

Abstract
Both apoptotic and autophagic pathways are activated in cells during anticancer treatment using DNA-damaging agents. Thus, the outcome is balanced between apoptotic cell death and enhanced autophagy, with the possibility of prolonged cell survival. It seems intuitively obvious that this survival mechanism might interfere with the desired tumor cell killing. We addressed this question by tipping the balance in favor of autophagy, using etoposide or cisplatin at low, sublethal doses. Over 4 days, only a little apoptosis was observed, but both drugs sharply increased autophagic flux. Surprisingly, cells underwent a cell cycle arrest at G2/M, followed later by mitotic catastrophe with formation of multipolar spindles, missegregated chromosomes, or enlarged, irregular, sometimes multiple nuclei. Why? The answer is that even a low level of DNA damage not only upregulates autophagy, but also provokes the recruitment of an autophagy-related protein, ATG5, to the nucleus, where it binds BIRC5/survivin, thereby interfering with correct assembly of the chromosome passenger complex needed for cytokinesis.
Keywords: anticancer drugs, apoptosis, ATG5, autophagy, Aurora B, chromosomal passenger complex, DNA damage, mitotic catastrophe, survivin
Macroautophagy (hereafter autophagy) is a fundamental survival mechanism in stress. It allows cells to simply enclose cytosol or organelles in characteristic double- or multiple-membraned vesicles to form autophagosomes. Once formed, autophagosomes merge with lysosomes, becoming autolysosomes capable of degrading macromolecules, allowing recovery of metabolic precursors. This process depends upon the function of several autophagy-related (ATG) proteins. ATG5 is one such, conjugating with ATG12 to generate an E3 ubiquitin ligase-like enzyme required for the formation of autophagosomes. We found that DNA-damaging treatment upregulates ATG5 expression as well as autophagic flux (Fig. 1). The G2/M arrest and mitotic catastrophe we observed are ATG5 dependent, since short hairpin interfering RNAs able to block ATG5 expression in etoposide- or cisplatin-treated cells also prevent both autophagy and mitotic catastrophe. To test whether ATG5 expression alone is sufficient to induce mitotic catastrophe in untreated cells, a human ATG5 construct was ectopically overexpressed. We found that G2/M arrest and mitotic catastrophe ensue, just as with etoposide and cisplatin treatment. We then studied levels of cell cycle regulators in order to understand the G2/M arrest. Not surprisingly, strong phosphorylation of CDK1 (cyclin-dependent kinase 1, on tyrosine 15), and CHEK2/Chk2 (checkpoint kinase 2, on threonine 68) is elicited both by drug treatment and by ectopic ATG5 expression, a finding consistent with persisting arrest in G2 phase.
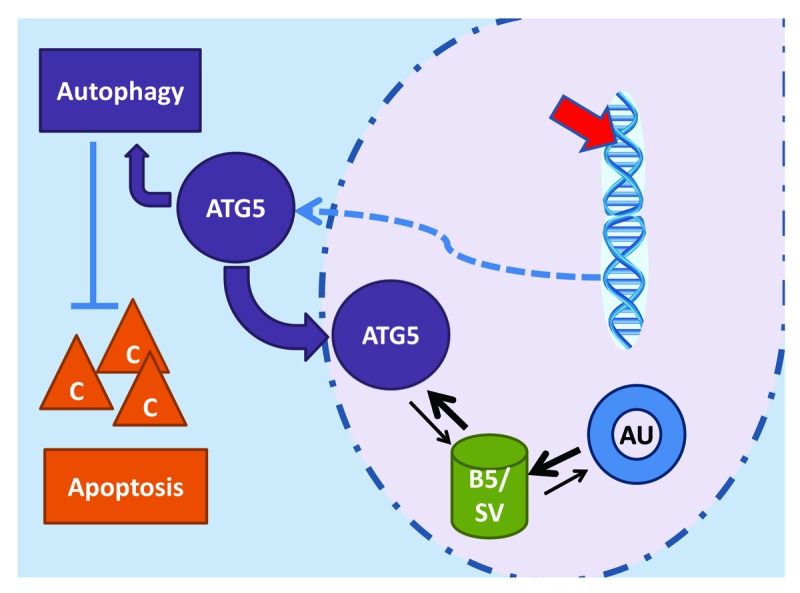
Figure 1. Low levels of DNA damage can be achieved with sublethal etoposide or cisplatin treatment, activating both apoptotic and autophagic pathways. The strongly upregulated ATG5 expression initiates autophagy, preventing caspase (C) activation and apoptosis. Much of the ATG5 is translocated to the nucleus, where it preferentially binds BIRC5/survivin (B5/SV), sharply reducing the availability for AURKB (AU) association, as needed for correct functioning of the chromosome passenger complex.
Speculating that ATG5 might itself somehow cause DNA damage, we analyzed several markers for the DNA damage response: H2AFX/H2AX phosphorylation, ATM phosphorylation (on Ser 1981) and ATR phosphorylation (on Ser 428) using flow cytometry, confocal microscopy, and immunoblotting. These measures of DNA damage are significantly elevated only after etoposide or cisplatin treatment. Hence, we concluded that ATG5 is not bringing about mitotic catastrophe by causing DNA damage.
When we realized that the ATG5 expression itself was linked to mitotic catastrophe, we wondered if the autophagic process was responsible. Experiments with autophagy inducers and with BECN1 (yeast Vps30/Atg6), however, produced no nuclear abnormalities. Interestingly, cells with high levels of ATG5, whether owing to ectopic expression or to DNA-damaging drug treatments, undergo massive, caspase-dependent apoptosis if the autophagic process is blocked pharmacologically. Thus, the effects of ATG5 in the nucleus seem to be independent of the autophagic process occurring in the cytoplasm, although autophagy is important if only to preclude apoptosis.
ATG5 had been considered a cytosolic protein, but in view of our evidence for its effects in the nucleus, we decided to look for its presence there. Cell fractionation revealed ATG5 in the nuclear fraction of both drug-treated and ATG5-overexpressing cells. Examining the sequence homology of ATG5 to BECN1, we found a homologous leucine-rich, putative nuclear export signal (NES). The presence of a cryptic nuclear localization motif in the same region can be inferred from the fact that a deletion mutant lacking this short sequence, ATG5-∆NES, is unable to accumulate in the nucleus when ectopically overexpressed and is, correspondingly, unable to produce either G2/M arrest or mitotic catastrophe.
We had two clues about the role of ATG5 recruited to the nucleus: G2/M arrest and mitotic catastrophe. Both point to a disturbance in chromosome segregation. Interestingly, we could show that following ectopic ATG5 expression, BIRC5 is upregulated. BIRC5 is a very special kind of protein, an apoptosis inhibitor in its cytoplasmic incarnation, and, in the nucleus, an essential component of the chromosome passenger complex. Thus, like ATG5, BIRC5 plays 2 completely distinct roles in 2 different compartments.
To test the possibility of a physical interaction between ATG5 and BIRC5, we performed reciprocal co-immunoprecipitation experiments with both etoposide-treated and ectopically ATG5-expressing cells. The result showed clearly that ATG5 in the nucleus physically associates with BIRC5 (Fig. 1), and that ATG5-∆NES, which is unable to accumulate in the nucleus, does not. Colocalization was also observed with immunofluorescence staining and confocal microscopy and could even be confirmed in vivo with examination of tumor sections from patients with lung and esophagus carcinomas.
Since, in the formation of the chromosome passenger complex, BIRC5 associates with AURKB (aurora kinase B; Fig. 1), we also performed co-immunoprecipitation assays for such binding. We found much less BIRC5 in association with AURKB than in untreated cells, even though BIRC5 had been upregulated. Thus, in sum, our observations indicate that ATG5 competes with AURKB for the available BIRC5. To study the consequences for the function of the chromosome passenger complex, we employed indirect immunofluorescence microscopy and obtained clear evidence of mislocalization in mitosis, both for AURKB and for BIRC5.
Taken together, these findings have important implications for development of an optimal cancer treatment strategy. On the one hand, DNA-damaging agents cause cell death by apoptosis, but on the other, they also induce an autophagic response. Due to the upregulation of ATG5, BIRC5 function in the nucleus is disturbed with the consequence that G2/M arrest occurs, then, due to mitotic slippage, a mitotic catastrophe takes place. Since the aim of treatment is to cause tumor cell death, one can hope that such catastrophe will finally lead to cell death. Unfortunately, however, while cell death is the most frequent outcome, one cannot ignore the reality that some few cells, through heroic efforts at repair, do survive, having established massive genetic modifications and stable aneuploidy. In a population of tumor cells, the resulting genetic diversity has negative consequences for the treatment outcome and for the patient. Thus, effort should be devoted to learning how to safely undermine autophagy during treatment with DNA-damaging agents.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.