

Abstract
Autophagy plays an important role in cellular responses to pathogens. However, the impact of the autophagy machinery on classical swine fever virus (CSFV) infection is not yet confirmed. In this study, we showed that CSFV infection significantly increases the number of autophagy-like vesicles in the cytoplasm of host cells at the ultrastructural level. We also found the formation of 2 ubiquitin-like conjugation systems upon virus infection, including LC3-I/LC3-II conversion and ATG12–ATG5 conjugation, which are considered important indicators of autophagy. Meanwhile, high expression of ATG5 and BECN1 was detected in CSFV-infected cells; conversely, degradation of SQSTM1 was observed by immunoblotting, suggesting that CSFV infection triggered a complete autophagic response, most likely by the NS5A protein. Furthermore, by confocal immunofluorescence analysis, we discovered that both envelope protein E2 and nonstructural protein NS5A colocalized with LC3 and CD63 during CSFV infection. Examination by immunoelectron microscopy further confirmed the colocalization of both E2 and NS5A proteins with autophagosome-like vesicles, indicating that CSFV utilizes the membranes of these vesicles for replication. Finally, we demonstrated that alteration of cellular autophagy by autophagy regulators and shRNAs affects progeny virus production. Collectively, these findings provide strong evidence that CSFV infection needs an autophagy pathway to enhance viral replication and maturity in host cells.
Keywords: autophagy, classical swine fever virus (CSFV), LC3, BECN1, replication
Introduction
Classical swine fever virus (CSFV) is a small enveloped virus with a single-stranded positive-sense genomic RNA and is classified as a member of the Pestivirus genus within the Flaviviridae family. 1 CSFV is the causative agent of classical swine fever (CSF), an OIE (World Organisation for Animal Health)-listed disease characterized by high fever, multiple hemorrhages, neurological disorders, and respiratory and gastrointestinal symptoms. 2 , 3 At present, treatment options for classical swine fever are limited; instead, prevention with vaccines against CSFV is usually used. 4 , 5 However, CSFV has evolved mechanisms that prevent apoptosis and induce immune depression, and is, therefore, able to establish persistent infection. 6 - 8 Albeit indirectly, these changes usually lead to huge economic losses worldwide. 9 - 11 Thus, it is essential to clarify the relationship between host and virus during CSFV infection to develop new vaccines or specific drugs for effectively controlling infection. Although many studies have investigated the pathogenesis of CSFV, 3 , 12 - 14 the underlying mechanism of CSFV replication remains poorly understood.
Autophagy is an intracellular degradation process that maintains the metabolic balance and homeostasis of cells. 15 More than 36 autophagy-related (ATG) genes have been identified in yeast, and many of these genes have counterpart orthologs in mammalian cells. 15 , 16 These genes are involved in a multistep mechanism to regulate autophagy, including the formation of autophagic vesicles and fusion with lysosomes. 16 - 19 Previous studies suggest that autophagy not only serves a protective function in cell survival under stress, but also plays a role in pathogen infection. 20 - 22 In Sindbis virus and tobacco mosaic virus infections, autophagy successfully restricts intracellular pathogen replication. 23 , 24 Conversely, herpes simplex virus type 1 and human immunodeficiency virus type 1 can inhibit autophagy to facilitate replication. 25 - 27 Nevertheless, several viruses such as coxsackievirus, dengue virus, influenza A virus, and hepatitis C virus have evolved strategies to use autophagic vesicles for replication. 28 - 31 Autophagy is virus-specific; thus, understanding the details of the relationship between autophagy and viral infection is critical for controlling disease transmission. No study has shown the functions of autophagy during CSFV replication. In the present study, we reveal the relationship between autophagy and CSFV infection.
By detecting the formation of autophagic vesicles and the expression of key autophagy molecules, we provide the first evidence that CSFV infection induces autophagy in vitro. Moreover, we explored the effect of the autophagy machinery on CSFV replication by disturbing the autophagy pathway with autophagy regulators and short hairpin RNAs (shRNAs).
Results
CSFV infection increases the levels of autophagic markers in host cells
To determine whether CSFV infection regulates cellular autophagy, we examined the formation of autophagosome-like vesicles in CSFV-infected PK-15 and 3D4/2 cells by using transmission electron microscopy (TEM) and quantitative analysis. We found that the number of double- or single-membrane vesicles increased in the cytoplasm of CSFV-infected PK-15 cells (Fig. 1A , b). Similar results were obtained in CSFV-infected 3D4/2 cells (Fig. 1A , e). Compared with CSFV-infected cells, autophagosome-like vesicles were rarely seen in the mock-infected cells (Fig. 1A , a and d). Quantitative analysis also showed a significant increase in the quantity of single- and double-membrane vesicles in the CSFV-infected cells compared with the uninfected cells (Fig. 1B). To determine whether the formation of double-membrane vesicles is linked to autophagy during CSFV infection, immunoelectron microscopy (IEM) was performed on CSFV-infected cells. The results showed the expression of autophagy protein microtubule-associated protein 1 light chain 3 (LC3) during virus infection, and the immunogold labeling localized the infection-associated membranes (Fig. 1A , c and f). The negative controls, which were incubated with normal rabbit immunoglobulin G and without the primary antibody, showed no positive signals (data not shown). These results show that CSFV infection can induce autophagosome formation in vitro.

Figure 1. CSFV infection increases the formation of autophagosome-like vesicles. (A) PK-15 (a–c) and 3D4/2 (d–f) cells were mock-infected (a and d) or infected with CSFV (b and e) at an MOI of 1 for 48 h and studied by electron microscopy. White arrows indicate the structures with the characteristics of autophagosomes (b and e). The cells were also processed for IEM analysis. LC3 was visualized with specific antibodies and detected with a secondary antibody conjugated to 18-nm colloidal gold particles. LC3 protein immunogold labeling of CSFV-infected cells is shown in (c and f). The immunogold labeling localized to the infection-associated membranes is indicated by black arrows in the infected cells. Scale bar: 500 nm. (B) Quantification of the autophagosome-like vesicles per cell image. Average number of the vesicles in each cell was obtained from at least 10 cells undergoing each treatment. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; ***P < 0.001.
To further analyze if the autophagy machinery can be triggered by CSFV infection, we examined the levels of autophagy marker proteins in CSFV-infected cells by using immunoblotting, including LC3 conversion, ATG12–ATG5 conjugation, and ATG5 and BECN1 expression. LC3 is widely used as a marker for assessing autophagy and correlates well with the formation of the autophagosome. 32 , 33 Our results showed that LC3-II expression was upregulated in CSFV-infected PK-15 and 3D4/2 cells, with a decrease in LC3-I expression relative to mock-infected cells (Fig. 2A and B, lanes 1 to 10). We next examined the autophagy conjugation system by detecting the expression of ATG12–ATG5 and ATG5 by using a specific antibody to ATG5, which associates with the membranes of precursor autophagosomes. 34 , 35 The CSFV-infected cells had an increased level of ATG12–ATG5 and ATG5 relative to mock-infected cells (Fig. 2A and B, lanes 1 to 10). We further detected the level of BECN1, which is involved in the early steps of the autophagy pathway. 36 The results showed that CSFV infection generated the overexpression of BECN1 in host cells relative to mock-infected cells (Fig. 2A and B, lanes 1 to 10). Meanwhile, the detection of CSFV envelope protein E2 was used to estimate the progression of infection (Fig. 2A and B, lanes 6 to 10). More importantly, the level of autophagy marker proteins was near the detection limit in mock-infected cells (Fig. 2A and B, lanes 1 to 5), suggesting that a basal level of autophagy is present in normal PK-15 and 3D4/2 cells. In addition, the densitometric ratios of autophagy marker proteins (relative to ACTB) not only increased in CSFV-infected cells compared with uninfected (mock) cells, but also increased progressively with increase in infection time (Fig. 2A and B, lower part). These findings indicate that the early stages of autophagy are induced upon CSFV infection in host cells.

Figure 2. Expression of autophagy marker proteins in CSFV-infected cells. (A) PK-15 cells were mock-infected or infected with CSFV (MOI = 1) or UV-inactivated CSFV (MOI = 1) for 6, 12, 24, 36, and 48 h. At the end of the infection, the expression of LC3, ATG12–ATG5, ATG5, BECN1, E2, and ACTB (loading control) were analyzed by immunoblotting with specific antibodies as described in Materials and Methods. (B) 3D4/2 cells were infected and analyzed as in (A). The relative levels of the targeted proteins were estimated by densitometry, and the ratios were calculated relative to the ACTB control. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001.
To further confirm that CSFV infection-induced autophagy requires viral replication, we used ultraviolet (UV)-inactivated CSFV to detect whether autophagy can be induced following treatment. The results showed that the expression of autophagy marker proteins, including LC3-II, ATG12–ATG5, ATG5, and BECN1, was near the detection limit in treated cells (Fig. 2A and B, lanes 11 to 15). However, the densitometry ratios of autophagy marker proteins to ACTB had no significant changes relative to mock-infected cells (Fig. 2A and B, lower part), suggesting that viral replication is required for CSFV infection-induced autophagy.
CSFV infection enhances autophagic flux
To determine whether a complete autophagic response is triggered by CSFV infection, we first measured the degradation of SQSTM1, a marker for autophagy-mediated protein degradation pathway, by using immunoblotting analysis. 33 , 37 - 39 As shown in Figure 3A and B, CSFV-infected cells had decreased levels of the SQSTM1 protein, from 12 to 48 h post-infection (hpi) (Fig. 3A and B, lanes 2, 4, and 6). In contrast, the level of SQSTM1 protein in mock-infected cells was higher than that in infected cells (Fig. 3A and B, lanes 1, 3, and 5). To confirm autophagic flux with viral infection, the levels of LC3-II and SQSTM1 were determined following treatment with or without the protease inhibitor E64d in infected cells, a widely used method for assessing autophagic flux. 33 , 39 , 40 As shown in Figure 3C and D, E64d increased the levels of LC3-II and SQSTM1 in both PK-15 and 3D4/2 cells, at 24 and 48 hpi, compared with control groups. Interestingly, E64d treatment also increased the level of the E2 protein during CSFV infection (Fig. 3C and D). These data indicate that a complete autophagic response can be induced in host cells following CSFV infection.

Figure 3. CSFV infection enhances autophagic flux. (A and B) PK-15 (A) and 3D4/2 (B) cells were mock-infected or infected with CSFV (MOI = 1) for 12, 24, and 48 h. The cell samples were then analyzed by immunoblotting with anti-SQSTM1 and anti-ACTB (loading control) antibodies. (C and D) PK-15 (C) and 3D4/2 (D) cells were infected with CSFV (MOI = 1) in the presence or absence of E64d (10 μg/ml). At 24 and 48 hpi, cell lysates were prepared and analyzed by immunoblotting using anti-LC3B, anti-SQSTM1, anti-E2, and anti-ACTB (for detection of ACTB as loading control) antibodies. The relative levels of the targeted proteins were estimated by densitometric scanning, and the ratios were calculated relative to the ACTB control. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001; # P > 0.05.
The membranes of autophagosome-like vesicles are required for CSFV replication
To verify the correlation between the autophagy and viral replication during the progression of CSFV infection, we first detected the subcellular localizations of viral proteins and LC3 in PK-15 and 3D4/2 cells that had been infected with CSFV, by using immunofluorescence analysis with antibodies specific to E2, NS5A, and LC3B. As illustrated in Figure 4A and B, significant fluorescence signals corresponding to the LC3 and CSFV proteins were detected in CSFV-infected PK-15 and 3D4/2 cells, with puncta accumulation (Fig. 4A and B , b and c, g and h). The fluorescence puncta of both E2 and NS5A were highly colocalized with the fluorescence puncta of LC3 (Fig. 4A and B , d and i). In contrast, the LC3 puncta accumulation was not observed in mock- and UV-CSFV-infected cells (Fig. 4A and B , k and m), indicating that UV-inactivated CSFV had lost its capability of inducing the formation of the autophagosome, consistent with the data shown in Figure 2. Additionally, the average number of LC3 puncta in the infected cells was significantly greater than that in rapamycin-treated cells (Fig. 4A and B, lower right part). These results indicate that CSFV infection induces the redistribution of autophagy marker LC3 in host cells, and the autophagosome could be involved in viral replication.

Figure 4. CSFV infection induces the redistribution of the autophagy marker LC3 in host cells. (A) PK-15 cells were mock-infected or infected with CSFV (MOI = 1) or UV-inactivated CSFV (MOI = 1) or treated with rapamycin (Rapa, 100 nM) for 48 h. The cells were then fixed and processed for indirect immunofluorescence using antibodies against LC3B and the CSFV proteins (E2 or NS5A), followed by the corresponding secondary antibodies conjugated to FITC and TRITC as described in Materials and Methods. The cell nucleus were counterstained with DAPI. The fluorescence signals were visualized by confocal immunofluorescence microscopy. In the images, the nucleus staining is shown in blue (a and f), E2 and NS5A staining is shown in green (b and g), LC3 staining is shown in red (c and h), and the signals of colocalization are shown in yellow in merged images (d and i). The higher magnification images are shown in (e and j) from the white-square frame-enclosed region in (d and i). The mock- and UV-CSFV-infected cells as well as the rapamycin treatment group were analyzed with anti-LC3B antibodies and used as the control groups (k–m). Scale bar: 10 μm. The average number of LC3 puncta in each cell was determined from at least 100 cells in each group. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA: *P < 0.05; **P < 0.01; ***P < 0.001; # P > 0.05. (B) 3D4/2 cells were infected and analyzed as in (A). Scale bar: 10 μm. The data represent the mean ± SD of 3 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; # P > 0.05.
CD63 is a widely used marker for lysosomes, and correlates well with the formation of autolysosomes. 41 To confirm the relationship between autophagosome-like vesicles and viral replication, we examined the distribution of CD63 in virus-infected cells (Fig. 5A and B , b and f). We found that the CD63 protein colocalized with the E2 and NS5A proteins in CSFV-infected PK-15 and 3D4/2 cells (Fig. 5A and B , c and g), confirming that autophagosome-like vesicles are involved in the replication of CSFV and that the membranes of these structures could be required for CSFV replication.

Figure 5. RNA replication of CSFV occurs on the membranes of autophagosome-like vesicles. (A) PK-15 cells infected with CSFV (MOI = 1) for 48 h. The cells were fixed and processed for indirect immunofluorescence using antibodies against the CD63 and CSFV proteins (E2 or NS5A), and the targeted protein staining was detected with secondary antibodies conjugated to FITC and TRITC as described in Materials and Methods. The fluorescence signals were visualized by confocal immunofluorescence microscopy. In the images, E2 and NS5A staining is shown in green (a and e), CD63 staining is shown in red (b and f), and the signals of colocalization are shown in yellow in the merged images (c and g). The higher magnification images are shown in (d and h) from the white-square frame-enclosed region in (c and g). Scale bar: 10 μm. One of 3 independent experiments is shown. (B) 3D4/2 cells were infected and analyzed as in (A). Scale bar: 10 μm. One of 3 independent experiments is shown. (C) PK-15 (a and c) and 3D4/2 (b and d) cells were infected with CSFV at an MOI of 1 for 48 h. Subsequently, IEM analysis was performed using specific monoclonal antibodies against the CSFV NS5A and E2 proteins, and the targeted proteins were detected with a secondary antibody conjugated to 12-nm colloidal gold particles. The immunogold labeling showing the localization of CSFV proteins on the membranes of autophagosome-like vesicles is indicated by black arrows. Scale bar: 500 nm. One of 3 independent experiments is shown.
To determine the sites of CSFV replication, IEM was performed on CSFV-infected cells by using specific monoclonal antibodies against the CSFV NS5A and E2 proteins and secondary antibodies conjugated to 12-nm colloidal gold particles. As shown in Figure 5C, immunogold labeling showed localization of the CSFV proteins on the membranes of autophagosome-like vesicles (arrows in Fig. 5C). Interestingly, IEM also showed the presence of gold particles within the autophagosome-like vesicles, likely representing degraded CSFV proteins. In contrast, the mock-infected cells and the negative controls showed no positive signals (data not shown). These data strongly suggest that RNA replication of CSFV occurs on the membranes of autophagosome-like vesicles.
CSFV nonstructural protein NS5A induces autophagy in host cells
NS5A is an important nonstructural protein of CSFV that is associated with virus replication. To further analyze whether CSFV replication is required for the induction of autophagy and determine the effect of NS5A on autophagy induction during CSFV infection, we expressed NS5A as a fusion protein with enhanced green fluorescent protein (EGFP) and transfected the cells with pEGFP-N1 as an empty vector control. We found that the fluorescence signals of LC3 and EGFP-NS5A were clearly enhanced in PK-15 and 3D4/2 cells after transfection with pEGFP-NS5A (Fig. 6A and B , a and b). Furthermore, the puncta accumulation and colocalization signals of the targeted proteins were detected by immunofluorescence analysis (Fig. 6A and B , c). In contrast, weak signals of LC3 and EGFP were observed in the control groups (Fig. 6A and B , e and f), and no colocalization signals were detected for the targeted proteins (Fig. 6A and B , g). Meanwhile, the average number of LC3 puncta in cells transfected with pEGFP-NS5A was significantly greater than in cells transfected with pEGFP-N1. Additionally, immunoblotting analysis was used to quantify the expression of LC3, SQSTM1, and NS5A. We found that pEGFP-NS5A significantly increased the level of LC3-II and promoted the degradation of SQSTM1 in host cells compared with pEGFP-N1-transfected and mock-infected cells (Fig. 6C and D). These data indicate that the replicase NS5A can induce autophagosome formation and activate a complete autophagic response.

Figure 6. CSFV nonstructural protein NS5A induces autophagy in host cells. (A) PK-15 cells were transfected with vector pEGFP-N1 or plasmid pEGFP-NS5A expressing the EGFP-NS5A fusion protein. At 48 hpi, cells were analyzed as described in the legend to Figure 4, but only with an antibody against LC3B and TRITC-conjugated goat anti-rabbit IgG. In the images, the expression of EGFP-NS5A is shown in green (a and e), LC3 staining is shown in red (b and f), and the signals of colocalization are shown in yellow in the merged images (c and g). The higher magnification image is shown in (d) from the white-square frame-enclosed region in (c). Scale bar: 10 μm. The average number of LC3 puncta in each cell was determined from at least 100 cells in each group. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; ***P < 0.001. (B) 3D4/2 cells were transfected and analyzed as in (A). Scale bar: 10 μm. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; ***P < 0.001. (C) PK-15 cells and 3D4/2 cells were transfected as described in (A), and mock-infected cells were used as the control. The cell samples were analyzed by immunoblotting with anti-LC3B and anti-ACTB (loading control) antibodies, and the efficiency of transfection was measured using an antibody against GFP. One of 3 independent experiments is shown. (D) PK-15 cells and 3D4/2 cells were transfected plasmid pEGFP-NS5A expressing the EGFP-NS5A fusion protein for 24, 36, and 48 h. The cell samples were analyzed by immunoblotting with anti-SQSTM1 and anti-ACTB (loading control) antibodies. One of 3 independent experiments is shown.
Induction of autophagy with rapamycin increases virus yield
Rapamycin, an inducer of autophagy, can activate autophagy by blocking the mechanistic target of rapamycin (MTOR) pathway. 42 - 44 To analyze the role of autophagy in the replication of CSFV, we examined viral envelope protein E2 expression and viral progeny yields following rapamycin treatment. We found that the induction of autophagy with rapamycin not only upregulated the expression of LC3-II and CSFV-E2 at 24 and 48 hpi in CSFV-infected PK-15 cells (Fig. 7A), but also increased the yields of CSFV progeny (Fig. 7B and C). Notably, the effect of rapamycin treatment on the extracellular viral load and titer was greater than the intracellular load and titer (Fig. 7B and C). Similar results for viral envelope protein E2 expression and viral progeny yields were also obtained in treated 3D4/2 cells (Fig. 7D–F). These data suggest that the autophagy mechanism may support CSFV replication.

Figure 7. Induction of autophagy with rapamycin enhances CSFV replication. (A and D) PK-15 (A) and 3D4/2 (D) cells were pretreated with rapamycin (100 nM) or DMSO (Control) for 1 h, followed by CSFV adsorption for 1 h at an MOI of 0.5. The cells were further cultured in fresh medium in the absence or presence of rapamycin (100 nM). At 24 and 48 hpi, cell samples were analyzed by immunoblotting with antibodies against LC3B, CSFV-E2, and ACTB (loading control). The relative levels of the targeted proteins were estimated by densitometric scanning, and the ratios were calculated relative to ACTB. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; **P < 0.01. (B and E) PK-15 (B) and 3D4/2 (E) cells were pretreated and infected as described in (A and D). At 24 and 48 hpi, both the extracellular and intracellular copy numbers of CSFV were detected by qRT-PCR. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001. (C and F) PK-15 (C) and 3D4/2 (F) cells were pretreated and infected as described in (A and D). At 24 and 48 hpi, both the extracellular and intracellular virus titers were measured by endpoint dilution titrations by using an immunofluorescence assay as described in Materials and Methods. Results are expressed in units of TCID50/ml. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001.
Inhibition of autophagy with 3-methyladenine (3-MA) decreases virus yield
To further determine the effect of autophagy on CSFV replication, we exposed the host cells to 3-MA, which inhibits autophagy by blocking the formation of autophagosomes, 45 , 46 and analyzed the capability of CSFV replication by detecting viral envelope protein E2 expression and viral progeny yield by using relevant assays. As shown in Figure 8, 3-MA treatment reduced the expression of LC3-II and CSFV-E2 at 24 and 48 hpi in CSFV-infected PK-15 cells and decreased the virus copy number and titer (Fig. 8A, C, and D). Notably, the effect of 3-MA treatment on the extracellular viral load and titer was greater than the intracellular load and titer (Fig. 8C and D). Importantly, the LC3-positive puncta and the colocalization of LC3 and E2 decreased in the presence of 3-MA (Fig. 8B). Similar results for viral envelope protein E2 expression and viral progeny yield were obtained in treated 3D4/2 cells (Fig. 8E–H). Furthermore, because autophagosome formation can be inhibited by 3-MA, the decreased densitometry ratio of LC3-II (relative to ACTB) in 3-MA-treated cells further verified that the enhanced autophagy shown in Figures 1 and 2 was triggered by the CSFV infection. These findings suggest that the induction of autophagy during CSFV infection promotes virus replication.

Figure 8. Inhibition of autophagy with 3-MA reduces CSFV replication. (A and E) PK-15 (A) and 3D4/2 (E) cells were pretreated with 3-MA (5 mM) or DMSO (Control) for 4 h. After 1 h of virus absorption at an MOI of 0.5, the cells were further cultured in fresh medium in the absence or presence of 3-MA (5 mM). At 24 and 48 hpi, cell samples were analyzed as described in the legend to Figure 7A and D. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001. (B and F) PK-15 (B) and 3D4/2 (F) cells were pretreated with 3-MA for 4 h. After 1 h of virus absorption at an MOI of 0.5, the cells were further cultured in fresh medium in the presence of 3-MA (5 mM). At 48 hpi, the cells were analyzed with antibodies against LC3B and E2, as described in the legend to Figure 4. The fluorescence signals were visualized with confocal immunofluorescence microscopy. In the images, E2 staining is shown in green, LC3 staining is shown in red, and the signals of colocalization are shown in the merged images. Scale bar: 10 μm. One of 3 independent experiments is shown. (C and G) PK-15 (C) and 3D4/2 (G) cells were pretreated and infected as described in (A and E). At 24 and 48 hpi, both the extracellular and intracellular copy numbers of CSFV were detected by qRT-PCR. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; **P < 0.01; ***P < 0.001. (D and H) PK-15 (D) and 3D4/2 (H) cells were pretreated and infected as described in (A and E). At 24 and 48 hpi, both the extracellular and intracellular virus titers were measured by endpoint dilution titrations by using the immunofluorescence assay described in Materials and Methods. Results are expressed in units of TCID50/ml. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; **P < 0.01; ***P < 0.001.
Depletion of endogenous BECN1 and LC3 reduces viral replication
Our results using pharmacological regulators highlighted the crucial role of the autophagy machinery in CSFV replication. To further explore the relationship between the autophagy machinery and virus replication, shRNA knockdown experiments were performed to specifically deplete endogenous BECN1 and MAP1LC3B (LC3B) proteins. As shown in Figures 9 and 10, PK-15 and 3D4/2 cells transfected with shBECN1 and shLC3B presented significantly decreased levels of endogenous BECN1 and LC3 proteins compared with cells transfected with nontargeting (scrambled) shRNAs comprising the control group (Figs. 9 and 10, A and E). Importantly, suppression of BECN1 and LC3 expression strongly reduced the expression of viral envelope protein E2 and the viral progeny yield in CSFV-infected PK-15 cells compared with the control group (Figs. 9 and 10, A, C, and D). Similar results were also obtained in infected 3D4/2 cells (Figs. 9 and 10, E, G, and H). Notably, the LC3-positive puncta and the colocalization of LC3 and E2 disappeared when depleting endogenous BECN1 and LC3 in both PK-15 and 3D4/2 cells (Figs. 9 and 10, B and F). These data further reveal that autophagy plays an important role in the replication of CSFV.

Figure 9. Inhibition of autophagy with specific shRNA targeting BECN1 reduces CSFV replication. (A and E) PK-15 (A) and 3D4/2 (E) cells were transfected with shRNAs targeting BECN1 or scrambled shRNAs for 48 h, followed by mock infection and CSFV infection at an MOI of 0.5. At 24 hpi, the silencing efficiency of BECN1 shRNA and the expression of autophagy marker proteins and CSFV-E2 were analyzed as described in the legend to Figure 2A. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; **P < 0.01; ***P < 0.001. (B and F) PK-15 (B) and 3D4/2 (F) cells were transfected with BECN1 shRNA for 48 h, followed by CSFV infection at an MOI of 0.5. At 24 hpi, the cells were analyzed as described in the legend to Figure 8B and F. Scale bar: 10 μm. One of 3 independent experiments is shown. (C and G) PK-15 (C) and 3D4/2 (G) cells were transfected as described in (A and E), followed by CSFV infection at an MOI of 0.5 for 24 h. Both the extracellular and intracellular copy numbers of CSFV were detected by qRT-PCR. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; **P < 0.01; ***P < 0.001. (D and H) PK-15 (D) and 3D4/2 (H) cells were transfected and infected as described in (C) and (G). At 24 hpi, both the extracellular and intracellular virus titers were measured by endpoint dilution titrations by using the immunofluorescence assay described in Materials and Methods. Results are expressed in units of TCID50/ml. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; **P < 0.01.

Figure 10. Inhibition of autophagy with LC3B-targeting shRNA reduces the replication of CSFV. (A and E) PK-15 (A) and 3D4/2 (E) cells were transfected with shRNAs targeting LC3B or scrambled shRNAs for 48 h, followed by mock infection and CSFV infection at an MOI of 0.5. At 24 hpi, the silencing efficiency of LC3B shRNA, as well as the expression of autophagy marker proteins and CSFV-E2 were analyzed as described in the legend to Figure 2A. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; *P < 0.05; ***P < 0.001. (B and F) PK-15 (B) and 3D4/2 (F) cells were transfected with LC3B shRNA for 48 h, followed by CSFV infection at an MOI of 0.5. At 24 hpi, the cells were analyzed as described in the legend to Figure 8B and F. Scale bar: 10 μm. One of 3 independent experiments is shown. (C and G) PK-15 (C) and 3D4/2 (G) cells were transfected as described in (A and E), followed by CSFV infection at an MOI of 0.5 for 24 h. Both the extracellular and intracellular copy numbers of CSFV were detected by qRT-PCR. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; **P < 0.01; ***P < 0.001. (D and H) PK-15 (D) and 3D4/2 (H) cells were transfected and infected as described in (C and G). At 24 hpi, both the extracellular and intracellular virus titers were measured by endpoint dilution titrations by using the immunofluorescence assay described in Materials and Methods. Results are expressed in units of TCID50/ml. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; . *P < 0.05; **P < 0.01; ***P < 0.001.
Modulation of autophagy activity with autophagy regulators does not affect cell viability
To determine whether the pharmacological alteration of autophagy with rapamycin and 3-MA affected the capability of CSFV replication by changing the cell viability, we performed the 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay to analyze the effects of these autophagic reagents on cell viability. Statistical analyses revealed no significant effects on the viability of cells treated with rapamycin or 3-MA (P > 0.05) (Fig. 11).
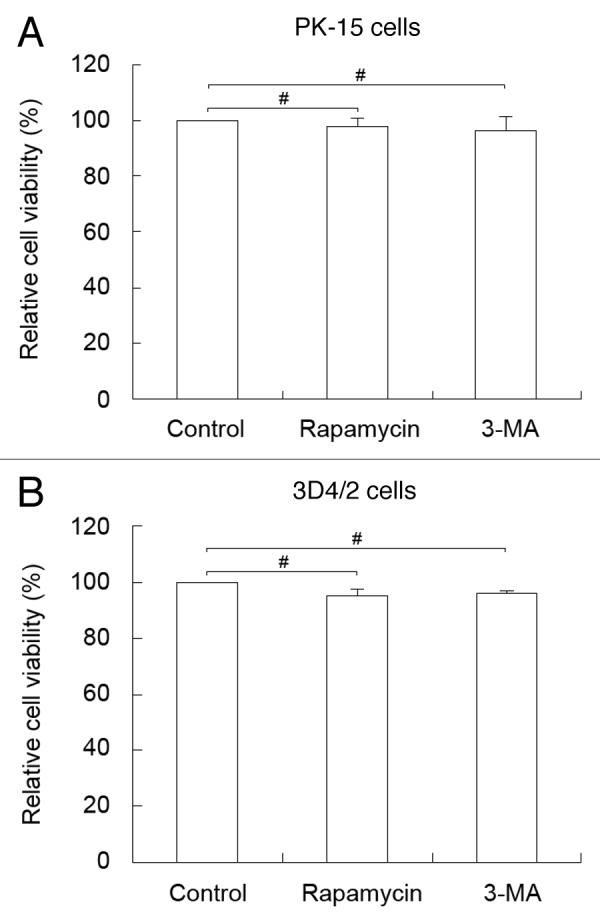
Figure 11. Pharmacological alteration of autophagy does not affect cell viability. The cell viability of PK-15 (A) and 3D4/2 (B) cells were determined by the MTT assay after treatments with rapamycin (100 nM) or 3-MA (5 mM) for 48 h. The data represent the mean ± SD of 3 independent experiments. Two-way ANOVA; # P > 0.05.
Discussion
The membrane-associated replication complex is a hallmark of all positive-strand RNA viruses during the infection process. 47 CSFV, a positive-stranded RNA virus, also requires intracellular membrane structures for replication. 48 Notably, envelope protein E2 was observed in the enrichment zone of the endoplasmic reticulum (ER), and the source of the membranes is likely the ER, which plays an important role in viral maturation and release. 48 Nevertheless, the specific mechanism by which these membrane structures are utilized during CSFV replication in host cells has not been elucidated. Autophagy is involved in the mechanism of balance between cells and stimuli, playing a key role in pathogenic infection. Interestingly, opposite effects of autophagy on the replication of different viruses have been noted, by blocking or inducing autophagosome formation. For instance, several studies showed that autophagy is activated upon viral infection, protecting cells by restricting viral replication. 23 - 25 Conversely, several reports reveal a positive role of autophagy in virus replication. 28 - 31 Based on previous findings on the intracellular membrane structures that participate in CSFV replication and the impact of autophagy on virus infection, autophagy is likely involved in the replication of CSFV. However, the relationship between CSFV infection and autophagy had not been reported. In this study, we provide the first strong evidence that CSFV infection induces autophagy to promote viral replication and maturation in vitro. We also show that viral replication is required for CSFV-induced autophagy.
Autophagy is involved in immunity by directly modulating pattern recognition receptor-mediated type I interferon production or by contributing to antigen presentation. 49 - 51 To define the role of autophagy in CSFV replication and provide a basis for further study of the relationship between autophagy and immune evasion of CSFV, we analyzed the physiological significance of autophagy in both infected PK-15 and 3D4/2 cells in vitro. PK-15 is usually used as the model cell line for studying CSFV infection. 52 , 53 The 3D4/2 cell line is representative of the macrophage that is the target for CSFV infection. 12 , 14 Considering the noncytopathogenic nature of CSFV, 13 , 54 identical multiplicities of infection (MOIs) and infection time points were used for both PK-15 and 3D4/2 cells.
When the target cells were infected with the Shimen strain of CSFV, we found using TEM that CSFV infection promoted the formation of autophagosome-like vesicles in host cells (see Fig. 1). This observation was further supported by immunoblotting analysis of an increased expression of autophagic markers in host cells infected with CSFV, and the expression exhibited a trend of time-dependent upregulation post-infection (Fig. 2A and B, lanes 6 to 10). In contrast, low basal levels of autophagy were present in mock- and UV-treated CSFV-infected cells, indicating that a basal autophagy in normal cells that is necessary for maintaining homeostasis (Fig. 2A and B, lanes 1 to 5 and 11 to 15). These data imply that UV-inactivated CSFV could lose its capability to induce autophagy. Recent studies have demonstrated that induction of autophagy not only increases autophagosome formation and expression of autophagy proteins, but also increases autophagic flux, which can be measured by detecting the levels of SQSTM1 and LC3-II in the presence or absence of lysosome inhibitors. 33 , 38 , 39 Our results showed that CSFV infection decreased the expression of SQSTM1 protein, and lysosome inhibitor E64d increased the levels of LC3-II and SQSTM1 in virus-infected cells (Fig. 3), indicating that CSFV infection enhanced the autophagic flux in both cell types. Interestingly, E64d treatment also increased the level of the E2 protein during CSFV infection (Fig. 3C and D), suggesting that a small amount of virus proteins degraded during autophagy. These findings demonstrate that CSFV infection can induce a complete autophagic response in host cells and that virus replication is involved in the induction of autophagy. However, recent studies describe that West Nile virus infection dose not involve the degradation of SQSTM1. 55 , 56 These findings imply that the role of autophagy is virus-specific, and the precise mechanism remains to be elucidated.
Colocalization of viral RNA replication complexes with the membrane of the autophagosome in infected cells has been described previously. This colocalization is beneficial for the replication of several different positive-stranded RNA viruses. 28 , 29 , 31 LC3 is a critical component of the mature autophagosome membrane, and CD63 is a widely used lysosomal marker for detecting the formation of autolysosomes. To understand how autophagy is involved in CSFV infection, we analyzed the localization of LC3-II or CD63 with viral proteins after infection with CSFV. We found that CSFV enhanced the redistribution of LC3 and CD63, and both envelope protein E2 and nonstructural protein NS5A colocalized with the positive puncta of LC3 and CD63 (Fig. 4; Fig. 5A and B), indicating that the membranes of autophagosome-like vesicles were required for CSFV replication. Previous studies demonstrated that some positive-stranded RNA viruses require membrane surfaces on which to assemble RNA replication complexes. To further determine the sites of CSFV replication, examination by IEM showed that viral protein E2 and NS5A localized on the membranes of autophagosome-like vesicles in the infected cells (Fig. 5C). Therefore, we provided evidence to support a mechanism of CSFV replication that is related to the autophagy pathway. Interestingly, IEM also showed the presence of gold particles within the autophagosome-like vesicles, likely representing degraded CSFV proteins. This observation may also explain a small increase in the expression of E2 following E64d treatment. Similar results are found in other virus infections, where autophagy can be utilized for viral replication. 57 , 58 Because autophagy is an intracellular degradation process in which viruses are directed to the lysosome through a membrane-mediated process, there is a small amount of virus proteins that will inevitably be degraded during autophagy, although autophagy can be triggered by viruses and utilized for viral replication. Additionally, UV-inactivated CSFV lost its capability of inducing the formation of the autophagosome (Fig. 4A and B , m), indicating that viral replication was required for CSFV infection-induced autophagy.
Prior studies demonstrate that NS5B and NS5A are essential components of the replication complex in Flaviviridae infections. 59 - 62 To further understand the relationship between autophagy and viral replication, we transfected both cell lines with pEGFP-NS5A and monitored the redistribution of LC3 and the colocalization of LC3 and EGFP-NS5A (Fig. 6A and B , a–d). Colocalization signals of the targeted protein were not observed in the control groups (Fig. 6A and B , e–g). To further examine the effect of NS5A on autophagy induction during CSFV infection, we analyzed the expression of LC3 and SQSTM1 and found that pEGFP-NS5A significantly increased the level of LC3-II and promoted the degradation of SQSTM1 compared with the pEGFP-N1-transfected and mock-infected groups (Fig. 6C and D). These data indicate that the replicase NS5A can induce autophagosome formation and activate a complete autophagic response. One recent observation is that CSFV NS5A is localized to the ER where it induces oxidative stress in vascular endothelial cells that are important for spreading the virus. 63 It is likely that viral replicase NS5A is important and necessary for the induction of autophagy by CSFV, although the mechanism remains to be elucidated. Clearly, these findings reinforce the theory that CSFV replication is required for the induction of autophagy; moreover, the autophagosome is involved in virus replication and maturation.
Recent studies show that several members of the Flaviviridae family can utilize an autophagy mechanism to facilitate replication, including dengue virus, hepatitis C virus, and Japanese encephalitis virus. 29 , 31 , 64 However, the West Nile virus, another member of the Flaviviridae family, can induce autophagy but does not require an autophagy mechanism for replication in host cells. 55 , 56 These results suggest that significant differences between replication mechanisms exist between different viruses of the same family, which may be related to host specificity and different pathogenic mechanisms of infections. To further determine what role autophagy plays in the replication of CSFV, we examined the capability of viral replication by exposing host cells to the autophagy-inducer rapamycin. Our data showed that rapamycin treatment not only upregulated the expression of the viral E2 protein but also increased the yield of CSFV progeny (Fig. 7). We also demonstrated that inhibition of autophagy with 3-MA or shRNA-based depletion of the essential autophagy proteins BECN1 and LC3 downregulated E2 expression and decreased the yield of CSFV progeny (Figs. 8–10). In addition, the LC3-positive puncta and the colocalization of LC3 with E2 were abolished upon inhibition of autophagy (Figs. 8–10 , B and F). These findings reveal that induction of autophagy during CSFV infection promotes virus replication. It is important to note that the effect of the mutative levels of autophagy on extracellular viral yields were more significant than on intracellular viral yields, indicating that autophagy is important for the release of cytoplasmic viruses. These results are similar to those form recent studies, which demonstrate that autophagy has a greater effect on extracellular than intracellular viral yields. 58 , 65 , 66 Meanwhile, the data from the MTT assay suggested that the pharmacological alteration of autophagy by rapamycin and 3-MA did not alter cell viability to affect the proliferation of the virus (Fig. 11). Based on these results, we conclude that the autophagy pathway serves a valuable role in CSFV infection rather than blocking virus spread in host cells, although the detailed relationship between autophagy and viral propagation still needs to be examined in depth.
In summary, our in vitro analysis demonstrates for the first time that CSFV needs to trigger a functional autophagy pathway to enhance virus replication and release in host cells. We postulate that autophagy may be a potential mechanism that CSFV uses to establish a persistent infection. Clearly, our understanding of the molecular mechanisms of the interplay between CSFV and autophagy is still insufficient. Therefore, further work investigating the immunological function of autophagy as well as its role in the immune escape of CSFV might be of importance to control viral infection.
Materials and Methods
Antibodies, chemicals, and plasmids
The primary antibodies used in the study were specific for LC3B (Cell Signaling Technology, 2775), ACTB (Beyotim, AA128), ATG5 (Novus Biologicals, NB110-53818), BECN1 (Novus Biologicals, NB110-87318), CD63 (Santa Cruz Biotechnology, sc-15363), and SQSTM1/p62 (Santa Cruz Biotechnology, sc-25575). Mouse anti-GFP antibody (AG281) and 4',6-diamidino-2-phenylindole (DAPI) (C1005) were purchased from Beyotim. Horseradish peroxidase (HRP)-labeled goat anti-rabbit (A0208) and anti-mouse (A0216) secondary antibodies were also obtained from Beyotim. Tetramethyl rhodamine isothiocyanate (TRITC)-conjugated goat anti-rabbit (BS10250) and fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse (BS50950) secondary antibodies were obtained from Bioworld Technology. Eighteen-nanometer colloidal gold-affinipure goat anti-rabbit (111-215-144) and 12-nm colloidal gold-affinipure goat anti-mouse (115-205-146) secondary antibodies were purchased from Jackson ImmunoResearch. protease inhibitor E64d (E8640) and 3-methyladenine (M9281) were purchased from Sigma-Aldrich. Rapamycin (9904) was procured from Cell Signaling Technology. pEGFP-NS5A, pEGFP-N1, and mouse anti-CSFV E2 antibodies were prepared in our laboratory, and the monoclonal antibody against the NS5A protein of CSFV was kindly provided by Dr. Xinglong Yu (Veterinary Department, Hunan Agricultural University, China). Additionally, 3 shRNAs targeting different sites within the coding sequences of BECN1 and LC3B, along with the scrambled shRNA, were designed by and obtained from Cyagen. shBECN1 sequence 5′-GAGCTTCAAG ATCCTGGATC GTGTTCTCGA GAACACGATC CAGGATCTTG AAGCTC-3′, shLC3B sequence 5′-GCTTGCAGCT CAATGCTAAC CCTCGAGGGT TAGCATTGAG CTGCAAGC-3′.
Cells and virus
The swine kidney cell line PK-15 (ATCC, CCL-33) was maintained in complete Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics. Porcine macrophage cell line 3D4/2 (ATCC, CRL-2845) was cultured in RPMI 1640 medium containing 10% FBS and 1% antibiotics. The cells were incubated at 37 °C with 5% CO2.
The CSFV strain (Shimen) used in the study was isolated in our laboratory from a typical symptom of the swine and propagated in 2 cell cultures with 2% FBS. To determine the virus titers, cells cultivated in 96-well plates were inoculated with 10-fold serial dilutions of virus and incubated at 37 °C for 5 d. Cells were fixed with 80% acetone at −20 °C for 30 min, and viruses were detected by an immunofluorescence assay using mouse anti-CSFV E2 antibody and FITC-conjugated goat anti-mouse secondary antibody. Virus titers were calculated according to Kaerber and expressed as 50% tissue culture infectious doses (TCID50) per milliliter. The multiplicity of infection (MOI) was confirmed according to the virus titer from the respective cell line. UV inactivation of CSFV was performed by irradiating with UV light for 30 min at room temperature. The infectivity of UV-treated CSFV was confirmed by detecting virus titers as described above, and the MOIs were the same as those for the untreated viruses.
Viral infection
For different experiments, viral stocks were prepared at an MOI of 1 for autophagy induction and an MOI of 0.5 for replication studies. PK-15 and 3D4/2 cells were grown to approximately 80% confluence in cell culture plates and were infected with CSFV at varying MOIs. The mock was infected with phosphate-buffered saline (PBS). After 1 h, the inoculum was removed by aspiration. The cells were then washed twice with PBS and incubated in complete medium at 37 °C for different times until harvesting.
Transmission electron microscopy
Cell cultures were washed twice with PBS and collected in the bottom of 1.5-ml Eppendorf tubes by centrifugation at 1,000 rpm for 5 min. The samples were fixed in 2.5% glutaraldehyde diluted with PBS for 4 h at room temperature and then postfixed in 1% osmium tetroxide, dehydrated with graded ethanol, and embedded in epoxy resin. Next, ultrathin sections were obtained and then stained with uranyl acetate and lead citrate. Finally, the autophagosome-like vesicles were examined under a JEM-2010HR TEM (JEOL).
To determine the autophagosome-like vesicles and the localization of CSFV particles in virus-infected cells, IEM was performed. Briefly, the frozen ultrathin sections were first incubated with antibodies against LC3B, NS5A, or E2 (1:100) and then incubated with 18-nm colloidal gold-affinipure goat anti-rabbit IgG or 12-nm colloidal gold-affinipure goat anti-mouse IgG (1:40). Finally, the gold labels were measured with the JEM-2010HR TEM (magnified 60,000×).
Immunoblotting
Cell monolayers were washed twice in cold PBS and were incubated on ice with RIPA lysis buffer (Beyotim, P0013B) containing 1 mM PMSF (Beyotim, ST506) for 10 min. The lysates were then clarified by centrifugation at 13,000 rpm for 20 min at 4 °C, and the protein concentration was quantified by the BCA protein assay kit (Beyotim, P0012). Equal amounts of protein samples were boiled for 5 min in 5 × SDS-PAGE loading buffer. Proteins (20 μg) were separated on 12% SDS-PAGE gels and then electrotransferred onto polyvinylidene fluoride (PVDF) membranes (Beyotim, FFP30), which were then blocked for 2 h at 25 °C in PBS containing 2% nonfat milk powder and 0.05% Tween 20. Next, the membranes were incubated with primary antibodies at 4 °C overnight and then with the corresponding secondary antibodies conjugated to HRP at 37 °C for 1 h at appropriate dilutions. The protein bands were detected by the ECL Plus kit (Beyotim, P0018). Protein blots were measured with the Image-Pro Plus 6.0 software (Media Cybernetics), and the images were obtained from a CanoScan LiDE 100 scanner (Canon).
Confocal immunofluorescence microscopy
After CSFV infection, cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. Cell monolayers were permeabilized with 0.2% Triton X-100 for 10 min. Then the cells were blocked with PBS containing 5% bovine serum albumin (BSA) for 30 min at room temperature. Next, cells were incubated for 1 h in the presence of a rabbit polyclonal antibody (anti-LC3B or anti-CD63) (1:400) and a mouse monoclonal antibody (anti-E2 or anti-NS5A) (1:100) in PBS buffer at 37 °C, followed by a 1 h incubation in PBS containing goat anti-mouse and anti-rabbit secondary antibodies conjugated to FITC and TRITC at a dilution of 1:200. The fluorescence signals were visualized with a TCS SP2 confocal fluorescence microscope (Leica). The average number of LC3 puncta per cell from at least 100 cells in each group was counted.
To further analyze the effect of autophagosomes on CSFV infection, PK-15 and 3D4/2 cells grown to approximately 80% confluence in a petri dish with a glass bottom (NEST, GBD-35-20) were transfected with plasmid pEGFP-NS5A by using an X-tremeGENE HP DNA transfection reagent (Roche, 06366236001) as described below. After 48 h, the following immunofluorescence analysis was performed as described above. An antibody against LC3B was used to determine the localization of targeted proteins in treated cells. Cells transfected with the empty vector (pEGFP-N1) served as the control group. The efficiency of transfection and the expression of autophagy proteins were measured using immunoblotting analysis.
Biochemical intervention
For detection of autophagic flux, 2 cell lines grown to 80% confluence in 6-well cell culture plates were pretreated with 10 μg/ml E64d for 4 h, followed by a 1 h absorption of CSFV. The cells were cultured in fresh medium in the presence of 10 μg/ml E64d for 24 and 48 h. For autophagy induction and inhibition experiments, cells grown to 60% confluence in 6-well cell culture plates were pretreated with 100 nM rapamycin for 1 h or 5 mM 3-MA for 4 h prior to viral infection. Viral adsorption was performed at 37 °C for 1 h. The inoculum was removed and washed twice with PBS, and the cells were then incubated in fresh medium containing rapamycin (100 nM) or 3-MA (5 mM) until harvesting of the cells or the culture medium. Moreover, the same amount of dimethyl sulfoxide (DMSO) was added as the control group.
Transfection and gene silencing with shRNA
PK-15 and 3D4/2 cells grown to 60% confluence in 6-well cell culture plates were transfected with BECN1 or LC3B shRNA by using the X-tremeGENE HP DNA transfection reagent. Briefly, 2 μg of shRNA was diluted in 200 μl of serum-free OptiMEM medium, and 6 μl of X-tremeGENE HP was directly pipetted into the medium containing the diluted plasmid of shRNA and incubated at 25 °C for 15 min. The cell culture medium was removed and replaced with 2 ml of OptiMEM containing the transfection complex and further cultured at 37 °C. After 5 h, the supernatant was removed, and the cells were further incubated with fresh complete medium for 48 h. Following 1 h of absorption of CSFV, the cells were incubated in fresh medium until harvesting of the cells or the culture medium. The gene knockdown efficiency was evaluated by immunoblotting, and a non-targeting vector was used as a negative control.
Quantification of viral RNA
Real-time quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was used to detect virus copies. Viral RNA was extracted using the MiniBEST Viral RNA/DNA Extraction Kit Ver. 4.0 (TAKARA, DV819), and the synthesis of cDNA was performed using the PrimeScript® RT reagent Kit (TAKARA, DRR037) according to the manufacturer’s protocol. For CSFV-specific detection, a primer pair (CSFV1: CCTGAGGACC AAACACATGT TG/CSFV2: TGGTGGAAGT TGGTTGTGTC TG) reported previously in reference 67, targeting a region corresponding to the NS5B gene, was used in the study. The real-time RT-PCR was performed using SYBR® Premix Ex Taq™ II (TAKARA, DRR081) on an iQ5 iCycler detection system (Bio-Rad), by using previously described reaction conditions in reference 68. The recombinant plasmid containing the CSFV NS5B gene was used to construct a standard curve for calculating the viral copy number in different samples.
Cell viability assay
Cell viability was determined by the MTT assay according to the manufacturer’s instructions. In brief, cells were seeded in 96-well culture plates at a density of 1 × 104 cells per well and cultured for 24 h at 37 °C. The culture medium was replaced with fresh medium containing the corresponding concentrations of autophagy regulators or the same amount of DMSO, and the cells were continuously incubated for 48 h. Next, the medium was replaced with 100 μl of fresh medium supplemented with 10 μl of MTT, and the plates were further cultured for 4 h at 37 °C. Subsequently, 100 μl of formazan solution was added to each well without removing the culture medium to dissolve the precipitate at 37 °C. The optical density was measured at 570 nm by using a model 680 microplate reader (Bio-Rad).
Statistical analysis
The data are expressed as the mean ± standard deviation (SD) and were analyzed by 2-way ANOVA using the SPSS software (version 17.0). A P value of < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Dr Xinglong Yu for providing anti-NS5A antibody. This work was supported by grants from the National Natural Science Foundation of China (Nos. 31072137 and 31172321), the Key Project of Natural Science Foundation of Guangdong Province, China (No. S2011020001037), the Special Project for Scientific and Technological Innovation in Higher Education of Guangdong, China (No. 2012CXZD0013), the Special Fund for Agro-Scientific Research in the Public Interest (No. 201203056) and the Research Fund for the Doctoral Program of Higher Education of China (No. 20114404110015).
Glossary
Abbreviations:
- 3-MA
3-methyladenine
- ATG
autophagy-related
- CSFV
classical swine fever virus
- DAPI
4',6-diamidino-2-phenylindole
- DMSO
dimethyl sulphoxide
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- hpi
hours post-infection
- HRP
horseradish peroxidase
- IEM
immunoelectron microscopy
- LC3
microtubule-associated protein 1 light chain 3
- PBS
phosphate-buffered saline
- qRT-PCR
real-time quantitative reverse transcriptase polymerase chain reaction
- shRNA
short hairpin RNA
- TEM
transmission electron microscopy
- UV
ultraviolet
- MOI
multiplicity of infection
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- TCID50
50% tissue culture infectious doses
- TRITC
tetramethyl rhodamine isothiocyanate
References
- 1. Becher P, Avalos Ramirez R, Orlich M, Cedillo Rosales S, König M, Schweizer M, Stalder H, Schirrmeier H, Thiel HJ. . Genetic and antigenic characterization of novel pestivirus genotypes: implications for classification. Virology 2003; 311:96 - 104; http://dx.doi.org/ 10.1016/S0042-6822(03)00192-2; PMID: 12832207 [DOI] [PubMed] [Google Scholar]
- 2. Kleiboeker SB. . Swine fever: classical swine fever and African swine fever. Vet Clin North Am Food Anim Pract 2002; 18:431 - 51; http://dx.doi.org/ 10.1016/S0749-0720(02)00028-2; PMID: 12442576 [DOI] [PubMed] [Google Scholar]
- 3. Lohse L, Nielsen J, Uttenthal A. . Early pathogenesis of classical swine fever virus (CSFV) strains in Danish pigs. Vet Microbiol 2012; 159:327 - 36; http://dx.doi.org/ 10.1016/j.vetmic.2012.04.026; PMID: 22608103 [DOI] [PubMed] [Google Scholar]
- 4. König M, Lengsfeld T, Pauly T, Stark R, Thiel HJ. . Classical swine fever virus: independent induction of protective immunity by two structural glycoproteins. J Virol 1995; 69:6479 - 86; PMID: 7666549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moormann RJ, Bouma A, Kramps JA, Terpstra C, De Smit HJ. . Development of a classical swine fever subunit marker vaccine and companion diagnostic test. Vet Microbiol 2000; 73:209 - 19; http://dx.doi.org/ 10.1016/S0378-1135(00)00146-2; PMID: 10785329 [DOI] [PubMed] [Google Scholar]
- 6. Bensaude E, Turner JL, Wakeley PR, Sweetman DA, Pardieu C, Drew TW, Wileman T, Powell PP. . Classical swine fever virus induces proinflammatory cytokines and tissue factor expression and inhibits apoptosis and interferon synthesis during the establishment of long-term infection of porcine vascular endothelial cells. J Gen Virol 2004; 85:1029 - 37; http://dx.doi.org/ 10.1099/vir.0.19637-0; PMID: 15039545 [DOI] [PubMed] [Google Scholar]
- 7. Sun J, Shi Z, Guo H, Tu C. . Changes in the porcine peripheral blood mononuclear cell proteome induced by infection with highly virulent classical swine fever virus. J Gen Virol 2010; 91:2254 - 62; http://dx.doi.org/ 10.1099/vir.0.022020-0; PMID: 20463149 [DOI] [PubMed] [Google Scholar]
- 8. Johns HL, Bensaude E, La Rocca SA, Seago J, Charleston B, Steinbach F, Drew TW, Crooke H, Everett H. . Classical swine fever virus infection protects aortic endothelial cells from pIpC-mediated apoptosis. J Gen Virol 2010; 91:1038 - 46; http://dx.doi.org/ 10.1099/vir.0.016576-0; PMID: 20007358 [DOI] [PubMed] [Google Scholar]
- 9. Edwards S, Fukusho A, Lefèvre PC, Lipowski A, Pejsak Z, Roehe P, Westergaard J. . Classical swine fever: the global situation. Vet Microbiol 2000; 73:103 - 19; http://dx.doi.org/ 10.1016/S0378-1135(00)00138-3; PMID: 10785321 [DOI] [PubMed] [Google Scholar]
- 10. Stegeman A, Elbers A, de Smit H, Moser H, Smak J, Pluimers F. . The 1997-1998 epidemic of classical swine fever in the Netherlands. Vet Microbiol 2000; 73:183 - 96; http://dx.doi.org/ 10.1016/S0378-1135(00)00144-9; PMID: 10785327 [DOI] [PubMed] [Google Scholar]
- 11. Moennig V. . Introduction to classical swine fever: virus, disease and control policy. Vet Microbiol 2000; 73:93 - 102; http://dx.doi.org/ 10.1016/S0378-1135(00)00137-1; PMID: 10785320 [DOI] [PubMed] [Google Scholar]
- 12. Ressang AA. . Studies on the pathogenesis of hog cholera. II. Virus distribution in tissue and the morphology of the immune response. Zentralbl Veterinarmed B 1973; 20:272 - 88; http://dx.doi.org/ 10.1111/j.1439-0450.1973.tb01127.x; PMID: 4751662 [DOI] [PubMed] [Google Scholar]
- 13. Tautz N, Meyers G, Thiel HJ. . Pathogenesis of mucosal disease, a deadly disease of cattle caused by a pestivirus. Clin Diagn Virol 1998; 10:121 - 7; http://dx.doi.org/ 10.1016/S0928-0197(98)00037-3; PMID: 9741637 [DOI] [PubMed] [Google Scholar]
- 14. Knoetig SM, Summerfield A, Spagnuolo-Weaver M, McCullough KC. . Immunopathogenesis of classical swine fever: role of monocytic cells. Immunology 1999; 97:359 - 66; http://dx.doi.org/ 10.1046/j.1365-2567.1999.00775.x; PMID: 10447754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reggiori F. . 1. Membrane origin for autophagy. Curr Top Dev Biol 2006; 74:1 - 30; http://dx.doi.org/ 10.1016/S0070-2153(06)74001-7; PMID: 16860663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klionsky DJ. . Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931 - 7; http://dx.doi.org/ 10.1038/nrm2245; PMID: 17712358 [DOI] [PubMed] [Google Scholar]
- 17. Klionsky DJ, Emr SD. . Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/ 10.1126/science.290.5497.1717; PMID: 11099404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin LT, Dawson PW, Richardson CD. . Viral interactions with macroautophagy: a double-edged sword. Virology 2010; 402:1 - 10; http://dx.doi.org/ 10.1016/j.virol.2010.03.026; PMID: 20413139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tanida I. . Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal 2011; 14:2201 - 14; http://dx.doi.org/ 10.1089/ars.2010.3482; PMID: 20712405 [DOI] [PubMed] [Google Scholar]
- 20. Shintani T, Klionsky DJ. . Autophagy in health and disease: a double-edged sword. Science 2004; 306:990 - 5; http://dx.doi.org/ 10.1126/science.1099993; PMID: 15528435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Levine B, Kroemer G. . Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/ 10.1016/j.cell.2007.12.018; PMID: 18191218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deretic V. . Autophagy in infection. Curr Opin Cell Biol 2010; 22:252 - 62; http://dx.doi.org/ 10.1016/j.ceb.2009.12.009; PMID: 20116986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. . Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586 - 96; PMID: 9765397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y, Schiff M, Czymmek K, Tallóczy Z, Levine B, Dinesh-Kumar SP. . Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005; 121:567 - 77; http://dx.doi.org/ 10.1016/j.cell.2005.03.007; PMID: 15907470 [DOI] [PubMed] [Google Scholar]
- 25. Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. . HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007; 1:23 - 35; http://dx.doi.org/ 10.1016/j.chom.2006.12.001; PMID: 18005679 [DOI] [PubMed] [Google Scholar]
- 26. Alirezaei M, Kiosses WB, Flynn CT, Brady NR, Fox HS. . Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One 2008; 3:e2906; http://dx.doi.org/ 10.1371/journal.pone.0002906; PMID: 18682838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, et al. . Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 2009; 186:255 - 68; http://dx.doi.org/ 10.1083/jcb.200903070; PMID: 19635843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. . Autophagosome supports coxsackievirus B3 replication in host cells. J Virol 2008; 82:9143 - 53; http://dx.doi.org/ 10.1128/JVI.00641-08; PMID: 18596087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, Lin YS, Yeh TM, Liu CC, Liu HS. . Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008; 374:240 - 8; http://dx.doi.org/ 10.1016/j.virol.2008.02.016; PMID: 18353420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou Z, Jiang X, Liu D, Fan Z, Hu X, Yan J, Wang M, Gao GF. . Autophagy is involved in influenza A virus replication. Autophagy 2009; 5:321 - 8; http://dx.doi.org/ 10.4161/auto.5.3.7406; PMID: 19066474 [DOI] [PubMed] [Google Scholar]
- 31. Dreux M, Gastaminza P, Wieland SF, Chisari FV. . The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 2009; 106:14046 - 51; http://dx.doi.org/ 10.1073/pnas.0907344106; PMID: 19666601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klionsky DJ, Cuervo AM, Seglen PO. . Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181 - 206; PMID: 17224625 [DOI] [PubMed] [Google Scholar]
- 33. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. . Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. . A protein conjugation system essential for autophagy. Nature 1998; 395:395 - 8; http://dx.doi.org/ 10.1038/26506; PMID: 9759731 [DOI] [PubMed] [Google Scholar]
- 35. Wang CW, Klionsky DJ. . The molecular mechanism of autophagy. Mol Med 2003; 9:65 - 76; PMID: 12865942 [PMC free article] [PubMed] [Google Scholar]
- 36. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. . Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/ 10.1016/j.cell.2005.07.002; PMID: 16179260 [DOI] [PubMed] [Google Scholar]
- 37. Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. . p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/ 10.1083/jcb.200507002; PMID: 16286508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. . p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/ 10.1074/jbc.M702824200; PMID: 17580304 [DOI] [PubMed] [Google Scholar]
- 39. Mizushima N, Yoshimori T. . How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390 [DOI] [PubMed] [Google Scholar]
- 40. Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. . Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1:84 - 91; http://dx.doi.org/ 10.4161/auto.1.2.1697; PMID: 16874052 [DOI] [PubMed] [Google Scholar]
- 41. Metzelaar MJ, Wijngaard PL, Peters PJ, Sixma JJ, Nieuwenhuis HK, Clevers HC. . CD63 antigen. A novel lysosomal membrane glycoprotein, cloned by a screening procedure for intracellular antigens in eukaryotic cells. J Biol Chem 1991; 266:3239 - 45; PMID: 1993697 [PubMed] [Google Scholar]
- 42. Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ. . The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 1997; 243:240 - 6; http://dx.doi.org/ 10.1111/j.1432-1033.1997.0240a.x; PMID: 9030745 [DOI] [PubMed] [Google Scholar]
- 43. Noda T, Ohsumi Y. . Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273:3963 - 6; http://dx.doi.org/ 10.1074/jbc.273.7.3963; PMID: 9461583 [DOI] [PubMed] [Google Scholar]
- 44. Hay N, Sonenberg N. . Upstream and downstream of mTOR. Genes Dev 2004; 18:1926 - 45; http://dx.doi.org/ 10.1101/gad.1212704; PMID: 15314020 [DOI] [PubMed] [Google Scholar]
- 45. Seglen PO, Gordon PB. . 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 1982; 79:1889 - 92; http://dx.doi.org/ 10.1073/pnas.79.6.1889; PMID: 6952238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. . Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275:992 - 8; http://dx.doi.org/ 10.1074/jbc.275.2.992; PMID: 10625637 [DOI] [PubMed] [Google Scholar]
- 47. Miller S, Krijnse-Locker J. . Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 2008; 6:363 - 74; http://dx.doi.org/ 10.1038/nrmicro1890; PMID: 18414501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Z, Lu Y, Zhou P, Zhai Z, Ding M. . [The morphological structure of classical swine fever virus and some characteristics of its multiplication]. Wei Sheng Wu Xue Bao 1999; 39:189 - 95; PMID: 12555533 [PubMed] [Google Scholar]
- 49. Levine B, Deretic V. . Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007; 7:767 - 77; http://dx.doi.org/ 10.1038/nri2161; PMID: 17767194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmid D, Münz C. . Innate and adaptive immunity through autophagy. Immunity 2007; 27:11 - 21; http://dx.doi.org/ 10.1016/j.immuni.2007.07.004; PMID: 17663981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. . Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005; 307:593 - 6; http://dx.doi.org/ 10.1126/science.1104904; PMID: 15591165 [DOI] [PubMed] [Google Scholar]
- 52. Grummer B, Fischer S, Depner K, Riebe R, Blome S, Greiser-Wilke I. . Replication of classical swine fever virus strains and isolates in different porcine cell lines. Dtsch Tierarztl Wochenschr 2006; 113:138 - 42; PMID: 16716048 [PubMed] [Google Scholar]
- 53. Sun J, Jiang Y, Shi Z, Yan Y, Guo H, He F, Tu C. . Proteomic alteration of PK-15 cells after infection by classical swine fever virus. J Proteome Res 2008; 7:5263 - 9; http://dx.doi.org/ 10.1021/pr800546m; PMID: 19367723 [DOI] [PubMed] [Google Scholar]
- 54. Mittelholzer C, Moser C, Tratschin JD, Hofmann MA. . Porcine cells persistently infected with classical swine fever virus protected from pestivirus-induced cytopathic effect. J Gen Virol 1998; 79:2981 - 7; PMID: 9880012 [DOI] [PubMed] [Google Scholar]
- 55. Vandergaast R, Fredericksen BL. . West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS One 2012; 7:e45800; http://dx.doi.org/ 10.1371/journal.pone.0045800; PMID: 23029249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Beatman E, Oyer R, Shives KD, Hedman K, Brault AC, Tyler KL, Beckham JD. . West Nile virus growth is independent of autophagy activation. Virology 2012; 433:262 - 72; http://dx.doi.org/ 10.1016/j.virol.2012.08.016; PMID: 22939285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. . Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol 2008; 82:2241 - 9; http://dx.doi.org/ 10.1128/JVI.02093-07; PMID: 18077704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang Y, Li Z, Ge X, Guo X, Yang H. . Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy 2011; 7:613 - 28; http://dx.doi.org/ 10.4161/auto.7.6.15267; PMID: 21460631 [DOI] [PubMed] [Google Scholar]
- 59. Shirota Y, Luo H, Qin W, Kaneko S, Yamashita T, Kobayashi K, Murakami S. . Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J Biol Chem 2002; 277:11149 - 55; http://dx.doi.org/ 10.1074/jbc.M111392200; PMID: 11801599 [DOI] [PubMed] [Google Scholar]
- 60. Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. . Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 2003; 77:5487 - 92; http://dx.doi.org/ 10.1128/JVI.77.9.5487-5492.2003; PMID: 12692249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tellinghuisen TL, Paulson MS, Rice CM. . The NS5A protein of bovine viral diarrhea virus contains an essential zinc-binding site similar to that of the hepatitis C virus NS5A protein. J Virol 2006; 80:7450 - 8; http://dx.doi.org/ 10.1128/JVI.00358-06; PMID: 16840325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen Y, Xiao J, Xiao J, Sheng C, Wang J, Jia L, Zhi Y, Li G, Chen J, Xiao M. . Classical swine fever virus NS5A regulates viral RNA replication through binding to NS5B and 3’UTR. Virology 2012; 432:376 - 88; http://dx.doi.org/ 10.1016/j.virol.2012.04.014; PMID: 22795973 [DOI] [PubMed] [Google Scholar]
- 63. He L, Zhang YM, Lin Z, Li WW, Wang J, Li HL. . Classical swine fever virus NS5A protein localizes to endoplasmic reticulum and induces oxidative stress in vascular endothelial cells. Virus Genes 2012; 45:274 - 82; http://dx.doi.org/ 10.1007/s11262-012-0773-2; PMID: 22718084 [DOI] [PubMed] [Google Scholar]
- 64. Li JK, Liang JJ, Liao CL, Lin YL. . Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect 2012; 14:159 - 68; http://dx.doi.org/ 10.1016/j.micinf.2011.09.001; PMID: 21946213 [DOI] [PubMed] [Google Scholar]
- 65. Jackson WT, Giddings TH Jr., Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. . Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005; 3:e156; http://dx.doi.org/ 10.1371/journal.pbio.0030156; PMID: 15884975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O’Donnell V, Pacheco JM, LaRocco M, Burrage T, Jackson W, Rodriguez LL, Borca MV, Baxt B. . Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 2011; 410:142 - 50; http://dx.doi.org/ 10.1016/j.virol.2010.10.042; PMID: 21112602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Díaz de Arce H, Nuñez JI, Ganges L, Barreras M, Frías MT, Sobrino F. . An RT-PCR assay for the specific detection of classical swine fever virus in clinical samples. Vet Res 1998; 29:431 - 40; PMID: 9779556 [PubMed] [Google Scholar]
- 68. Pérez LJ, Díaz de Arce H, Tarradas J, Rosell R, Perera CL, Muñoz M, Frías MT, Nuñez JI, Ganges L. . Development and validation of a novel SYBR Green real-time RT-PCR assay for the detection of classical swine fever virus evaluated on different real-time PCR platforms. J Virol Methods 2011; 174:53 - 9; http://dx.doi.org/ 10.1016/j.jviromet.2011.03.022; PMID: 21458490 [DOI] [PubMed] [Google Scholar]