

Abstract
A main function of the heart is to pump blood to the tissues and organs of the body. Although formed by different types of cells, the cardiomyocytes are the ones responsible for the coordinated and synchronized heart contraction. Given their low mitotic activity, cardiomyocytes largely depend on protein degradation mechanisms to maintain proteostasis and energetic balance. Autophagy, one of the main pathways whereby cells eliminate damaged, nonfunctional, or obsolete proteins, and organelles, is vital to ensure cell function, including in cardiomyocytes, both in rest and stress conditions. However, the impact of autophagy activation in the heart, being either protective or harmful, is not consensual and likely depends upon the severity of the stimuli and consequently the autophagy players involved. One of the signals that direct proteins for autophagy degradation, namely in the context of heart disorders, is ubiquitin. Indeed, the attachment of ubiquitin moieties to a target substrate and further recognition by autophagy adaptors constitute a main regulatory pathway that directs proteins to the lysosome. Therefore, a better understanding of the mechanisms and signals that regulate the autophagy process in the heart, including substrates targeting, is of utmost importance to design new approaches directed to this degradation pathway. We have previously shown that ubiquitination of the gap junction (GJ) protein Connexin43 (Cx43) triggers its degradation by autophagy through a process that requires the ubiquitin adaptors epidermal growth factor receptor substrate 15 (Eps15) and p62. This is particularly relevant in the heart because GJs, that form intercellular channels, are responsible for the rapid and efficient anisotropic propagation of the electrical impulse through the cardiomyocytes, essential for synchronized contraction of the cardiac muscle. In this review, we present recent studies devoted to the involvement of autophagy in heart homeostasis, with a particular focus on ubiquitin and GJs.
Introduction
The maintenance of protein homeostasis, essential for cell survival, relies on a myriad of mechanisms and players that include molecular chaperones and protein degradation systems. These strategies are particularly important in cells with a low mitotic activity, in which the accumulation of misfolded and/or toxic protein aggregates can result in cell damage and death, ultimately leading to organ and system failure. One good example of an organ that largely depends upon the mechanisms of protein degradation is the heart. Indeed, given the postmitotic nature of cardiomyocytes, which constitute the main contractile unit of the heart, these cells require a competent proteolytic machinery to maintain the proteostasis. Therefore, a deregulation of the proteolytic systems in the heart is often associated with cardiac diseases. In this review, we will describe some of the mechanisms and pathways involved in ubiquitin-dependent protein degradation in the heart, giving particular attention to autophagy.
Protein Degradation and Ubiquitination
Protein homeostasis is the process by which an integrated network of biological processes that maintain a functional group of proteins in their native state and degrade damaged, misfolded, or unwanted proteins, ensuring normal functioning of cells, tissues and organs. Protein degradation or proteolysis relies on two different systems: (i) the lysosomes, which are membrane-bound organelles with an acidic lumen containing proteolytic enzymes and (ii) the proteasome, which constitutes a barrel-like structure, with a limited access of substrates to its catalytically active interior.
The ubiquitin–proteasome system (UPS) is the main cytosolic proteolytic system in eukaryotes, being implicated in a wide variety of cell functions with relevance to health and disease. In general, degradation of a protein mediated by the UPS consists of two successive steps. The first step involves the covalent attachment of a polyubiquitin chain to the target protein, in a process generally called ubiquitination, while in the second step, the ubiquitin-tagged protein is recognized and degraded in the proteasome complex, with the prior release of ubiquitin, which is subsequently used in new cycles of protein ubiquitination (Glickman and Ciechanover, 2002; Schmidt and Finley, 2014). Conjugation of ubiquitin to a substrate is a highly regulated and orchestrated three-step cascade of enzymatic reactions catalyzed by a ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), and ubiquitin ligase (E3), sometimes acting as a ubiquitin chain elongation factor. In brief, ubiquitin is first activated by the E1 through an ATP-dependent energy reaction originating an E1-thioester intermediate, E1-S∼ubiquitin. Next, the activated ubiquitin is transferred to an E2 that finally shuttles ubiquitin to a substrate, either by itself or in cooperation with an E3 that specifically binds to the target protein (Pickart and Eddins, 2004). The covalent binding of ubiquitin to a substrate occurs, usually, through an isopeptide bond formed between the -COOH group of the glycine terminal of ubiquitin and the free ɛ-NH2 group of a lysine (Lys) residue present in the target protein. More recently, it was demonstrated that ubiquitin can also be conjugated to the terminal amino group of the substrate (Komander and Rape, 2012).
In successive reactions, a polyubiquitin chain is synthesized by the addition of ubiquitin moieties to the Lys of the previously conjugated ubiquitin molecule. Given the presence of seven potentially ubiquitinable Lys residues in ubiquitin, a complex variety of polyubiquitin chains with different topologies can be putatively formed. It has been widely shown that different types of polyubiquitin chains are associated with diverse biological functions, including the regulation of DNA repair, endocytosis, autophagy, protein activity, and proteasomal degradation. Importantly, the role of ubiquitin in such processes has not only to do with targeting proteins for degradation but also with the modulation of protein activity (Welchman et al., 2005; Mukhopadhyay and Riezman, 2007; Komander and Rape, 2012). Although ubiquitin-dependent degradation was initially described as a signal for the proteasome, more recent studies have ascribed a role to protein ubiquitination in targeting lysosomal degradation.
Similar to many other biological processes that are reversible, protein ubiquitination may also be regulated by deubiquitinating enzymes (DUBs), which cleave ubiquitin isopeptide linkages, selectively removing ubiquitin moieties from tagged proteins. Being ubiquitin considered a signal for degradation, an immediate intuitive role for DUBs is to rescue proteins from degradation. Although initially thought as two independent events, it is now widely accepted that ubiquitination and deubiquitination processes happen alongside. Indeed, it has been shown for different substrates that the effect of ubiquitin on protein turnover and/or activity, largely depending on the length and type of ubiquitin modifications, relies on a complex chain editing process carried out by the orchestrated action of E3 ligases and DUBs. Therefore, DUB-catalyzed deubiquitination of proteins can not only prevent ubiquitin-dependent degradation but also modulate protein activity. For example, the activity of several components of autophagy and endocytic machinery is regulated by ubiquitination/deubiquitination. On the other hand, the removal of ubiquitin from substrates can also promote degradation. In fact, both in lysosomal and proteasomal degradation, before the entrance into the proteolytic chamber (lysosome or proteasome), DUBs catalyze the removal of ubiquitin from the substrates, thus avoiding the degradation of ubiquitin and allowing the recycling to free ubiquitin that will be further used in new ubiquitination cycles. Another function of DUBs is the processing of inactive transcribed ubiquitin precursors, producing active forms available to be reused in protein ubiquitination. An emergent idea in the field is that E3 ubiquitin ligases and DUBs compete along the degradation pathway, thus determining the ultimate fate of substrates, them being either degraded or recycled (Komander et al., 2009; Eletr and Wilkinson, 2014).
Despite being first described as a signal for UPS degradation, ubiquitin and ubiquitin chains have also been shown to play an important role in protein degradation in the lysosome. The most prevalent idea in the field is that depending on the type of ubiquitin chain, a substrate can be degraded either in the proteasome or in the lysosome. For example, globular polyubiquitin chains formed through the Lys48 are usually considered a signal for proteasomal degradation, whereas monoubiquitination, multimonoubiquitination, and linear Lys63-linked ubiquitin chains have been implicated in the internalization of membrane channels and receptors and lysosomal degradation of both cytosolic and membrane proteins.
Autophagy
The degradation of cellular components by the lysosome can occur through endocytosis, in which, after a complex maturation process, endosomes converge in the lysosome to deliver their cargo, or by autophagy. Based on the route by which a substrate enters the lysosome, three autophagy-related pathways have been described, namely macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), which can promote bulk and selective degradation of organelles and cytosolic components (Mizushima et al., 2008). In macroautophagy, proteins, cytosolic regions, or organelles are sequestered inside double-membrane vesicles (autophagosomes) that then fuse with the lysosome for cargo degradation. This process, which can be highly selective, is mediated by a number of proteins encoded by autophagy-related genes (ATGs) that regulate the biogenesis of autophagosome and its subsequent fusion with the lysosome. The core macroautophagy machinery can be divided into four functional subgroups according to the different dynamic stages of the autophagic process: (i) the induction of autophagosome formation, mediated by the mammalian homolog of the yeast autophagy-related gene 1 (Atg1)/UNC-51-like kinase 1 (ULK1) complex (Atg1, Atg11, Atg13, Atg17, Atg29, and Atg31), (ii) the membrane delivery to the expanding phagophore, by Atg9 and its cycling system (Atg2, Atg9, and Atg18), (iii) the vesicle nucleation, mediated by class III phosphatidylinositol 3-kinase (PI3K-III) complex (Vps34, Vps15, Vps30/Atg6/Beclin 1, and Atg14), and finally, (iv) the vesicle expansion, by the concerted action of two ubiquitin-like conjugation systems: the Atg12 (Atg5, Atg7, Atg10, Atg12, and Atg16) and Atg8 (Atg3, Atg4, Atg7, and Atg8) (Ravikumar et al., 2010). At the stage of phagophore elongation, the lipidation of the cytosolic microtubule-associated protein 1 light chain 3 (Atg8/LC3)—LC3-I, occurs with the subsequent formation of LC3-II that translocates to the autophagosome membrane (Feng et al., 2013).
Cellular energy levels are strikingly related with the autophagy machinery by means of three major energy-sensing pathways—(i) protein kinase B (Akt), (ii) adenosine monophosphate-activated protein kinase (AMPK), and (iii) mammalian target of rapamycin (mTOR) (Nishida et al., 2009). Oxygen or nutrient deprivation can activate AMPK through ADP:AMP accumulation, which negatively regulates mTOR, either by AMPK-mediated phosphorylation of mTOR or by activation of its upstream repressor tuberous sclerosis complex. In the absence of oxygen, the suppression of the inactivation of mTOR signaling can also be caused by amino acid withdrawal and by the lack of oxygen through the upregulation of hypoxia-responsive genes.
Another less understood type of autophagy, called microautophagy, involves the nonselective engulfment of cytoplasmic material by inward invagination of the lysosomal.
In contrast to the in-bulk sequestration of cytosolic components characteristic of macro- and microautophagy, soluble cytosolic proteins can be targeted selectively for degradation in lysosomes by a process called CMA in which a KFERQ sequence in the substrate protein is recognized by a cytosolic chaperone, heat shock cognate protein of 70 kDa (Hsc70), which then delivers them to the surface of the lysosomes for internalization through a translocation complex formed by lysosomal-associated membrane protein-2A (LAMP-2A) (Kaushik and Cuervo, 2012).
Ubiquitin and Autophagy
Degradation by autophagy is critical not only in the maintenance of protein homeostasis in basal conditions but also to overcome stress inducing phenomena, such as nutrient deprivation, oxidative stress, and hypoxia (Kroemer et al., 2010; Ravikumar et al., 2010; Mizushima and Komatsu, 2011). In this case, autophagy may act as a protective mechanism that eliminates potentially toxic aggregates, limits the accumulation of ubiquitinated proteins, and allows the selective removal of dysfunctional or obsolete organelles, such as mitochondria, which release proapoptotic factors and generate reactive oxygen species (Youle and Narendra, 2011). Moreover, when nutrients are scarce, the protective effect of autophagy is associated with the replenishing of the intracellular pool of amino acids required to ensure energy demand and synthesis of vital proteins essential for cell survival. Besides its role in protecting cells in adverse conditions, autophagy can also constitute a mechanism of protein quality control, by selectively degrading unwanted cellular components (Sarkar et al., 2009; Yao, 2010). Selective degradation of ubiquitinated substrates is conferred by autophagy adaptors, such as p62/SQSTM1 (sequestosome 1) and neighbor of BRCA1 (NBR1), which recognize ubiquitin moieties bound to target proteins and then direct them to the autophagosome, through the interaction with LC3 (Lamark et al., 2009; Stolz et al., 2014). Additionally, and as described for endocytosis, ubiquitination has been established to play an essential role on the regulation of autophagy itself, through the control of activity, recruitment, and turnover of autophagy components (Kirkin et al., 2009). Recently, Nazio et al. (2013) demonstrated that upon autophagy activation, the autophagy/Beclin 1 regulator 1 (AMBRA1) interacts with the E3-ligase TNF receptor associated factor 6 (TRAF6), promoting the ubiquitination of ULK1 and, consequently, modulating its stabilization, self-association, and function. Moreover, several studies have reported that autophagy activity can be regulated at the levels of Beclin 1 ubiquitination, with several E3 ligases, including the neural precursor cell expressed developmentally downregulated protein 4 (Nedd4), AMBRA1, and TRAF6, implicated in this process (Shi and Kehrl, 2010; Platta et al., 2011; Xia et al., 2013). While AMBRA1 was described to mediate Beclin 1 Lys63-ubiquitination during starvation-induced autophagy (Xia et al., 2013), TRAF6 ubiquitinates Beclin 1, also through Lys63 chains, in response to Toll-like receptor 4 (TLR4)-induced autophagy (Shi and Kehrl, 2010). In both cases, ubiquitination of Beclin 1 enhances its binding to PI3K-III, which leads to increased PI3K-III activity, and consequently, autophagy activation. On the other hand, inhibition of deubiquitination activity and the maintenance of ubiquitination levels can promote the degradation of key players of autophagy, namely Vps34 complexes, inhibiting autophagy (Liu et al., 2011).
The relationship between the three types of autophagy and UPS remains largely unknown, but several evidences point to the existence of an interplay between them (Wong and Cuervo, 2010; Park and Cuervo, 2013). For example, it has been frequently suggested that the impairment of a particular type of degradation pathway can result in the activation of another one, as part of a compensatory mechanism. Indeed, it has been widely shown in different biological models that the impairment of the UPS, with the consequent accumulation of polyubiquitinated proteins, leads to an increase in macroautophagy activity (Du et al., 2009; Zhu et al., 2010; Zheng et al., 2011b). Moreover, the putative communication between the autophagy degradation pathways is highlighted by the studies showing that activation of CMA and macroautophagy occurs sequentially, with CMA being activated in prolonged stress conditions, whereas macroautophagy is triggered in the earlier stages (Kaushik et al., 2008; Kaushik and Cuervo, 2012; Ferreira et al., 2013; Cuervo and Wong, 2014).
Ubiquitin, Autophagy, and the Heart
The requirement of proteolysis to maintain protein homeostasis is particularly important in nondiving cells, in which the persistent and chronic accumulation of potentially toxic proteins and aggregates may lead to cell death and, consequently, compromise the function of organs and tissues. As postmitotic cells, cardiomyocytes largely depend on efficient mechanisms of protein quality control to maintain the energetic balance (Terman and Brunk, 2005). Therefore, protein degradation processes, including both UPS and autophagy, play a vital role in the regulation of cardiac remodeling and response to stress. Not surprisingly, an impairment of the proteolytic pathways has been often associated with cardiomyopathy. Although initially thought as two independent pathways, it is now widely accepted that UPS and autophagy cross talk in the heart. Some studies suggest a synergic role for UPS and autophagy allowing degradation of larger substrate pools needed in both physiological and pathological conditions. Other studies show that autophagy can be activated as a compensatory mechanism following UPS dysfunction. For instance, during myocardial ischemia, UPS is impaired, likely as a consequence of ATP depletion, which leads to the accumulation of ubiquitinated proteins and aggregates that can subsequently lead to cytotoxicity (Zheng et al., 2011a). Under these conditions, autophagy adaptors, such as p62 and NBR1, have been described as important players linking ubiquitination machinery and autophagic degradation. Indeed, under these circumstances, p62 can act as a shuttle, diverting ubiquitinated proteasomal substrates to the autophagy pathway (Su and Wang, 2011). Also, p62 assumes important roles in cardiac proteinopathies and myocardial ischemia, participating in aggresome formation and autophagy activation by misfolded proteins in cardiomyocytes (Bhuiyan et al., 2013).
As stated above, the autophagy activity can be regulated by the ubiquitination of autophagy components (Kirkin et al., 2009). Consistently, several E3 ligases have been associated with autophagy regulation in the heart, and besides their activity-dependent role upon ubiquitination and regulation of autophagy machinery stability, E3 ligases can also have activity-independent roles, by facilitating the recruitment of autophagy adaptors, namely p62 and NBR1 (Kirkin et al., 2009). Recent studies have shown that the HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1 (HACE1) play a protective role in the heart, by controlling autophagic clearance of ubiquitinated protein aggregates, independently of its E3 ligase activity (Zhang et al., 2014). Another E3 ligase, Atrogin-1, was demonstrated to have a role upon regulation of autophagy in cardiomyocytes (Zaglia et al., 2014). Additionally, Atrogin-1 is an important mediator of the cross talk between UPS and autophagy, once it targets the endosomal sorting complex required for transport-III (ESCRT-III) protein charged multivesicular body protein 2B (CHMP2B), a key effector of autophagy, for proteasomal degradation. Studies from Zaglia et al. (2014) have also shown that knockout of Atrogin-1 in mice hearts leads to accumulation of CHMP2B, causing autophagosome accumulation, apoptosis, and ultimately, cardiomyopathy.
Ubiquitin, Autophagy, the Heart, and Gap Junctions
Increasing amount of evidence has suggested that both the UPS and autophagy machinery have a specific and well-regulated subcellular localization throughout heart compartments, which is of extreme importance in the regulation of cardiac proteome turnover and consequently for heart function. For instance, several studies have demonstrated the presence of ubiquitin conjugates, autophagosomes, and autophagy machinery proteins, such as p62, at the intercalated discs that constitute plasma membrane domains enriched in adhering junctions, including gap junctions (GJs) (Lange et al., 2005; Lyon et al., 2013). These results suggest that intercalated discs act as anchors to hold autophagy and UPS machinery, thus implicating these domains in highly regulated proteolytic activities.
The specific localization of GJs at intercalated discs allows the rapid and anisotropic propagation of the cardiac impulse required for a coordinated contraction of the heart muscle, and since a normal heart function relies on the proper electric and metabolic coupling between cardiomyocytes, an efficient gap junction intercellular communication (GJIC) has to be maintained. Indeed, GJs allow the cardiomyocytes to form an integrated network of cells with low resistance for the spreading of the electrical impulse, enabling the heart to work as a syncytium. GJs are intercellular channels formed by the docking of two hemichannels, each one being composed by the assembly of six subunits of a transmembrane protein called Connexin (Cx) (Thevenin et al., 2013). Several members of the Cx family are expressed in the heart, with Cx43 being the main protein of ventricular GJs. Unsurprisingly, degradation of Cx43 and impairment of GJIC have been associated with several cardiomyopathies, such as heart failure and myocardial ischemia (Beardslee et al., 2000; Severs et al., 2008). The molecular mechanisms and signaling pathways whereby Cx43 is degraded have been a matter of intense research in the last years and had already contributed to develop some promising therapeutic strategies against heart disorders. Several studies demonstrated that the E3 ligase Nedd4 mediates ubiquitination of Cx43, triggering GJ internalization and degradation of Cx43 (Girao et al., 2009; Catarino et al., 2011). Furthermore, we showed that Cx43 ubiquitination signals the degradation of GJ through autophagy and endocytic pathways, both being dependent on the canonical endocytic adaptor epidermal growth factor receptor substrate 15 (Eps15) (Bejarano et al., 2012) (Fig. 1). More recently, we established that deubiquitination of Cx43 catalyzed by associated molecule with the SH3-domain of STAM (AMSH) stabilizes GJs at the plasma membrane (Ribeiro-Rodrigues et al., 2014). Therefore, it is plausible that the modulation of the ubiquitin-mediated mechanisms whereby Cx43 is degraded may constitute privileged targets to maintain an efficient GJIC between cardiac cells, thus preventing loss of heart function.
FIG. 1.

Proposed model for ischemia-induced degradation of Connexin43 (Cx43). Under ischemic conditions, Cx43 is ubiquitinated by Nedd4, a recognized signal for interaction with epidermal growth factor receptor substrate 15 (Eps15) and proteins of the autophagy machinery. Consequently, Cx43 is internalized and degraded through autophagy, after fusion with the lysosomes.
Although degradation of Cx43 by autophagy has only been demonstrated in starved cells, it is conceivable that activation of autophagy in the ischemic heart triggers the degradation of Cx43. Recent studies carried out in our laboratory suggest that ischemia-induced remodeling of GJs is mediated by the ubiquitination of Cx43 localized at the intercalated discs. Indeed, using the Langendorff heart perfusion model, we showed that rat hearts subjected to 30 min of no-flow ischemia present dephosphorylation and lateralization of Cx43, with the concomitant recruitment of ubiquitin to intercalated discs (Fig. 2, unpublished data).
FIG. 2.
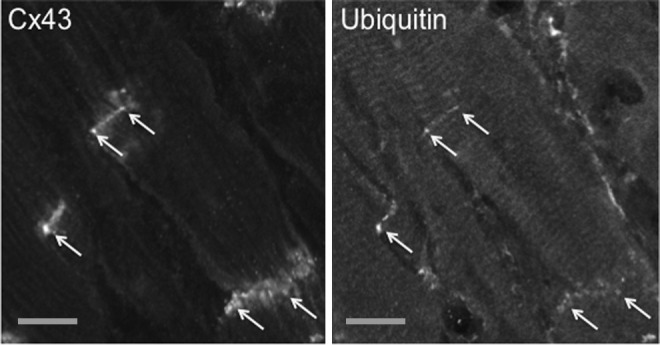
Ischemia induces colocalization of ubiquitin with Cx43 at the intercalated discs. Hearts from 10-week-old Wistar rats were maintained using a Langendorff apparatus for 10 min, followed by 30 min of no-flow ischemia. Cryosections of ischemic hearts were immunostained using antibodies directed against Cx43 and ubiquitin. Arrows indicate the colocalization of Cx43 with ubiquitin at discrete points of intercalated discs localized on the opposing ends of cardiomyocytes. Scale bars, 25 μm.
Although the involvement of autophagy in degradation of Cx43 in cardiomyocytes has never been demonstrated, these data strongly suggest that the increased ubiquitination of proteins localized at the intercalated discs upon ischemia may constitute a triggering signal for further autophagic degradation and protein remodeling usually associated with cardiac disorders.
As stated above, in the normal myocardium, autophagy is essential for the maintenance of proteostasis, including an accurate regulation of cardiac structure and function. In agreement, several studies have demonstrated that a failure of autophagy activity underlies some cardiac malfunctions. Nakai et al. (2007) showed that Atg5-deficient hearts present altered sarcomeric structure, mitochondrial misalignment and aggregation, and accumulation of ubiquitinated proteins, which likely contribute to the phenotype of contractile dysfunction and cardiac hypertrophy in these animals. Moreover, ultraviolet irradiation resistance-associated gene (UVRAG)-deficient hearts display autophagic flux impairment and autophagosome accumulation, which can trigger, at least partially, the age-related cardiomyopathy and enhanced inflammatory response developed by these mice (Song et al., 2014). Additionally, in Danon disease patients, a condition clinically characterized by cardiomyopathy, LAMP-2 deficiency emerges as a primary cause (Nishino et al., 2000). In the absence of LAMP-2, fusion of autophagosomes with lysosomes is blocked leading to accumulation of long-lived proteins, ultimately resulting in myopathy. Recent studies by Pedrozo et al. (2013) demonstrated that the ryanodine receptor type 2 (RyR2), the main cardiac sarcoplasmic reticulum calcium-release channel required for cardiomyocyte excitation–contraction and often associated with myocardial remodeling after ischemia/reperfusion (I/R) and cardiac failure, is degraded by CMA. Furthermore, the authors demonstrated that CMA-mediated degradation of RyR2 induced by oxidative stress depends on presenilins (Pedrozo et al., 2013). These data envision a new model for the pathophysiology of Danon disease, where the deregulation of the RyR2 receptor due to the loss of LAMP-2A and the impairment of CMA may contribute to hypertrophic cardiomyopathy associated with the disorder.
In addition to the housekeeping role of constitutive autophagy under basal conditions, an upregulation of autophagy is also known to be involved in response to stress stimuli, such as cardiac ischemia, hypertrophy, and heart failure (Nishida et al., 2009). Although several cardiomyopathies have been associated with an upregulation of autophagy, the consequences of such increased autophagic activity have been a matter of intense debate. If on one hand autophagy seems to play an important role by overriding apoptosis and protecting cardiomyocytes from programed death (Przyklenk et al., 2012), on the other hand, autophagy enhancement could also be detrimental, leading to exacerbated cell damage (Gottlieb et al., 2010).
Regarding myocardial ischemia, studies carried out in mice showed that 30 min of cardiac ischemia leads to activation of autophagy, and that its inhibition upon treatment with 3-methyladenine (3-MA) or Bafilomycin A1 (Baf) increases cardiomyocyte death, showing that ischemia-induced autophagy may have a protective role (Hamacher-Brady et al., 2006). It is rather consensual that autophagy-mediated heart protection is conferred by a downregulation of apoptosis. Indeed, it has been consistently shown that activation of autophagy and the formation of autophagosomes might help the clearance of damaged mitochondria, preventing the release of cytochrome c and inhibiting the formation of apoptotic bodies. Moreover, studies carried out in a cardiac cell line demonstrated that knockdown of Beclin 1 results in increased cardiomyocyte apoptosis upon I/R, suggesting that a fully active autophagy is required to protect cardiomyocytes from ischemia-induced damage (Hamacher-Brady et al., 2006). However, other studies demonstrated that attenuation of autophagy can be protective. For example, Matsui et al. (2007) demonstrated that heart-specific knockdown of Beclin 1 results in smaller infarct sizes and less apoptotic cell death in mice subject to cardiac I/R, suggesting that autophagy plays a detrimental role under these circumstances (Takagi et al., 2007). These apparent discrepancies may be explained by the fact that Beclin 1, besides interaction with Vps34, needed for autophagosome nucleation, also interacts with Rubicon, which slows down the process of autophagosome–lysosome fusion. For that reason, Beclin 1+/− mice may be more resistant to I/R injury because they clear autophagosomes more efficiently through enhanced lysosomal fusion, instead of less formation of vesicles, as can be initially assumed (Matsui et al., 2007). In fact, it is important to notice that impaired clearance of autophagosomes, which is called frustrated autophagy, can constitute one of the worst-case scenarios. Indeed, stopping the autophagic flux may lead to exosome secretion, accompanied by release of metalloproteinases and activation of inflammation, which could promote cardiac rupture (Gottlieb et al., 2010).
These observations highlight the importance of unraveling the molecular partners of the main autophagy mediators all the way through ischemia and I/R. The initiators mTOR and AMPK, and Beclin 1, as the main trigger of downstream events, are the most widely studied members of the autophagy machinery under I/R. However, more recently, it was also reported that alterations on the phosphorylation state of glycogen synthase kinase 3β (GSK-3β) modulate autophagy in a phase-dependent manner, both in ischemia and I/R (Zhai and Sadoshima, 2012). Dephosphorylation of GSK-3β at Serine9 leads to activation of autophagy; however, the protective roles of GSK-3β can also be ascribed to increased glucose availability for glycolysis during acute ischemia. Additionally, during I/R, phosphorylation of GSK-3β can have an impact upon inhibition of apoptosis and mitochondrial permeability transition pore (Zhai and Sadoshima, 2012). To reconcile all these apparently contradictory observations, it has been proposed that the dichotomous role of autophagy likely depends on the nature, extent, and severity of the stimuli, as well as on the autophagy players involved. Thus, a cardioprotective role of ischemia-induced autophagy has been attributed to AMPK, a ubiquitous sensor of cellular energy status that promotes autophagy upon ATP depletion, helping to reestablish energy stores, whereas detrimental autophagy has been associated with upregulation of Beclin 1 (Matsui et al., 2007).
In the hypertrophied myocardium, despite in the initial stages, anabolic processes override degradative ones rendering cardiomyocyte growth, sustained hypertrophy is characterized by an upregulation of autophagy. Indeed, by the use of different animal models, it has been demonstrated that in short-term treatments with isoproterenol or pressure overload after transverse aortic constriction (TAC), autophagy is suppressed (Wang et al., 2010). On the other hand, other groups have shown that long-term pressure overload after TAC (Nakai et al., 2007), in mice, and treatment with angiotensin II, in cell-based models (Porrello et al., 2009), lead to upregulation of autophagy, which corelate with the hypertrophied phenotype observed in those models. To elucidate the role of autophagy in cardiac hypertrophy, Nakai et al. (2007) developed an animal model with cardiac-specific knockout of Atg5. These authors demonstrated that knockout of Atg5 in adult mice induces cardiac hypertrophy and heart failure, suggesting that an impairment of autophagy contributes to hypertrophy development. However, when the Atg5 knockout is performed in embryonic mice that were further submitted to a TAC procedure in adult age, similar levels of cardiac hypertrophy were observed in both wild-type and knockout animals, although the Atg5 knockout mice developed a lethal heart failure (Nakai et al., 2007). Therefore, it is conceivable that autophagy plays a major role in controlling broad protein turnover and energy generation, rather than have a direct causal effect upon hypertrophy. However, it remains unclear both the role and the kinetics of autophagy activation during cardiac hypertrophy, and whether autophagy can modulate the progression from hypertrophy to heart failure. It is likely that, similarly to what happens during the set of ischemia and I/R, autophagy may have different purposes during the hypertrophic process, being either protective or detrimental depending on the stage of disease.
Conclusions and Future Perspectives
The main function of the heart is to pump blood to the various organs of the organism. For that, the heart depends on the correct and efficient contractility of cardiac muscle cells, namely cardiomyocytes. Due to their low mitotic activity, these cells require a capable system of protein degradation, to ensure a correct protein quality control. An active proteolytic network, including UPS and autophagy, is important both in resting conditions, to eliminate misfolded and/or obsolete proteins and organelles, and also as part of a stress response, to degrade damaged and aggregation prone proteins. Not surprisingly, the deregulation of autophagy activity has been often associated with the development of several cardiomyopathies. If, conceptually, the activation of autophagy would be beneficial to eliminate damaged proteins and organelles upon stress conditions, avoiding the accumulation of toxic material, an overactivation of autophagy can be harmful and lead to cell death. This dual role of autophagy constitutes a hot topic in the field, since it remains unclear whether autophagy is protective or detrimental in the context of heart maladies. It is rather consensual that the consequences of autophagy activation largely depend on the severity of the stimuli and also on the autophagy players involved. This issue is well exemplified in ischemic heart, where activation of protective autophagy in early periods of ischemia involves AMPK, whereas in later periods and reperfusion, the activation of detrimental autophagy relies on Beclin 1. Taking into account these evidences, the use of autophagy modulators in the context of heart diseases has to be approached with caution. A better understanding of the mechanisms, pathways, and players involved in lesion-triggered autophagy is vital to develop efficient and targeted autophagy-based therapeutic approaches. For example, a pharmacological intervention directed to autophagy, in the context of ischemic insult, has to consider the stage of the lesion and, consequently, the players involved. Furthermore, it is conceivable that the autophagy degradation of different substrates under stress conditions relies on different players and mechanisms. Indeed, the autophagy process is not identical for all the substrates, making some proteins more prone to degradation in certain circumstances, in comparison to others that are protected. One of the proteins that is essential for the efficient propagation of the action potential through the heart, permitting it to beat in a coordinated and synchronized manner, is Cx43. We have previously demonstrated that Cx43 is a substrate for autophagy; however, the pathophysiological implications of such mechanism have never been established. Therefore, it is crucial to elucidate whether degradation of Cx43, usually associated with heart disorders, occurs by autophagy. It is likely, for example, that autophagy activation during ischemia account for degradation of Cx43, with the consequent impairment of GJIC and loss of heart function. Future studies are required to unveil the mechanisms whereby Cx43 is degraded in the heart disorders, which may open new avenues toward the development of new pharmacological strategies that aim to preserve GJIC.
Acknowledgments
This work was supported by the Portuguese Foundation for Science and Technology (FCT) grants PTDC/SAU-ORG/119296/2010 and PEST-C/SAU/UI3282/2013-COMPETE. T.M.M. was supported by the FCT grant PTDC/SAU-ORG/119296/2010. T.R.R. was supported by the FCT PhD grant SFRH/BD/52294/2013.
Disclosure Statement
No competing financial interests exist.
References
- Beardslee M.A., Lerner D.L., Tadros P.N., Laing J.G., Beyer E.C., Yamada K.A., Kleber A.G., Schuessler R.B., and Saffitz J.E. (2000). Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 87,656–662 [DOI] [PubMed] [Google Scholar]
- Bejarano E., Girao H., Yuste A., Patel B., Marques C., Spray D.C., Pereira P., and Cuervo A.M. (2012). Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell 23,2156–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan M.S., Pattison J.S., Osinska H., James J., Gulick J., McLendon P.M., Hill J.A. (2013). Sadoshima J., and Robbins J.Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123,5284–5297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino S., Ramalho J.S., Marques C., Pereira P., and Girao H. (2011). Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem J 437,255–267 [DOI] [PubMed] [Google Scholar]
- Cuervo A.M., and Wong E. (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24,92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Yang D., Li L., Luo G., Li T., Fan X., Wang Q., Zhang X., Wang Y., and Le W. (2009). An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy 5,663–675 [DOI] [PubMed] [Google Scholar]
- Eletr Z.M., and Wilkinson K.D. (2014). Regulation of proteolysis by human deubiquitinating enzymes. Biochim Biophys Acta 1843,114–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., He D., Yao Z., and Klionsky D.J. (2013). The machinery of macroautophagy. Cell Res 24,24–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira J.V., Fofo H., Bejarano E., Bento C.F., Ramalho J.S., Girao H., and Pereira P. (2013). STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 9,1349–1366 [DOI] [PubMed] [Google Scholar]
- Girao H., Catarino S., and Pereira P. (2009). Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res 315,3587–3597 [DOI] [PubMed] [Google Scholar]
- Glickman M.H., and Ciechanover A. (2002). The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82,373–428 [DOI] [PubMed] [Google Scholar]
- Gottlieb R.A., and Mentzer R.M. (2010). Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol 72,45–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacher-Brady A., Brady N.R., and Gottlieb R.A. (2006). Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 281,29776–29787 [DOI] [PubMed] [Google Scholar]
- Kaushik S., and Cuervo A.M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22,407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S., Massey A.C., Mizushima N., and Cuervo A.M. (2008). Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 19,2179–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V., McEwan D.G., Novak I., and Dikic I. (2009). A role for ubiquitin in selective autophagy. Mol Cell 34,259–269 [DOI] [PubMed] [Google Scholar]
- Komander D., and Rape M. (2012). The ubiquitin code. Annu Rev Biochem 81,203–229 [DOI] [PubMed] [Google Scholar]
- Komander D., Clague M.J., and Urbe S. (2009). Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 10,550–563 [DOI] [PubMed] [Google Scholar]
- Kroemer G., Mariño G., and Levine B. (2010). Autophagy and the integrated stress response. Mol Cell 40,280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T., Kirkin V., Dikic I., and Johansen T. (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8,1986–1990 [DOI] [PubMed] [Google Scholar]
- Lange S., Xiang F., Yakovenko A., Vihola A., Hackman P., Rostkova E., Kristensen J., Brandmeier B., Franzen G., Hedberg B., Gunnarsson L.G., Hughes S.M., Marchand S., Sejersen T., Richard I., Edstrom L., Ehler E., Udd B., and Gautel M. (2005). The kinase domain of titin controls muscle gene expression and protein turnover. Science 308,1599–1603 [DOI] [PubMed] [Google Scholar]
- Liu J., Xia H., Kim M., Xu L., Li Y., Zhang L., Cai Y., Norberg H.V., Zhang T., Furuya T., Jin M., Zhu Z., Wang H., Yu J., Li Y., Hao Y., Choi A., Ke H., Ma D., and Yuan J. (2011). Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147,223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon R.C., Lange S., and Sheikh F. (2013). Breaking down protein degradation mechanisms in cardiac muscle. Trends Mol Med 19,239–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y., Takagi H., Qu X., Abdellatif M., Sakoda H., Asano T., Levine B., and Sadoshima J. (2007). Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100,914–922 [DOI] [PubMed] [Google Scholar]
- Mizushima N., and Komatsu M. (2011). Autophagy: renovation of cells and tissues. Cell 147,728–741 [DOI] [PubMed] [Google Scholar]
- Mizushima N., Levine B., Cuervo A.M., and Klionsky D.J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451,1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay D., and Riezman H. (2007). Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315,201–205 [DOI] [PubMed] [Google Scholar]
- Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M., Nishida K., Hori M., Mizushima N., and Otsu K. (2007). The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13,619–624 [DOI] [PubMed] [Google Scholar]
- Nazio F., Strappazzon F., Antonioli M., Bielli P., Cianfanelli V., Bordi M., Gretzmeier C., Dengjel J., Piacentini M., Fimia G.M., and Cecconi F. (2013). mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 15,406–416 [DOI] [PubMed] [Google Scholar]
- Nishida K., Kyoi S., Yamaguchi O., Sadoshima J., and Otsu K. (2009). The role of autophagy in the heart. Cell Death Differ 16,31–38 [DOI] [PubMed] [Google Scholar]
- Nishino I., Fu J., Tanji K., Yamada T., Shimojo S., Koori T., Mora M., Riggs J.E., Oh S.J., Koga Y., Sue C.M., Yamamoto A., Murakami N., Shanske S., Byrne E., Bonilla E., Nonaka I., DiMauro S., and Hirano M. (2000). Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406,906–910 [DOI] [PubMed] [Google Scholar]
- Park C., and Cuervo A.M. (2013). Selective autophagy: talking with the UPS. Cell Biochem Biophys 67,3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrozo Z., Torrealba N., Fernandez C., Gatica D., Toro B., Quiroga C., Rodriguez A.E., Sanchez G., Gillette T.G., Hill J.A., Donoso P., and Lavandero S. (2013). Cardiomyocyte ryanodine receptor degradation by chaperone-mediated autophagy. Cardiovasc Res 98,277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart C.M., and Eddins M.J. (2004). Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta 1695,55–72 [DOI] [PubMed] [Google Scholar]
- Platta H.W., Abrahamsen H., Thoresen S.B., and Stenmark H. (2011). Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem J 441,399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello E.R., Delbridge L.M., and Thomas W.G. (2009). The angiotensin II type 2 (AT2) receptor: an enigmatic seven transmembrane receptor. Front Biosci (Landmark Ed) 14,958–972 [DOI] [PubMed] [Google Scholar]
- Przyklenk K., Dong Y., Undyala V.V., and Whittaker P. (2012). Autophagy as a therapeutic target for ischaemia/reperfusion injury? Concepts, controversies, and challenges. Cardiovasc Res 94,197–205 [DOI] [PubMed] [Google Scholar]
- Ravikumar B., Sarkar S., Davies J.E., Futter M., Garcia-Arencibia M., Green-Thompson Z.W., Jimenez-Sanchez M., Korolchuk V.I., Lichtenberg M., Luo S., Massey D.C., Menzies F.M., Moreau K., Narayanan U., Renna M., Siddiqi F.H., Underwood B.R., Winslow A.R., and Rubinsztein D.C. (2010). Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90,1383–1435 [DOI] [PubMed] [Google Scholar]
- Ribeiro-Rodrigues T.M., Catarino S., Marques C., Ferreira J.V., Martins-Marques T., Pereira P., and Girao H. (2014). AMSH-mediated deubiquitination of Cx43 regulates internalization and degradation of gap junctions. FASEB J 28,4629–4641 [DOI] [PubMed] [Google Scholar]
- Sarkar S., Ravikumar B., and Rubinsztein D.C. (2009). Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol 453,83–110 [DOI] [PubMed] [Google Scholar]
- Schmidt M., and Finley D. (2014). Regulation of proteasome activity in health and disease. Biochim Biophys Acta 1843,13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severs N.J., Bruce A.F., Dupont E., and Rothery S. (2008). Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res 80,9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.S., and Kehrl J.H. (2010). TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 3,ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z., An L., Ye Y., Wu J., Zou Y., He L., and Zhu H. (2014). Essential role for UVRAG in autophagy and maintenance of cardiac function. Cardiovasc Res 101,48–56 [DOI] [PubMed] [Google Scholar]
- Stolz A., Ernst A., and Dikic I. (2014). Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16,495–501 [DOI] [PubMed] [Google Scholar]
- Su H., and Wang X. (2011). p62 Stages an interplay between the ubiquitin-proteasome system and autophagy in the heart of defense against proteotoxic stress. Trends Cardiovasc Med 21,224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H., Matsui Y., and Sadoshima J. (2007). The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid Redox Signal 9,1373–1381 [DOI] [PubMed] [Google Scholar]
- Terman A., and Brunk U.T. (2005). Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res 68,355–365 [DOI] [PubMed] [Google Scholar]
- Thevenin A.F., Kowal T.J., Fong J.T., Kells R.M., Fisher C.G., and Falk M.M. (2013). Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology (Bethesda) 28,93–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.V., Rothermel B.A., and Hill J.A. (2010). Autophagy in hypertensive heart disease. J Biol Chem 285,8509–8514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welchman R.L., Gordon C., and Mayer R.J. (2005). Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol 6,599–609 [DOI] [PubMed] [Google Scholar]
- Wong E., and Cuervo A.M. (2010). Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol 2,a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P., Wang S., Du Y., Zhao Z., Shi L., Sun L., Huang G., Ye B., Li C., Dai Z., Hou N., Cheng X., Sun Q., Li L., Yang X., and Fan Z. (2013). WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J 32,2685–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T.P. (2010). The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer 1,779–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle R.J., and Narendra D.P. (2011). Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12,9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaglia T., Milan G., Ruhs A., Franzoso M., Bertaggia E., Pianca N., Carpi A., Carullo P., Pesce P., Sacerdoti D., Sarais C., Catalucci D., Kruger M., Mongillo M., and Sandri M. (2014). Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J Clin Invest 124,2410–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai P., and Sadoshima J. (2012). Glycogen synthase kinase-3beta controls autophagy during myocardial ischemia and reperfusion. Autophagy 8,138–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Chen X., Sharma P., Moon M., Sheftel A.D., Dawood F., Nghiem M.P., Wu J., Li R.K., Gramolini A.O., Sorensen P.H., Penninger J.M., Brumell J.H., and Liu P.P. (2014). HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat Commun 5,3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q., Su H., Ranek M.J., and Wang X. (2011a). Autophagy and p62 in cardiac proteinopathy. Circ Res 109,296–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q., Su H., Tian Z., and Wang X. (2011b). Proteasome malfunction activates macroautophagy in the heart. Am J Cardiovasc Dis 1,214–226 [PMC free article] [PubMed] [Google Scholar]
- Zhu K., Dunner K., Jr., and McConkey D.J. (2010). Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29,451–462 [DOI] [PMC free article] [PubMed] [Google Scholar]