

Abstract
Secretory immunoglobulin (Ig) A (SIgA), comprised of dimeric IgA and secretory component (SC), is believed to provide a defense mechanism on the mucosal surface. Influenza A virus (IAV) hemagglutinin (HA)-specific SIgA is thought to play an important role in the prevention of IAV infection. However, the topical application of preformed IAV-specific SIgA has not been shown to prevent IAV infection. This is due to the difficulty in the production of antigen-specific IgA monoclonal antibodies (mAbs) and monoclonal SIgA. Here, a recombinant hybrid IgA (HIgA) was established that utilizes variable regions of an HA-specific mouse IgG mAb and the heavy chain constant region of a mouse IgA mAb. We expressed the dimeric HIgA in Chinese hamster ovary-K1 (CHO-K1) cells. When in vitro IAV infection of Madin–Darby canine kidney (MDCK) cells was tested, 10 times lower concentrations of HIgA were able to inhibit it as compared with an HA-specific IgG with the same variable regions. A functional hybrid secretory IgA (HSIgA) was also produced through incubation of the dimeric HIgA with recombinant mouse SC in vitro. It was demonstrated that HSIgA could be separated from the dimeric HIgA on size exclusion chromatography. This study provides a basic strategy for investigating the role of SIgA upon IAV infection on the mucosal surface.
Introduction
Influenza A virus (IAV) is responsible for annual worldwide epidemics. Seasonal influenza kills many people annually (28). To control seasonal influenza infections, an IAV vaccine is administered. Hemagglutinin (HA) is a major envelope protein of IAV and is involved in the initiation of IAV infection (24). When an IAV vaccine is administered subcutaneously, HA-specific immunoglobulin (Ig) G is induced in the serum, but IgA production on the mucosal surface is not sufficient (43). Because IAV infects individuals through the mucosal surface of the upper respiratory tract, the efficacy of intramuscular or subcutaneous inactivated influenza vaccines is thought to be limited. On the other hand, an intranasal live-attenuated seasonal influenza vaccine, FluMist®, which has been approved since 2003 in the United States, likely provides protection through secretory IgA (SIgA) not by serum IgG. Protection was shown to correlate with the level of IAV-specific IgA in nasal washes but not with that of serum hemagglutination inhibition antibodies (1,3,4).
Animal models have revealed the importance of IgA. When IAV-HA vaccine was subcutaneously administered to mice, HA-specific IgG was induced in the serum. On the other hand, when IAV-HA vaccine was intranasally administered to mice, HA-specific SIgA on the mucosal surface was produced in addition to IgG in the serum. Upon IAV challenge, mice that received intranasal IAV-HA vaccine remained in a good state compared with ones that received the subcutaneous IAV-HA vaccine (37). For these reasons, HA-specific SIgA is thought to play an important role in the prevention of IAV infection. In addition, when a polymeric form of IAV-specific IgA monoclonal antibodies (mAbs) was intravenously administered to mice, IAV-specific IgA was secreted in nasal washes (30). This passive IgA mAb immunization protected mice from intranasal challenge of IAV. However, topical treatment with preformed SIgA has not been examined.
IgA is the most abundant immunoglobulin on the mucosal surface (45). SIgA, which comprises dimeric IgA (dIgA) and secretory component (SC), is thought to serve as a defense mechanism against invasion by bacteria (8,9,44) and pathogens on the mucosal surface (6,22,25,31,42). By association with SC, IgA acquires resistance to proteases and the ability to be localized on the mucosal surface (18,26). For these reasons, SIgA is thought to be suitable for inhibiting pathogens on the mucosal surface.
Methods for the production of antigen-specific IgG mAbs have been established. It is still difficult, however, to produce antigen-specific IgA mAbs to any given antigen. Regarding HA, there have been two reports on the production of IgA mAbs that bind to HA (33,40). In the first study, IgA mAbs were produced by conventional methods generally used for IgG mAbs (33). Among many clones producing IgG, only one IgA-producing hybridoma was established. In other words, the probability of obtaining an IgA-producing hybridoma is considerably low compared with IgG ones. Although there have been several reports of the use of HA-specific IgA since 1983, the same clone was used, including in studies that involved intravenous injection of IgA (29,30,32). IgA mAbs were previously established that are specific for Shiga toxin 1 B subunit (Stx1B) (11) or ovalbumin (39). However, this particular Stx1B-specific IgA did not neutralize the toxin activity (38). Although repeated attempts were made to produce Stx1B-specific IgA mAbs, no clone was obtained that could neutralize the toxin.
An Stx1B-specific hybrid-IgG/IgA was previously produced, of which the heavy chain consisted of a variable region, the Ch1 domain and the hinge region from Stx1B-specific IgG, and the Ch2 and Ch3 domains from IgA (41). The light chain was from the IgG (41). Here, an HA-specific hybrid IgA (HIgA) was produced using the methods previously described for Stx1B-specific hybrid IgG/IgA. HIgA inhibited IAV infection of Madin–Darby canine kidney (MDCK) cells. Hybrid secretory IgA (HSIgA) was also produced by incubating HIgA with SC. HSIgA was shown to bind to HA of IAV as well as to IAV particles. This study will lead to the development of a new tool for examining the role of SIgA through topical application on the mucosal surface.
Materials and Methods
Reagents
2-[(4-Hydroxyethyl)-1-piperazine]-ethanesulfonic acid (HEPES), Tween-20, kanamycin sulfate, hygromycine B, methanol, 4-chloro-1-naphthol, ortho-phenylenediamine, and 2,2′-azinobis (3-ethylbenzothiazoline-6-sulfonic acid) were purchased from Wako Pure Chemicals; Dulbecco's modified Eagle's medium (DMEM) and Ham's F12 (F12) from Nissui Pharmaceuticals; Hybridoma-SFM, Eagle's Minimum Essential Medium, zeocin, and MagicMark™ XP Western Protein standards from Life Technologies; G418 from Nacalai Tesque; HyClone fetal bovine serum (FBS) from GE Healthcare; and a DNA size marker kit (100-bp DNA Ladder) from Takara. Horseradish peroxidase (HRP)-rabbit antimouse IgG, goat antimouse IgG, rabbit antimouse IgA (α chain-specific), and HRP-goat antirabbit IgG were purchased from Zymed; HRP-goat antimouse IgA (α chain-specific), HRP-goat antimouse kappa, goat antimouse kappa, and HRP-donkey antigoat IgG (H+L) from Southern Biotech; TEPC 15, MOPC 21, and bovine serum albumin (BSA) from Sigma; rabbit antimouse J chain and HRP-donkey antigoat IgG from Santa Cruz Biotechnology; goat antimouse polymeric Ig receptor (pIgR) and recombinant mouse pIgR from R&D Systems; rabbit antimouse pIgR from Sino Biological, Inc.; and HRP-goat antimouse IgG+M from Jackson Immuno Research. Can Get Signal Solution 1, Can Get Signal Solution 2, and PVDF Blocking Reagent were purchased from TOYOBO; and N,N-diethyl-para-phenylendiamine from Kanto Chemicals. Mouse 4E6 mAb, which recognizes the nucleoprotein (NP) of IAV, was established as previously described (36).
Cell culture
Chinese hamster ovary-K1 (CHO-K1) cells were obtained from the American Type Culture Collection. CHO-K1 cells were cultured in DMEM/F12 supplemented with 10 mM HEPES (pH 7.4), 10% FBS, and 60 μg/mL kanamycin. Mouse mAb-producing hybridomas were cultured in Hybridoma-SFM containing 60 μg/mL kanamycin. HEK293T cells were cultured in DMEM containing 10 mM HEPES (pH 7.4), 4.5 mg/mL glucose, 10% FBS, and 60 μg/mL kanamycin. MDCK cells were cultured in Eagle's Minimum Essential Medium containing 10 mM HEPES (pH 7.4), 5% FBS, and 60 μg/mL kanamycin. All cell cultures were performed at 37°C under a humidified atmosphere of 5% CO2/95% air.
Production of antibodies
Mouse anti-HA (H3 specific) mAb 2E10 (IgG1, κ) (36) and mouse anti-Stx1B mAb G2G7 (IgA, κ) (11) were established as previously described, respectively. A cDNA fragment of the 2E10 heavy chain variable region and the full-length light chain were isolated by the 5′-rapid amplification of cDNA ends (5′-RACE) method using a Gene Racer™ kit (Life Technologies), and the following primers: GeneRacer™ 5′primer forward, 5′-CGA CTG GAG CAC GAG GAC ACT GA-3′; IgG1CH1RACE reverse, 5′-GGA TAG ACA GAT GGG GGT GTC GTT TTG G-3′ (heavy chain variable region); or ALC-RACE1 reverse, 5′-CTA ACA CTC ATT CCT GTT GAA GCT C-3′ (light chain). cDNA for the G2G7 heavy chain was isolated as previously described (41). Anti-HA HIgA heavy chain was produced using the recombinant polymerase chain reaction (PCR) method as previously described (10,41). In brief, the cDNA fragment corresponding to the 2E10 heavy chain variable region and that to the G2G7 constant region were joined using the following primers: 2E10-Hc-F-KpnI, 5′-ACG GTA CCC AGG ACC TCA CAA TGG-3′; 2E10-Hc-R-rPCR, 5′-ATT TCT CGC AGA CTC TGA GGA GAC GGT GAC C-3′ (variable region); IgA-Hc-F-rPCR, 5′-GGT CAC CGT CTC CTC AGA GTC TGC GAG AAA TCC-3′; and IgA-Hc-R-BamHI, 5′-CTG GGA TCC TCA GTA GCA GAT GCC AT-3′ (constant region). The cDNAs encoding the hybrid heavy, light, and J chains were subcloned into pcDNA3.1 (+), pcDNA3.1 (+)/Hygro and pcDNA3.1 (+)/Zeo (Life Technologies), respectively. CHO-K1 cells were transfected with these three expression vectors by means of FuGENE6 (Roche Diagnostics). Stable transfectants were selected in the presence of G418, hygromycine B, and zeocin, and the best producing clone was isolated by limiting dilution. The best producing clone was cultured in DMEM/F12 supplemented with 10 mM HEPES (pH 7.4), 10% FBS, 60 μg/mL kanamycin, 800 μg/mL G418, 400 IU/mL hygromycin B, and 1,000 μg/mL zeocin. Under subconfluent conditions, the medium was replaced with a serum-free BMpro medium (Cell Science & Technology Institute) supplemented with 60 μg/mL kanamycin, and then the culture was continued for 3 days. The culture supernatant was recovered and concentrated with Viva Flow 50, Viva Spin 20, and Viva Spin 500 (MWCO: 100 kDa; Sartorius Stedim Biotech). The concentrated HIgA was purified on a column of Sephacryl S-300 HR (1.5 cm×60 cm; GE Healthcare) equilibrated with phosphate-buffered saline (PBS) containing 0.02% NaN3. For further purification, TSK gel G3000SWXL (7.8 mm×30 cm; TOSOH) equilibrated with 0.1 M phosphate buffer (pH 6.7) containing 0.1 M sodium sulfate was used. For the TSK gel, proteins were monitored at UV280 nm (UV/VIS-151; Gilson). The purified HIgA was filtrated using a Milex GV 0.22 μm (Millipore) and stored at 4°C.
Preparation of SC
Mouse pIgR cDNA was kindly provided by Dr. C. S. Kaetzel (University of Kentucky, College of Medicine) (27). A cDNA fragment corresponding to SC that consists of the extracellular domain of pIgR was amplified using the following primers: SC-F-XhoI forward, 5′-CCT CTC GAG TCT CTT TAG TTG GCA AAA GGC-3′; SC-R-HindIII reverse, 5′-CCC AAG CTT CAC AAG CAA TGA GGC TCT-3′. The SC cDNA was subcloned into pcDNA3.1 (+). CHO-K1 cells were transfected with this expression vector by means of FuGENE6. Stable transfectants were selected with G418, and the best producing clone was isolated by limiting dilution. The best producing clone was cultured in DMEM/F12 supplemented with 10 mM HEPES (pH 7.4), 10% FBS, 60 μg/mL kanamycin, and 800 μg/mL G418. Under subconfluent conditions, the medium was replaced with a serum-free CD CHO-A medium (Life Technologies), and then the culture supernatant was harvested after 3 days. The supernatant was concentrated with Viva Spin 20 (MWCO: 30 kDa; Sartorius). The concentrated SC was purified on a column of Sephacryl S-300 HR (1.5 cm×60 cm) equilibrated with 0.02% NaN3-PBS. For further purification, Sephacryl S-200 (1.5 cm×60 cm) equilibrated with PBS, and TSK gel G3000SWXL (7.8 mm×30 cm) equilibrated with 0.1 M phosphate buffer (pH 6.7) containing 0.1 M sodium sulfate were used. In some experiments, the concentration of SC was calculated with a BCA protein assay kit (Thermo Scientific) using BSA (Thermo Scientific) as a standard. The purified SC was filtrated using a Milex GV 0.22 μm and stored at 4°C.
Virus
Human influenza virus A/Memphis/1/71 (H3N2) used in this study was propagated in 10-day-old embryonated chicken eggs. The virus particles were purified by sucrose density gradient centrifugation as previously described (34).
Enzyme-linked immunosorbent assay
A human influenza virus A/Memphis/1/71 (H3N2) suspension (2 hemagglutinin units [HAU]) in PBS was added (50 μL/well) to and dried on the wells of a 96-well flat-bottomed plate (Maxisorp NUNC-Immunoplate; NUNC). The wells were washed with PBS and then blocked with PBS containing 1% BSA for 2 h at room temperature. Samples containing immunoglobulins were added to the wells, followed by incubation for 1 h at room temperature. After the wells had been washed, HRP-goat antimouse IgA (1:1,000 dilution) was added to the wells, and incubated for 1 h at room temperature to detect the IgA heavy chain. To detect SC on HSIgA using antimouse pIgR antibodies, the wells were incubated with goat antimouse pIgR (1:400) followed by HRP-donkey antigoat IgG (1:500). All antibodies were diluted in PBS containing 0.1% BSA and 0.1% Tween-20. As a substrate, 0.4 mg/mL ortho-phenylenediamine dissolved in 0.1 M citrate phosphate buffer (pH 5.6) containing 0.03% H2O2 was added. After incubation for 15 min at room temperature, absorbance readings were made with a microplate reader (InfiniteM200-SPHOS; TECAN).
To determine the total recombinant immunoglobulin concentrations in samples, a sandwich enzyme-linked immunosorbent assay (ELISA) was performed (41). Appropriately diluted samples containing HIgA were captured with rabbit antimouse IgA that had been coated onto the wells of an ELISA plate at 1:500 dilution in PBS (Costar #9018; Corning). The captured antibodies were detected with HRP-goat antimouse kappa chain (1:500). In some experiments, goat antimouse kappa (1:1,000) was used for capture and HRP-goat antimouse IgA (α-chain specific; 1:1,000) for detection. To detect IgG, rabbit antimouse IgG (1:1,000) was used for capture and HRP-goat antimouse IgG (1:1,000) for detection. To determine recombinant SC concentrations, rabbit antimouse pIgR (1:2,000) was used for capture, and goat antimouse pIgR (1:1,000) plus HRP-donkey antigoat IgG (1:4,000; Santa Cruz) for detection. All antibodies except those immobilized on the wells were diluted in PBS containing 0.1% BSA and 0.1% Tween-20. As a substrate, 1 mM 2,2′-azinobis (3-ethylbenzothiazoline-6-sulfonic acid) dissolved in 0.1 M citrate buffer (pH 4.2) containing 0.03% H2O2 was added. After incubation for 20 min at room temperature, absorbance readings were made with a microplate reader (SUNRISE Rainbow RC-R; TECAN) at 405 nm. As standards, IgA myeloma proteins (TEPC 15) or IgG myeloma proteins (MOPC 21) were used for each immunoglobulin class, and recombinant mouse pIgR for SC.
In vitro preparation of HSIgA
Partially purified HIgA (2.5 μg of IgA) was incubated with partially purified SC (0, 0.5, or 1 μg of SC) in 50 μL of PBS for 2 h at 37°C. The reaction mixture was analyzed by SDS-PAGE and Western blotting. In some experiments, HIgA and SC were incubated in 50 mM sodium phosphate buffer (pH 7.2). The resultant HSIgA preparation was used for binding assays involving cell surface HA or IAV particles, and further separation by TSK gel G3000SWXL.
Western blotting
Proteins separated by SDS-PAGE on 7.5% or 12% polyacrylamide gels were transferred to 0.22 μm polyvinylidene difluoride (PVDF) membranes (Bio-Rad). The membranes were blocked with PVDF blocking reagent. The membranes were then incubated with HRP-conjugated antibodies in Can Get Signal Solution 2 and washed with PBS containing 0.05% Tween-20. For the indirect method, the membranes were incubated with primary antibodies in Can Get Signal Solution 1 and washed with PBS containing 0.05% Tween-20. The membranes were then incubated with HRP-conjugated antibodies in Can Get Signal Solution 2 and washed with PBS containing 0.05% Tween-20. The antibody dilutions are specified in the figure legends. As a substrate, SuperSignal West Pico Chemiluminescent Substrate (Thermo) was added. Bands were observed using an imaging analyzer (LAS3000UV mini; Olympus). MagicMark™ XP Western Protein standards were used as molecular weight standards.
Expression of HA on the cell surface
cDNA for HA of A/Memphis/1/71 (H3N2) was subcloned into a plasmid expression vector, pCAGGS/MCS, to construct pCAGGS/HA (35). HEK293T cells on a Millicell EZ Slide (Millipore) were transiently transfected with pCAGGS/HA by means of FuGENE6. Cells were used after 24 h of culture.
Immunostaining
HA-expressing HEK293T cells were fixed in methanol and then blocked with PBS containing 3% BSA. The cells were incubated with an HA-specific immunoglobulin at 1 μg/mL for 1 h at room temperature, and then washed with PBS. The cells were then incubated with HRP-goat antimouse IgA (1:1,000) for 1 h at room temperature. To detect SIgA using anti-pIgR antibodies, goat anti-pIgR (1:100) followed by HRP-donkey antigoat IgG (H+L) (1:500) were used. All antibodies were diluted in PBS containing 1% BSA. To visualize antibody binding, 0.06 M N,N-diethyl-para-phenylendiamine and 0.11 M 4-chloro-1-naphthol in 50 mM citrate buffer (pH 6.0) containing 0.03% H2O2 were added. After incubation for 10 min at room temperature, the cells were washed with PBS. The cells were then observed under a microscope (IX71; Olympus).
Virus neutralization test
A human influenza virus strain A/Memphis/1/71 (H3N2) suspension (0.5 HAU) in Hybridoma-SFM medium (270 μL) was mixed with serially diluted antibodies (30 μL), followed by incubation for 30 min at room temperature. Confluent MDCK cells in the wells of a 24-well plate were incubated with the virus antibody mixture for 30 min at 37°C and then washed with PBS. The monolayers were cultured for 7 h at 37°C in Hybridoma-SFM, and then the production of viral proteins was assessed by immunostaining using mouse anti-NP mAb (undiluted supernatant of 4E6), followed by HRP-goat antimouse IgG+M (1:3,000) in PBS. Infected cells were observed and counted under a microscope. The cells producing NP were designated as infected cells.
Results
To produce recombinant SIgA, it was considered that IgA and SC are produced by different types of cells. Although IgA is produced by B-cells, SC is the product of epithelial cells. Thus, cDNAs were expressed for HIgA and SC in different CHO-K1 cells. As for HIgA, CHO-K1 cells were stably transfected with vector constructs for the HIgA heavy, light, and J chains to produce dimeric HIgA.
As for HIgA heavy chain, the cDNA of the IgG mAb 2E10 heavy chain variable region and that of the IgA heavy chain constant region were joined by means of recombinant PCR. The cDNA products corresponding to the 2E10 heavy chain variable region (lane 1), the IgA heavy chain constant region (lane 2), and the recombinant HIgA heavy chain (lane 3) were observed by means of agarose gel electrophoresis (Fig. 1A). The nucleotide sequence of the HIgA variable region was identical to that of the original RACE PCR product obtained from the 2E10 hybridoma cell line. Full-length cDNA was also prepared encoding the 2E10 light chain by means of the 5′-RACE PCR method.
FIG. 1.
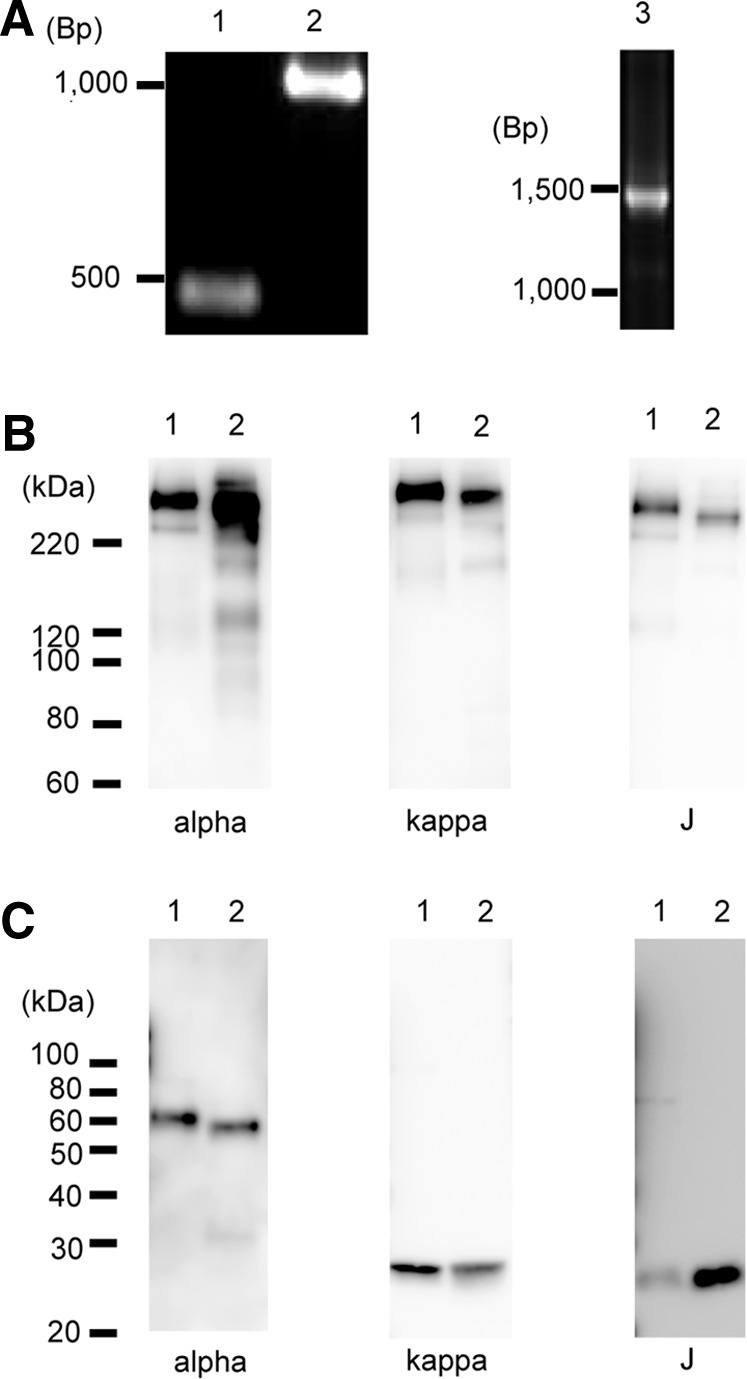
Production of hybrid immunoglobulin (Ig) A (HIgA). (A) Polymerase chain reaction (PCR) products of the variable region (lane 1), constant region (lane 2), and hybrid heavy chain (lane 3) were detected by agarose gel electrophoresis. A 100 base pairs (bp) DNA ladder was used as DNA size markers. On the left, the electrophoretic mobility of markers is shown in bp. (B) and (C) The hybrid IgA (lane 1) and IgA mAb G2G7 (lane 2) were subjected to SDS-PAGE on 7.5% gel under nonreducing conditions (B) or on 12% gel under reducing conditions (C), and visualized by Western blotting, on which the heavy chain (alpha), light chain (kappa), or J chain (J) was detected. HRP-goat antimouse IgA (1:1,000), HRP-goat antimouse kappa (1:1,500), and rabbit antimouse J chain (1:1,000) plus HRP-goat antirabbit IgG (1:5,000) were employed. G2G7 represents mouse dimeric IgA as a control. The positions of the molecular weight standards are shown on the left in kDa.
CHO-K1 cells were co-transfected with expression vectors harboring the hybrid heavy chain, the 2E10-associated light chain, and the J chain, respectively. The best producing clone was isolated by limiting dilution. Production of HIgA proteins was checked by Western blotting. Anti-IgA heavy chain, anti-kappa chain, and anti-J chain antibodies detected a band (lane 1) corresponding to a molecular mass >220 kDa, which is located at the same position as an IgA mAb G2G7 (lane 2) under nonreducing conditions (Fig. 1B). Due to the difference in sensitivity as to detection on Western blotting, 2 ng/lane, 10 ng/lane, and 10 ng/lane of protein were electrophoresed to detect the heavy, light, and J chains, respectively, under nonreducing conditions. There is essentially no monomeric HIgA that would be electrophoresed to a position around 160–180 kDa. Under reducing conditions, HIgA gave a band of IgA heavy, light, and J chains with similar mobility to that of IgA mAb (Fig. 1C). Under reducing conditions, 2 ng/lane, 10 ng/lane, and 20 ng/lane of protein were electrophoresed to detect heavy, light, and J chains, respectively. These data indicate that HIgA forms a dimer composed of heavy, light, and J chains.
The virus neutralization activity of the dimeric HIgA in vitro was then determined. The virus neutralization activity of 2E10 was determined previously (unpublished results). Because HIgA was designed to have the same paratope as that of 2E10, it was expected that HIgA could neutralize IAV infection (Fig. 2). IAV was pretreated with various doses of antibodies, and then each mixture was added to MDCK cells. During cell culture, infected cells synthesized viral proteins such as NP protein. Thus, the infected cells were identified by means of viral NP proteins using anti-NP antibodies. Pre-incubation with HIgA inhibited IAV infection of MDCK cells in vitro. Dose–response curves revealed that a 10 times lower concentration of HIgA than that of IgG mAb 2E10, which only forms monomers, gave rise to the same level of inhibition (Fig. 2).
FIG. 2.

Neutralizing activity of hemagglutinin (HA)-specific antibodies in vitro. Inhibition of influenza A virus (IAV) infection by 2E10 (filled squares) and the hybrid IgA (open circles). IAV was pretreated with various concentrations of antibodies (abscissa: final concentrations), and then added to Madin–Darby canine kidney cells. Cells were infected with IAV for 30 min at 37°C and then washed with phosphate-buffered saline. After 7 h of culture, cells were immunostained with antibodies specific for viral nucleoprotein proteins (mAb 4E6). The infected cells were counted under a microscope. Control represents when IAV was added without antibody pretreatment, and the data were expressed as the relative numbers of infected cells compared with the control (ordinate). Data are expressed as means±standard deviation (SD).
An attempt was then made to produce HSIgA in vitro by the incubation of HIgA with recombinant SC. First recombinant mouse SC was expressed in CHO-K1 cells. cDNA was prepared for SC, which consists of the extracellular domain of pIgR, using a cDNA for pIgR as a template, by means of PCR. The cDNA fragment corresponding to the SC was subcloned into an expression vector, pcDNA3.1(+), and CHO-K1 cells were transfected with this vector construct. Recombinant SC proteins were partially purified from a supernatant of the CHO-K1 cells stably expressing SC by means of gel filtration on Sephacryl S-300. Based on comparison of the results on sandwich ELISA and those of protein determination, 30% of the protein was estimated to be recombinant SC in this preparation. Dimeric HIgA was also partially purified from a supernatant of the CHO-K1 cells expressing HIgA by means of gel filtration on Sephacryl S-300. In this preparation, 90% of the protein was estimated to be recombinant HIgA.
HSIgA was then produced by in vitro incubation of the partially purified HIgA with SC. The quantities of HIgA and SC used were based on the determination by sandwich ELISA using appropriate standard proteins. HIgA was incubated with increasing amounts of SC for 2 h at 37°C. Western blotting was performed to determine the association between HIgA and SC (Fig. 3). Under nonreducing conditions, a high molecular weight band (arrow) was observed with anti-IgA (Fig. 3A, left) only in the presence of SC. The band intensity was higher when the ratio of HIgA to SC was 5:2 as compared with when it was 5:1. This band was also revealed by anti-pIgR antibodies, indicating the incorporation of SC (Fig. 3A, right). A band representing HIgA (arrowhead) with a little smaller molecular mass was observed only with anti-IgA antibodies, that is, not with anti-pIgR. Under reducing conditions, HIgA heavy chains were observed with anti-IgA (Fig. 3B, right), and SC was observed with anti-pIgR (Fig. 3B, left). With increasing amounts of SC added, the signal for SC increased, while there was no signal in the absence of added SC.
FIG. 3.
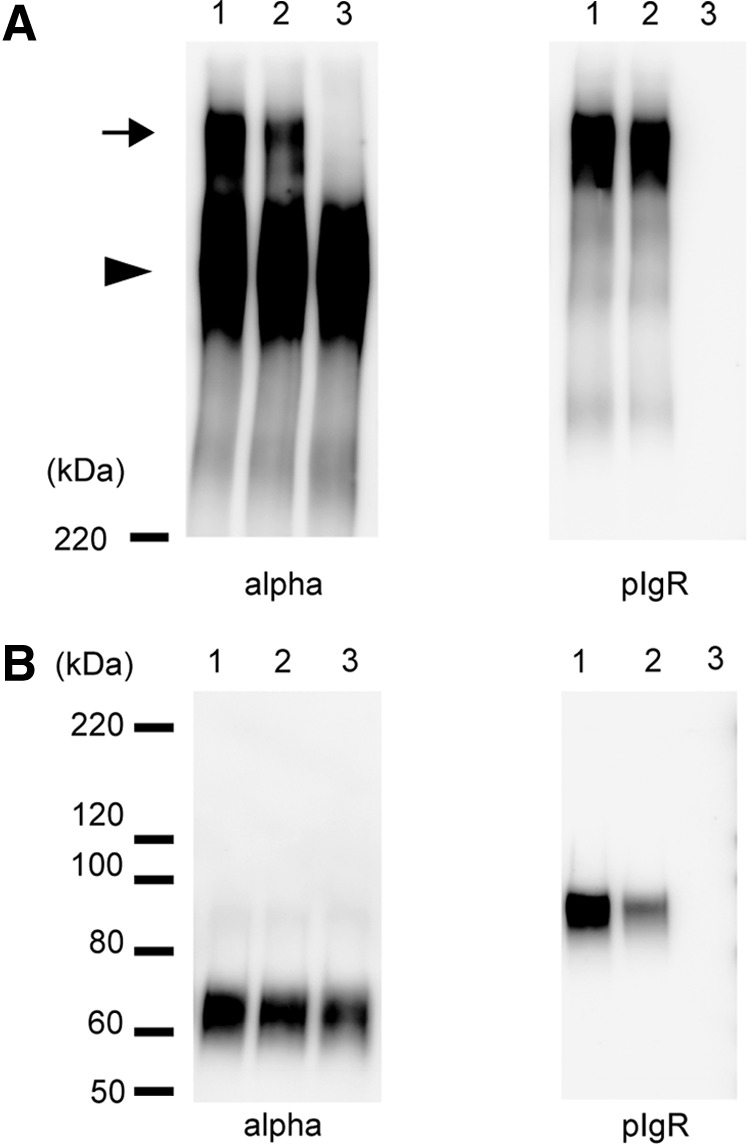
Production of hybrid secretory IgA (HSIgA) through in vitro association of the HIgA with SC. Partially purified dimeric HIgA and SC were incubated for 2 h at 37°C at a ratio of 5:2 (lane 1), 5:1 (lane 2), or 5:0 (lane 3) on a weight basis. In each lane, the amount of HIgA was 100 ng. The concentrations of HIgA and SC were determined by enzyme-linked immunosorbent assay (ELISA). The reassociated proteins were visualized by Western blotting after SDS-PAGE on a 7.5% gel under nonreducing (A) or reducing (B) conditions. The left panels (alpha) show the presence of heavy chains, while the right ones (pIgR) show that of SC. HRP-goat antimouse IgA (1:1,000) was used in the left panels, and goat anti-pIgR (1:1,000) plus HRP-donkey antigoat IgG (H+L; 1:3,000) in the right ones. The arrow indicates the band of HSIgA and the arrowhead the band of HIgA. The positions of molecular weight standards are shown on the left in kDa.
To test the binding ability of in vitro produced HSIgA, antibodies were employed against mouse pIgR. The binding of 2E10 mAb to HA of IAV was determined previously (35,36). First, it was examined whether HSIgA bind to immobilized IAV by ELISA. Anti-IgA antibodies revealed that HSIgA as well as HIgA bound to IAV with similar dose–response relationships (Fig. 4A). On the other hand, anti-pIgR antibodies detected the binding of antibodies only with the HSIgA preparation, that is, not with the HIgA one (Fig. 4D). Despite the fact that the HSIgA preparation comprised a mixture of HSIgA and HIgA (Fig. 3), the results indicated that the binding of HSIgA to IAV could be verified with specific antibodies reactive to SC. These data indicated that not only HIgA but also HSIgA retained the ability of binding to IAV.
FIG. 4.

Both anti-IgA and anti-pIgR can detect the binding of HSIgA to IAV and HA. As a control, HIgA was used. (A) Binding to immobilized IAV was detected with anti-IgA by ELISA. Varying concentrations of HSIgA (filled squares) or HIgA (open circles) were allowed to bind to immobilized IAV. (B) and (C) Binding to cell surface HA was immunohistochemically detected with anti-IgA. HSIgA (B) or HIgA (C) was allowed to bind to HEK293 cells expressing HA. (D) Binding to immobilized IAV was detected with anti-pIgR by ELISA. Binding of HSIgA (filled squares) but not HIgA (open circles) was detected with anti-pIgR. (E) and (F) Binding to cell surface HA was immunohistochemically detected with anti-pIgR. Again, the binding of HSIgA (E) but not HIgA (F) to HEK293 cells expressing HA was detected with anti-pIgR. Data are expressed as means±SD in (A) and (D). The bars represent 200 μm in (B), (C), (E), and (F).
Because complete IAV particles were used as an antigen in this study, it is not clear whether the binding activity was directed to HA or other virus components. To determine whether the specificity is directed to HA, the binding of antibodies to HEK293T cells transiently expressing HA was examined. Cells were allowed to express HA for 24 h culture, and then were immunostained with the HSIgA (Fig. 4B and E) or HIgA (Fig. 4C and F) preparation. The binding of HSIgA was detected not only with anti-IgA (Fig. 4B), but also with anti-pIgR (Fig. 4E). In contrast, the binding of HIgA was detected with anti-IgA (Fig. 4C), but not with anti-pIgR (Fig. 4F). Collectively, these data suggest that HSIgA bound to HA of IAV.
Incubation of HIgA with SC gave rise to HSIgA, but the preparation comprised a mixture of HIgA and HSIgA. For better separation of HSIgA and HIgA on HPLC, an attempt was made to obtain a cleaner preparation as a starting material. That is, HIgA was prepared by means of Sephacryl S-300 followed by TSKG3000 HPLC, and SC by means of Sephacryl S-300, Sephacryl S-200, and then TSKG3000 HPLC. After incubation of HIgA with SC in the ratio of 5:2 (w/w) in 50 mM sodium phosphate buffer (pH 7.2) for 2 h at 37°C, an attempt was made to separate HSIgA from HIgA by means of size exclusion chromatography HPLC on TSKG3000. The chromatogram contained two high molecular weight peaks (fractions 13 and 14) and a small lower molecular weight peak (fraction 18; arrowhead) (Fig. 5A). These fractions were examined by Western blotting (Fig. 5B). Frs. 13 and 14 each gave a high molecular weight band (arrow) that could be detected with anti-IgA (left panel) and anti-pIgR (right panel), respectively. Fraction 15 gave a dimeric HIgA band (arrowhead) that was detected with anti-IgA but not with anti-pIgR. Fraction 18 contained free SC (data not shown). Although the separation of the peaks is not perfect, fraction 13 contained only HSIgA. These data suggest that HSIgA could be prepared through in vitro association between HIgA and SC.
FIG. 5.

Separation of HSIgA from HIgA. (A) Chromatogram of size exclusion chromatography on TSK gel G3000SWXL. Proteins were detected at UV280 nm. The peaks containing HSIgA (fraction 13), HSIgA and HIgA (fraction 14), and free SC (arrowhead; fraction 18) are indicated. (B) Analysis of purified HSIgA by Western blotting. The number above each lane corresponds to the fraction number. Anti-IgA (left panel) or anti-pIgR (right panel) was used. SDS-PAGE was carried out on 7.5% gels under nonreducing conditions. The arrows indicate the band of HSIgA and the arrowheads the band of HIgA. The positions of molecular weight standards are shown on the left.
Discussion
The critical role of SIgA against IAV infection was challenged by a study using IgA knockout mouse (IgA–/–) model. In that model, IgA–/– mice were immunized with an IAV subunit vaccine together with cholera toxin B subunit and holotoxin. They exhibited a similar level of protection from IAV infection as wild-type mice, despite the absence of IgA (20). It was concluded that IgA is not required for the prevention of IAV infection. However, it was pointed out that leakage of serum IgG through irritated mucosal surface lining and secretory IgM in place of SIgA may compensate for the lack of SIgA (5). On the other hand, pIgR knockout mice (pIgR–/–) were utilized to test the roles of SIgA (2). After intranasal immunization with IAV vaccine, pIgR–/– mice exhibited a reduced degree of protection from IAV infection as compared with wild-type mice, despite the leakage of IgG, and some IgA from serum into mucosal surface was observed.
It is still not easy to produce antigen-specific IgA mAbs and monoclonal SIgA specific for given pathogens. In this study, an attempt was made to produce an antigen-specific monoclonal SIgA by means of a practical method. To determine which class of immunoglobulin is suitable for topical application on the mucosal surface in vivo, it is necessary to produce IgG, IgA, and SIgA specific for the same epitope. To produce an IgA specific for the same epitope as that recognized by IgG mAbs, HIgA was produced using the recombinant PCR method (10). HSIgA was also produced by incubating HIgA with SC in vitro. HIgA as well as HSIgA were found to form dimeric immunoglobulins (Figs. 1B and 5B) such as native IgA and SIgA. HIgA and HSIgA bound to HA of IAV as well as complete IAV particles (Fig. 4). Because the HSIgA preparation comprised a mixture of HSIgA and HIgA, two detection antibodies, anti-IgA and anti-pIgR, were used. The binding of HIgA was detected with anti-IgA but not with anti-pIgR. On the other hand, HSIgA was detected with both of them. These data suggest that both HIgA and HSIgA are functional.
HIgA inhibited IAV infection of MDCK cells in vitro. Sialic acid expressed on the cell surface constitutes the receptor for IAV. Human influenza viruses preferentially bind to α-2,6-linked sialic acids, whereas avian influenza viruses bind to α-2,3-linked ones (12). Because MDCK cells express both types of sialic acid moiety, almost all strains of IAV can infect these cells. Although the cleavage of HA into HA1 is critical for the process of IAV infection (21), trypsin was not added to this system. In addition, MDCK cells produce only a marginal level of intrinsic trypsin-like activity that can be utilized by IAV. Thus, MDCK cells were infected with IAV only once, and no virus progeny was produced in this study. Therefore, the inhibition of the initial step of virus infection by HIgA was demonstrated.
Although functional HSIgA can be generated through in vitro association of HIgA and SC, it is still challenging to obtain a sufficient amount of purified HSIgA to determine whether HSIgA inhibits IAV infection. Although HSIgA can be separated from HIgA by size exclusion chromatography, sufficient separation has not been achieved so far. In an HSIgA-enriched fraction (Fig. 5; fraction 14), there was also a large amount of HIgA. This result suggested the loss of HSIgA into the mixture fraction, leading to the reduced recovery of HSIgA in the preparation. The method of purification of HSIgA needs to be improved to examine its biological activity without the influence of HIgA.
In this study, in vitro IAV infection of MDCK cells was inhibited by HIgA at 10 times lower concentrations than those of IgG specific for the same epitope (Fig. 2). Renegar et al. established monomeric IgA, dIgA, and SIgA specific for the same epitope of IAV (29). In that study, the SIgA was composed of mouse IgA and rat SC because it was produced in vivo by the injection of mouse dIgA into rats. They showed that dIgA and SIgA neutralized the infectivity of IAV more effectively than monomeric IgA in vitro. Recently, toxin neutralization activity was examined using a recombinant IgG/IgA specific for Shiga toxin (13). A monomeric and a dimeric form were examined in different CHO-K1 cells. In this case, the dimeric IgG/IgA was more than 10 times more effective than the monomeric one in terms of toxin neutralization.
There have been many reports showing that SIgA is produced through the in vitro association of IgA and SC (14,17,19). Although it has not been determined whether the three-dimensional structure is the same for both in vitro associated SIgA and in vivo secreted SIgA, all reports demonstrated that the in vitro associated SIgA was functional. Because IgA and SC were joined by a disulfide bond, it was thought that an association might not be achieved by simple incubation of IgA with SC (15). As thought, when crude HIgA and SC were used without purification in advance, only a small amount of HSIgA was obtained (data not shown). This problem was solved by the presence of glutathione and glutathione disulfide (GSH/GSSG), as reported in the literature (14). However, the purified HIgA efficiently bound to SC in the absence of GSH/GSSG. These results indicate that purification of IgA and SC is a critical factor for the efficient production of in vitro associated SIgA.
It was shown that SIgA can be produced using antigen-specific IgG. Although more detailed functional analyses of HSIgA are needed, it was shown that SIgA can be produced by an available method. This method can be applied to SIgA specific for various pathogens that enter through mucosa other than the influenza virus. It is expected that HSIgA will become a useful tool for studying the role of SIgA in adaptive immunity on the mucosal surface. The authors are planning to perform studies on in vivo efficacy of topically applied HSIgA in the future. These plans will be focused on studies for in vivo protection from IAV infection, comparison of the efficacy of HSIgA, HIgA, and IgG upon topical application, and half-life of topically applied HSIgA.
IAV is responsible for annual influenza epidemics, sometimes on a worldwide scale. Antigenic changes of HA contribute to the epidemics and even to pandemics (24,28). Due to such antigen changes, the same IAV vaccine cannot be used every year. Unfortunately, 2E10 mAb cannot bind to HA of the current seasonal influenza virus (unpublished results). Regarding therapeutic antibodies, ones with the ability to bind to the epitopes shared by many virus strains will be useful. Virus neutralizing activity of such antibodies is also required. Recently, some groups reported HA-specific antibodies that can bind to various subtypes of HA (7,16,23). Although all of them are IgG, it is thought that a recombinant IgA with the variable regions of these cross-reactive antibodies may contribute to the prevention of seasonal influenza.
In conclusion, a recombinant dimeric IgA specific for HA of IAV was produced. The dimeric IgA neutralized in vitro IAV infection at 10 times lower concentrations than those of IgG that has the same variable regions as the dimeric IgA. SIgA produced through association between the dimeric IgA and SC in vitro was found to bind to target antigens. Size exclusion chromatography was shown to be applicable to the separation of the SIgA from dimeric IgA.
Acknowledgments
The authors thank Mr. Shingo Kunugi and Mr. Yasukazu Sasanuma (University of Shizuoka) for their technical assistance. This work was supported partly by a Grant-in-aid for Challenging Exploratory Research (grant number 23659067, 25670063) to Y.I., and by research funding for the Global COE Program from the Japan Society for the Promotion of Science.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Ambrose CS, Luke C, and Coelingh K. Current status of live attenuated influenza vaccine in the United States for seasonal and pandemic influenza. Influenza Other Respi Viruses 2008;2:193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asahi Y, Yoshikawa T, Watanabe I, et al. Protection against influenza virus infection in polymeric Ig receptor knockout mice immunized intranasally with adjuvant-combined vaccines. J Immunol 2002;168:2930–2938 [DOI] [PubMed] [Google Scholar]
- 3.Barría MI, Garrido JL, Stein C, et al. Localized mucosal response to intranasal live attenuated influenza vaccine in adults. J Infect Dis 2013;207:115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belshe RB, Gruber WC, Mendelman PM, et al. Correlates of immune protection induced by live, attenuated, cold-adapted, trivalent, intranasal influenza virus vaccine. J Infect Dis 2000;181:1133–1137 [DOI] [PubMed] [Google Scholar]
- 5.Brandtzaeg P. Role of secretory antibodies in the defence against infections. Int J Med Microbiol 2003;293:3–15 [DOI] [PubMed] [Google Scholar]
- 6.Clements ML, O'Donnell S, Levine MM, et al. Dose response of A/Alaska/6/77 (H3N2) cold-adapted reassortant vaccine virus in adult volunteers: role of local antibody in resistance to infection with vaccine virus. Infect Immun 1983;40:1044–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corti D, Voss J, Gamblin SJ, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011;333:850–856 [DOI] [PubMed] [Google Scholar]
- 8.Czinn SJ, Cai A, and Nedrud JG. Protection of germ-free mice from infection by Helicobacter felis after active oral or passive IgA immunization. Vaccine 1993;11:637–642 [DOI] [PubMed] [Google Scholar]
- 9.French MA, Denis KA, Dawkins R, et al. Severity of infections in IgA deficiency: correlation with decreased serum antibodies to pneumococcal polysaccharides and decreased serum IgG2 and/or IgG4. Clin Exp Immunol 1995;100:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higuchi R. Recombinant PCR. In: Innis MA, Gelfand DH, Sninsky JJ, and White TJ, eds. PCR Protocols: A Guide to Methods and Applications. San Diego, CA: Academic Press, 1990:177–183 [Google Scholar]
- 11.Imai Y, Ishikawa T, Tanikawa T, et al. Production of IgA monoclonal antibody against Shiga toxin binding subunits employing nasal-associated lymphoid tissue. J Immunol Methods 2005;302:125–135 [DOI] [PubMed] [Google Scholar]
- 12.Ito T, Suzuki Y, Takada A, et al. Differences in sialic acid-galactose linkages in the chicken egg amnion and allantois influence human influenza virus receptor specificity and variant selection. J Virol 1997;71:3357–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwata K, Kurohane K, Nakanishi K, et al. Stable expression and characterization of monomeric and dimeric recombinant hybrid-IgG/IgA immunoglobulins specific for Shiga toxin. Biol Pharm Bull 2014;37:1510–1515 [DOI] [PubMed] [Google Scholar]
- 14.Jones RM, Schweikart F, Frutiger S, et al. Thiol-disulfide redox buffers maintain a structure of immunoglobulin A that is essential for optimal in vitro binding to secretory component. Biochim Biophys Acta 1998;1429:265–274 [DOI] [PubMed] [Google Scholar]
- 15.Kerr MA. The structure and function of human IgA. Biochem J 1990;271:285–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lingwood D, McTamney PM, Yassine HM, et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 2012;489:566–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lüllau E, Heyse S, Vogel H, et al. Antigen binding properties of purified immunoglobulin A and reconstituted secretory immunoglobulin A antibodies. J Biol Chem 1996;271:16300–16309 [DOI] [PubMed] [Google Scholar]
- 18.Mantis NJ, Rol N, and Corthésy B. Secretory IgA's complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol 2011;4:603–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathias A, and Corthésy B. Recognition of gram-positive intestinal bacteria by hybridoma- and colostrum-derived secretory immunoglobulin A is mediated by carbohydrates. J Biol Chem 2011;286:17239–17247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mbawuike IN, Pacheco S, Acuna CL, et al. Mucosal immunity to influenza without IgA: an IgA knockout mouse model. J Immunol 1999;162:2530–2537 [PubMed] [Google Scholar]
- 21.Nayak DP, Balogun RA, Yamada H, et al. Influenza virus morphogenesis and budding. Virus Res 2009;143:147–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Offit PA, and Clark HF. Protection against rotavirus-induced gastroenteritis in a murine model by passively acquired gastrointestinal but not circulating antibodies. J Virol 1985;54:58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohshima N, Iba Y, Kubota-Koketsu R, et al. Naturally occurring antibodies in humans can neutralize a variety of influenza virus strains, including H3, H1, H2, and H5. J Virol 2011;85:11048–11057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palese P. Influenza: old and new threats. Nat Med 2004;10:S82–S87 [DOI] [PubMed] [Google Scholar]
- 25.Persson E, Eneroth P, and Jeansson S. Secretory IgA against herpes simplex virus in cervical secretions. Genitourin Med 1988;64:373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phalipon A, Cardona A, Kraehenbuhl JP, et al. Secretory component: a new role in secretory IgA-mediated immune exclusion in vivo. Immunity 2002;17:107–115 [DOI] [PubMed] [Google Scholar]
- 27.Piskurich JF, Blanchard MH, Youngman KR, et al. Molecular cloning of the mouse polymeric Ig receptor. Functional regions of the molecule are conserved among five mammalian species. J Immunol 1995;154:1735–1747 [PubMed] [Google Scholar]
- 28.Rappuoli R, and Dormitzer PR. Influenza: options to improve pandemic preparation. Science 2012;336:1531–1533 [DOI] [PubMed] [Google Scholar]
- 29.Renegar KB, Jackson GD, and Mestecky J. In vitro comparison of the biologic activities of monoclonal monomeric IgA, polymeric IgA, and secretory IgA. J Immunol 1998;160:1219–1223 [PubMed] [Google Scholar]
- 30.Renegar KB, and Small PA. Passive transfer of local immunity to influenza virus infection by IgA antibody. J Immunol 1991;146:1972–1978 [PubMed] [Google Scholar]
- 31.Renegar KB, and Small PA. Immunoglobulin A mediation of murine nasal anti-influenza virus immunity. J Virol 1991;65:2146–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renegar KB, Small PA, Boykins LG, et al. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J Immunol 2004;173:1978–1986 [DOI] [PubMed] [Google Scholar]
- 33.Staudt LM, and Gerhard W. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. I. Significant variation in repertoire expression between individual mice. J Exp Med 1983;157:687–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki Y, Matsunaga M, and Matsumoto M. N-Acetylneuraminyllactosylceramide, GM3-NeuAc, a new influenza A virus receptor which mediates the adsorption-fusion process of viral infection. Binding specificity of influenza virus A/Aichi/2/68 (H3N2) to membrane-associated GM3 with different molecular species of sialic acid. J Biol Chem 1985;260:1362–1365 [PubMed] [Google Scholar]
- 35.Takahashi T, Moriyama Y, Ikari A, et al. Surface localization of the nuclear receptor CAR in influenza A virus-infected cells. Biochem Biophys Res Commun 2008;368:550–555 [DOI] [PubMed] [Google Scholar]
- 36.Takahashi T, Murakami K, Nagakura M, et al. Sulfatide is required for efficient replication of influenza A virus. J Virol 2008;82:5940–5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura SI, Asanuma H, Ito Y, et al. Superior cross-protective effect of nasal vaccination to subcutaneous inoculation with influenza hemagglutinin vaccine. Eur J Immunol 1992;22:477–481 [DOI] [PubMed] [Google Scholar]
- 38.Tanikawa T, Ishikawa T, Maekawa T, et al. Characterization of monoclonal immunoglobulin A and G against Shiga toxin binding subunits produced by intranasal immunization. Scand J Immunol 2008;68:414–422 [DOI] [PubMed] [Google Scholar]
- 39.Tanikawa T, Kurohane K, and Imai Y. Production and characterization of IgA monoclonal antibody against ovalbumin. Hybridoma (Larchmt) 2007;26:328–332 [DOI] [PubMed] [Google Scholar]
- 40.Tanimoto T, Haredy AM, Takenaka N, et al. Comparison of the cross-reactive anti-influenza neutralizing activity of polymeric and monomeric IgA monoclonal antibodies. Viral Immunol 2012;25:433–439 [DOI] [PubMed] [Google Scholar]
- 41.Tobisawa Y, Maruyama T, Tanikawa T, et al. Establishment of recombinant hybrid-IgG/IgA immunoglobulin specific for Shiga toxin. Scand J Immunol 2011;74:574–584 [DOI] [PubMed] [Google Scholar]
- 42.Tomoda T, Morita H, Kurashige T, et al. Prevention of influenza by the intranasal administration of cold-recombinant, live-attenuated influenza virus vaccine: importance of interferon-gamma production and local IgA response. Vaccine 1995;13:185–190 [DOI] [PubMed] [Google Scholar]
- 43.van Riet E, Ainai A, Suzuki T, et al. Mucosal IgA responses in influenza virus infections; thoughts for vaccine design. Vaccine 2012;30:5893–5900 [DOI] [PubMed] [Google Scholar]
- 44.Winner L, 3rd, Mack J, Welzin R, et al. New model for analysis of mucosal immunity: intestinal secretion of specific monoclonal immunoglobulin A from hybridoma tumors protects against Vibrio cholerae infection. Infect Immun 1991;59:977–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woof JM, and Kerr MA. The function of immunoglobulin A in immunity. J Pathol 2006;208:270–282 [DOI] [PubMed] [Google Scholar]