-course study using these cell lines, we showed that WT SNCA inhibited autophagy in a time-dependent manner while SNCAA53T activated autophagy at the earlier time-points (1 to 6 h) and then inhibited autophagy after 24 h induction (Fig. 1A–C). However, no significant change in autophagy status was observed after 48 and 72 h induction of both WT and SNCAA53T (Fig. 2A–C). As discussed in a previous study, WT and SNCAA53T may impair autophagy in a similar way. 20 The earlier activation of autophagy by SNCAA53T may reflect a compensatory response to its inhibition of CMA. 27 Our results suggest that overexpression of SNCA triggers a dynamic change in autophagy process. The relative SNCA levels and the duration of overexpression as well as the interactions of CMA are important factors determining the autophagy status. These factors may contribute to the different outcomes of the previous studies. 20 , 26 Regardless of these factors, both WT and SNCAA53T inhibit autophagy at certain stages of overexpression.
At different stages of SNCA pathology, the successive failure of protein degradation pathways (UPS, CMA, and autophagy) ultimately contributes to neuronal cell death. 1 , 17 In our experimental settings without severe cell death, WT SNCA inhibits autophagy and increases lysosome numbers while SNCAA53T inhibits both autophagy and proteasomal activity at 24 h. However, the effects of WT and SNCAA53T on these proteolytic systems are not obvious at later time points (48 and 72 h), which may be the result of compensatory responses of surviving cells to the stress.
Despite the previous study demonstrating that WT SNCA compromises autophagy via Rab1a inhibition, 20 how SNCA overexpression inhibits autophagy is yet largely unknown. Our first observation is that SNCA overexpression inhibited the expression of BECN1 (Fig. 1A). In SNCA animal models of PD, Becn1 gene-transfer activates autophagy and ameliorate neurodegeneration. 12 In our cell model, Becn1 transfection activates basal autophagy and rescues SNCA-induced autophagy impairment (Fig. S2). Since the regulation of BECN1 expression is controlled by several factors, 7 how SNCA overexpression deregulates BECN1 needs to be further investigated.
Inspired by an early report showing that SNCA filaments bind to HMGB1 in brain tissues from PD patients, 21 we further demonstrated that both endogenous and overexpressed SNCA bind to HMGB1. Importantly, for the first time we determined that SNCA-HMGB1 interaction may contribute to autophagy inhibition. HMGB1, a nuclear DNA binding factor and a secreted protein important for cell death and survival, is directly involved in the positive regulation and maintenance of autophagy. 8 Depletion of HMGB1 inhibits autophagy while inhibition of autophagy limits the cytosolic translocation of HMGB1. 8 We first showed that SNCA overexpression inhibited basal (Fig. 3A and B) and starvation-triggered (Fig. 3D and E) cytosolic translocation of HMGB1. The deficient translocation of HMGB1 was further exacerbated by inhibition of autophagy by WM (Fig. 3B). Taken together, our data suggest the deficient HMGB1 translocation triggered by SNCA overexpression may contribute to autophagy impairment. However, the mechanism(s) how SNCA overexpression blocks the cytosolic translocation of HMGB1 is still unknown. Delineating the binding sites of SNCA-HMGB1 complex would be helpful for resolving this uncertainty.
Cytosolic HMGB1-BECN1 binding results in disassociation of the BCL2-BECN1 complex, 28 which is an important event during autophagy induction. 8 In our study, SNCA overexpression weakens HMGB1-BECN1 interaction and strengthens BECN1-BCL2 interaction in the cytosolic fractions (Fig. 4). The result may be interpreted from 2 perspectives. On the one hand, the decreased translocation of HMGB1 into cytosol may directly lead to decreased HMGB1-BECN1 binding. On the other, SNCA may competitively bind to HMGB1 and thus prevent the binding of BECN1 to HMGB1.
To further define the role of HMGB1 in SNCA-triggered autophagy impairment, we determined the effects of knockdown and overexpression of HMGB1 on this process. As expected, transfection of Hmgb1 siRNA inhibited basal autophagy. Importantly, SNCA overexpression failed to inhibit autophagy in HMGB1-defective cells (Fig. 5A and B), which indicate HMGB1 mediates SNCA-induced autophagy inhibition. Conversely, HMGB1 overexpression can restore the deficient autophagy. However, unlike BECN1, HMGB1 overexpression cannot promote the clearance of SNCA, indicating that autophagy induction by different factors may not necessarily contribute equally to the degradation of SNCA. Notably, a new study has identified HMGB1 as a chaperone-like molecule, which interacts with polyglutamine (polyQ) and reduces aggregation and toxicity of polyQ, the pathogenic cause of Huntington disease. 29 The finding, together with the previous report that HMGB1 interacts with SNCA filaments in PD brain samples, 21 revealed a possible connection between HMGB1 and the pathogenic protein aggregates associated with neurodegenerative diseases including PD and Huntington disease. Meanwhile, SNCA and polyQ have been shown to impair autophagy, without a fully understood mechanism. 20 , 30 Our study, for the first time, revealed the role of HMGB1 in the SNCA-mediated autophagy inhibition thus raising the possibility that sequestration of HMGB1 by pathogenic protein aggregates might be a common mechanism in the pathogenic protein aggregates-mediated autophagy dysfunction.
Finally, we tested the effects of autophagy inducers on SNCA-HMGB1 interaction. Our findings showed that although both rapamycin and Cory B restored the decreased expression levels LC3-II and BECN1 in SNCA-overexpressing cells, only Cory B restored the deficient cytosolic translocation of HMGB1 (Fig. 6). We further showed that SNCA-HMGB1 interaction was weakened in the presence of Cory B (Fig. 6). Therefore, the restoration of HMGB1 cytosolic translocation and subsequent autophagy by Cory B in cells overexpressing SNCA may be mediated by its ability to block SNCA-HMGB1 interaction. Our data indicates that specifically disrupting the binding of SNCA to HMGB1 with small molecules may be a rational approach to restore autophagy.
Overall, our study has identified the roles of SNCA-HMGB1 interaction in autophagy impairment. Based on our results, we propose HMGB1 as a new target for drug intervention to restore the deficient autophagy caused by SNCA. Given the central role of SNCA in PD pathogenesis, our data add new clues for the deregulation of autophagy in PD. Moving forward, in vivo studies using adenovirus-mediated SNCA overexpression animal models 31 will be performed to confirm our in vitro findings.
Materials and Methods
Reagents and antibodies
Chloroquine (C6628), anti-FLAG® M2 (F1804) antibody, MG132 (C2211) and doxycycline (D9891) were purchased from Sigma-Aldrich. Rapamycin (R5000) and wortmannin (W-2990) were purchased from LC Laboratories. Anti-GFP (sc-8334), ACTB/β-actin (sc-47778), LMNB1/Lamin B1 (H-90) (sc-20682), BCL2 (C-2) (sc-7382) antibodies, BCL2-agarose conjugate (sc-492 AC), rat Hmgb1 siRNA (sc270015) and control siRNA (sc37007) were purchased from Santa Cruz Biotechnology. Anti-LC3 (2775), anti-SQSTM1/p62 (5114), anti-ubiquitin (3936), mouse anti-rabbit IgG-HRP (conformation specific) (L27A9) (5127) and Protein A agarose beads (9863) were purchased from Cell Signaling Technology. α-Syn (SNCA) antibody (610786) was purchased from BD Transduction Laboratories. BECN1 (beclin 1, NB110-87318) and HMGB1 (NBP1-40650) antibodies were purchased from Novus Biologicals. Anti-HA (12CA5) antibody and LDH Cytotoxicity Detection Kit was purchased from Roche Applied Science. DMEM (11965-126), horse serum (16050-122), fetal bovine serum (FBS) (16000-044), hygromycin B (10687-010), G418 (10131-035), goat anti-mouse-HRP (626520), goat anti-rabbit-HRP (G21234), LysoTracker Red DND-99 (L-7528), Alexa Fluor® 488 goat anti-mouse IgG (A-11001) and Alexa Fluor® 594 goat anti-rabbit IgG (A-11012) were purchased from Invitrogen. Corynoxine B was purchased from Aktin Chemicals (IRH-121319).
Cell lines and cell culture
Inducible PC12 cell lines (iPC12) overexpressing SNCA (WT and A53T) 22 were generous gifts from Prof. David C. Rubinsztein at Cambridge University. They were grown in DMEM supplemented with 10% horse serum, 5% FBS, 100 units/ml penicillin/streptomycin, 50 μg/ml G418, and 150 μg/ml hygromycin B at 37 °C, 10% CO2. Cells were induced to express SNCA with 2 μg/ml doxycycline (Dox). PC12 cells constitutively expressing GFP-SNCA were selected using 800 μg/ml G418 and maintained in 400 μg/ml G418.
LDH assay
The cytotoxicity was determined by measurement of lactate dehydrogenase (LDH) release from damaged cells using LDH Kit (Roche) according to the manufacturer’s protocol.
Plasmids and transfection
pcDNA3.1 Flag hHMGB1 plasmid was a generous gift from Dr. Yasuhiko Kawakami (University of Minnesota, USA). 32 GFP-SNCA plasmids (WT and A53T) were kind gifts from Dr. Zhuo-Hua Zhang at Central South University (China). pEGFP-N3-BECN1 plasmid was provided by Prof. Yanxiang Zhao (Hong Kong Polytechnic University, Hong Kong). 33 Cells were transfected with the plasmids using Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer’s protocol.
Hmgb1 siRNA
Hmgb1 siRNA is a pool of 3 target-specific 19- to 25-nt siRNAs designed to knock down Hmgb1 gene expression. 25 Briefly, iPC12 cells were seeded in 12-well plates at 30% confluency. Hmgb1 siRNA or the control siRNA was mixed with Lipofectamine 2000 and added into each well. Cells were incubated at 37 °C in a CO2 incubator for 48 h to knock down the Hmgb1 gene and then treated with Dox (2 μg/ml) for 24 h to induce SNCA expression.
Immunofluorescent staining and visualization of lysosomes
Inducible PC12 cells were seeded on coverslips placed in 24-well plates. Cells were treated with (or without) 2 μg/ml doxycycline (Dox) for 24 h and then starved by EBSS treatment for 2 h. Thereafter, cells were fixed with 3.7% paraformaldehyde, blocked with 5% BSA and double stained with anti-HA (mouse, 1:200) and HMGB1 (rabbit, 1:500) antibodies overnight at 4 °C. Alexa Fluor® 488 (green) and Alexa Fluor® 594 (red) secondary antibodies (1:500) were added for 1 h at room temperature. After nuclear staining with DAPI, the slides were mounted with FluorSave reagent (Calbiochem, 345789). Cells were visualized using an Eclipse 80i (Nikon Instruments Inc.) fluorescence microscope (60×). Images of at least 400 cells for each treatment group were analyzed to obtain the mean nuclear and cytosolic HMGB1 intensity using a previously described protocol. 34 For labeling of lysosomes, cells were incubated with LysoTracker Red (100 nM) in PBS for 5 min at 37 °C. After fixation and nuclear staining, cells were visualized under fluorescent microscope (40×). LysoTracker intensity per cell was quantified by ImageJ software and data were presented as mean fluorescence value of each group compared with relative control cells. 35
Immunoprecipitations and western blot analyses
For whole cell lysates, cells were lysed on ice in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, 0.35% sodium deoxycholate, 1 mM EDTA, 1% NP40) with complete protease inhibitor mixture (Roche Applied Science, 04693124001). Cytosolic and nuclear fractions were isolated using protocols similar to those described previously. 36 HMGB1 antibody or BCL2-agarose conjugate were added to the lysates and rotated overnight at 4 °C, and then 20 μl of protein A agarose beads (for HMGB1 precipitation) were added for 3 h. Immunoprecipitates were washed 3 times with 1× cell lysis buffer. Whole cell lysates and immunoprecipitated proteins were boiled in sample buffer, separated by 10–15% SDS-PAGE, transferred, and blotted with the antibodies described. The blots were then incubated with secondary antibodies at room temperature for 1 h. For immunoprecipitation (IP) followed by western blotting, conformation-specific secondary antibodies which only react with native IgG were used where necessary to eliminate the denatured IgG light and heavy chains of the primary antibodies. The protein signals were detected by ECL kit (Pierce, 32106) and quantified using the ImageJ software.
Statistical analysis
Each experiment was performed at least 3 times, and the results were presented as mean ± SD. One-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls test using the SigmaPlot 11.0 software packages. A probability value of P < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
The authors would like to thank Prof. David C Rubinsztein for providing the PC12 inducible SNCA cell lines (WT and A53T), Dr Y Kawakami for providing the pcDNA3.1 Flag hHMGB1 plasmid, Prof Zhuo-Hua Zhang for providing the GFP-SNCA plasmids, and Dr Yan-xiang Zhao for providing the GFP-BECN1 plasmid. This study was supported by NSFC-RGC join research grant (RGC-N_HKBU 213/11) from the Hong Kong Government and research grants SCM/10-11/03, IRMS/12-13/1A and FRG II/12-13/038 from Hong Kong Baptist University. J-XS is supported by the Postdoctoral Fellowship Scheme of School of Chinese Medicine, Hong Kong Baptist University. The authors specially thank Ms Loretta Ho for her financial support for this study and Dr Han-Ming Shen for his valuable comments and revision of this manuscript. The authors would also like to thank Dr Martha Dahlen for her English editing of this manuscript.
Glossary
Abbreviations:
- ALP
autophagy-lysosomal pathway
- BECN1
Beclin 1, autophagy related
- CMA
chaperone-mediated autophagy
- Cory B
corynoxine B
- CQ
chloroquine
- Dox
doxycycline
- EBSS
Earle's balanced salt solution
- FBS
fetal bovine serum
- HA
hemagglutinin
- HMGB1
high mobility group box 1
- LC3
microtubule-associated protein 1 light chain 3
- PD
Parkinson disease
- Rap
rapamycin
- SNCA
synuclein, alpha (non A4 component of amyloid precursor)
- UPS
ubiquitin-proteasome system
- WM
wortmannin
- WT
wild type
Disclosure of Potential Conflicts of Interest
There are no potential conflicts of interest to be disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/26751
References
- 1. Ebrahimi-Fakhari D, Wahlster L, McLean PJ. . Protein degradation pathways in Parkinson’s disease: curse or blessing. Acta Neuropathol 2012; 124:153 - 72; http://dx.doi.org/ 10.1007/s00401-012-1004-6; PMID: 22744791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shacka JJ, Roth KA, Zhang J. . The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci 2008; 13:718 - 36; http://dx.doi.org/ 10.2741/2714; PMID: 17981582 [DOI] [PubMed] [Google Scholar]
- 3. Pan T, Kondo S, Le W, Jankovic J. . The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 2008; 131:1969 - 78; http://dx.doi.org/ 10.1093/brain/awm318; PMID: 18187492 [DOI] [PubMed] [Google Scholar]
- 4. Cook C, Stetler C, Petrucelli L. . Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb Perspect Med 2012; 2:a009423; http://dx.doi.org/ 10.1101/cshperspect.a009423; PMID: 22553500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ding WX, Yin XM. . Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008; 4:141 - 50; PMID: 17986870 [DOI] [PubMed] [Google Scholar]
- 6. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. . Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383 - 435; http://dx.doi.org/ 10.1152/physrev.00030.2009; PMID: 20959619 [DOI] [PubMed] [Google Scholar]
- 7. Kang R, Zeh HJ, Lotze MT, Tang D. . The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571 - 80; http://dx.doi.org/ 10.1038/cdd.2010.191; PMID: 21311563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, et al. . Endogenous HMGB1 regulates autophagy. J Cell Biol 2010; 190:881 - 92; http://dx.doi.org/ 10.1083/jcb.200911078; PMID: 20819940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. . Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/ 10.1038/nature04724; PMID: 16625204 [DOI] [PubMed] [Google Scholar]
- 10. Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. . Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/ 10.1038/nature04723; PMID: 16625205 [DOI] [PubMed] [Google Scholar]
- 11. Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. . Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J Neurosci 2012; 32:7585 - 93; http://dx.doi.org/ 10.1523/JNEUROSCI.5809-11.2012; PMID: 22649237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. . Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 2009; 29:13578 - 88; http://dx.doi.org/ 10.1523/JNEUROSCI.4390-09.2009; PMID: 19864570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E. . Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010; 5:e9313; http://dx.doi.org/ 10.1371/journal.pone.0009313; PMID: 20174468 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Harris H, Rubinsztein DC. . Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2012; 8:108 - 17; http://dx.doi.org/ 10.1038/nrneurol.2011.200; PMID: 22187000 [DOI] [PubMed] [Google Scholar]
- 15. Fleming A, Noda T, Yoshimori T, Rubinsztein DC. . Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol 2011; 7:9 - 17; http://dx.doi.org/ 10.1038/nchembio.500; PMID: 21164513 [DOI] [PubMed] [Google Scholar]
- 16. Lu JH, Tan JQ, Durairajan SS, Liu LF, Zhang ZH, Ma L, Shen HM, Chan HY, Li M. . Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. [Erratum in Autophagy. 2012;8(5)] Autophagy 2012; 8:98 - 108; http://dx.doi.org/ 10.4161/auto.8.1.18313; PMID: 22113202 [DOI] [PubMed] [Google Scholar]
- 17. Xilouri M, Brekk OR, Stefanis L. . Alpha-synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol 2013; 47:537 - 51; http://dx.doi.org/ 10.1007/s12035-012-8341-2; PMID: 22941029 [DOI] [PubMed] [Google Scholar]
- 18. Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Duff KE. . Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol 2009; 175:736 - 47; http://dx.doi.org/ 10.2353/ajpath.2009.080928; PMID: 19628769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A. . Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011; 286:10814 - 24; http://dx.doi.org/ 10.1074/jbc.M110.132514; PMID: 21252228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, et al. . α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 2010; 190:1023 - 37; http://dx.doi.org/ 10.1083/jcb.201003122; PMID: 20855506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lindersson EK, Højrup P, Gai WP, Locker D, Martin D, Jensen PH. . alpha-Synuclein filaments bind the transcriptional regulator HMGB-1. Neuroreport 2004; 15:2735 - 9; PMID: 15597044 [PubMed] [Google Scholar]
- 22. Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. . Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003; 278:25009 - 13; http://dx.doi.org/ 10.1074/jbc.M300227200; PMID: 12719433 [DOI] [PubMed] [Google Scholar]
- 23. Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. . Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem 2003; 278:11753 - 9; http://dx.doi.org/ 10.1074/jbc.M208641200; PMID: 12551928 [DOI] [PubMed] [Google Scholar]
- 24. Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. . Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 2001; 21:9549 - 60; PMID: 11739566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH. . Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A 2012; 109:7505 - 10; http://dx.doi.org/ 10.1073/pnas.1121146109; PMID: 22529378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang TF, Zhang YJ, Zhou HY, Wang HM, Tian LP, Liu J, Ding JQ, Chen SD. . Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J Neuroimmune Pharmacol 2013; 8:356 - 69; http://dx.doi.org/ 10.1007/s11481-012-9431-7; PMID: 23325107 [DOI] [PubMed] [Google Scholar]
- 27. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. . Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004; 305:1292 - 5; http://dx.doi.org/ 10.1126/science.1101738; PMID: 15333840 [DOI] [PubMed] [Google Scholar]
- 28. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. . Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/ 10.1016/j.cell.2005.07.002; PMID: 16179260 [DOI] [PubMed] [Google Scholar]
- 29. Min HJ, Ko EA, Wu J, Kim ES, Kwon MK, Kwak MS, Choi JE, Lee JE, Shin JS. . Chaperone-like activity of high-mobility group box 1 protein and its role in reducing the formation of polyglutamine aggregates. J Immunol 2013; 190:1797 - 806; http://dx.doi.org/ 10.4049/jimmunol.1202472; PMID: 23303669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. . Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 2010; 13:567 - 76; http://dx.doi.org/ 10.1038/nn.2528; PMID: 20383138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T, Unser M, Aebischer P. . Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum Mol Genet 2009; 18:872 - 87; PMID: 19074459 [DOI] [PubMed] [Google Scholar]
- 32. Taniguchi N, Caramés B, Kawakami Y, Amendt BA, Komiya S, Lotz M. . Chromatin protein HMGB2 regulates articular cartilage surface maintenance via beta-catenin pathway. Proc Natl Acad Sci U S A 2009; 106:16817 - 22; http://dx.doi.org/ 10.1073/pnas.0904414106; PMID: 19805379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li X, He L, Che KH, Funderburk SF, Pan L, Pan N, Zhang M, Yue Z, Zhao Y. . Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun 2012; 3:662; http://dx.doi.org/ 10.1038/ncomms1648; PMID: 22314358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vakkila J, DeMarco RA, Lotze MT. . Imaging analysis of STAT1 and NF-kappaB translocation in dendritic cells at the single cell level. J Immunol Methods 2004; 294:123 - 34; http://dx.doi.org/ 10.1016/j.jim.2004.09.007; PMID: 15604022 [DOI] [PubMed] [Google Scholar]
- 35. Bessadottir M, Egilsson M, Einarsdottir E, Magnusdottir IH, Ogmundsdottir MH, Omarsdottir S, Ogmundsdottir HM. . Proton-shuttling lichen compound usnic acid affects mitochondrial and lysosomal function in cancer cells. PLoS One 2012; 7:e51296; http://dx.doi.org/ 10.1371/journal.pone.0051296; PMID: 23227259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tu X, Batta P, Innocent N, Prisco M, Casaburi I, Belletti B, Baserga R. . Nuclear translocation of insulin receptor substrate-1 by oncogenes and Igf-I. Effect on ribosomal RNA synthesis. J Biol Chem 2002; 277:44357 - 65; http://dx.doi.org/ 10.1074/jbc.M208001200; PMID: 12202493 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.