

Abstract
Elevated levels of interferon-alpha (IFNα) in the central nervous system (CNS) are linked to cognitive dysfunction in patients with inflammatory CNS diseases such as HIV-associated neurocognitive disorders (HAND). Increased CNS IFNα has also been found to be associated with cognitive dysfunction in a HAND mouse model. Here, we corroborate previous studies showing a dose-dependent decrease in dendritic branching and length caused by IFNα treatment and extend those studies. Because both direct and indirect mechanisms of IFNα-induced neurotoxicity are likely involved, the cell signaling pathway involving the IFNα receptor (IFNAR) was initially evaluated. Rat neuronal cultures exposed to IFNα demonstrate increased phosphorylation of STAT1 and increased interferon stimulating gene 15 (ISG15) expression, indicators of IFNAR engagement. However, specific blocking antibodies to the IFNAR were found to only partially protect neurons from IFNα-induced neurotoxicity. Additionally, inhibiting the GluN2A subunit of N-methyl-D-asparate receptor (NMDAR) was also found to be partially protective against IFNα-induced neurotoxicity compared with the GluN2B subunit. Neurotoxicity is evident in neurons extracted from IFNAR KO mice treated with IFNα as well, further indicating that IFNAR signaling is not required for IFNα neurotoxicity. The neurotoxic actions of IFNα are mediated through both the IFNAR as well as the GluN2A subunit of the NMDAR to reduce dendritic arborization in neurons. Complete protection from IFNα-induced neurotoxicity was demonstrated when both pathways were blocked. Blocking these pathways could lead to potential therapies for cognitive dysfunction during neuroinflammation and specifically lead to better treatments for HAND.
Introduction
Interferon-alpha (IFNα) plays a critical role in early inhibition of viral replication within infected cells and establishes an antiviral state in the surrounding cells. However, excessive amounts of IFNα can cause neurotoxicity. IFNα is elevated in patients with neuroinflammatory diseases such as HIV-associated neurocognitive disorders (HAND) (Rho and others 1995), Aicardi–Goutieres syndrome (Goutieres and others 1998), neuropsychiatric systemic lupus erythematosus (Shiozawa and others 1992) and multiple sclerosis (Traugott and Lebon 1988; Fritz-French and Tyor 2012; Tzartos and others 2012). In addition, neuronal toxicity has been implicated in hepatitis and cancer patients being treated with high-dose IFNα therapy, where patients have shown neuropsychiatric side effects, such as subcortical dementia, which dissipate after terminating treatment (Valentine and others 1998; Kirkwood and others 2002; Schaefer and others 2002). Identifying pathways involved in IFNα neurotoxicity could lead to potential therapies for central nervous system (CNS) inflammatory diseases as well as treatments targeting the side effects of IFNα therapy.
Ample evidence of IFNα-induced neurotoxicity has been shown in both clinical studies of HAND (Rho and others 1995) and animal studies (Sas and others 2009). Importantly, IFNα levels in the cerebrospinal fluid of HIV-infected patients are significantly higher in patients with dementia compared with patients without dementia (Rho and others 1995). Studies using a SCID mouse model of HAND found that elevated IFNα levels in the brains of HAND mice correlated with cognitive and neuronal dysfunction (Sas and others 2007) that was prevented by treatment with neutralizing antibodies to IFNα (Sas and others 2009). Despite the advent of antiretroviral therapy, the prevalence of HAND continues to increase in HIV-infected patients, affirming the need for more specific treatments targeted to HAND. Therefore, we have focused on evaluating the mechanism of IFNα-induced neurotoxicity to identify potential targets for treatment in HAND.
The mechanism of IFNα-induced neurotoxicity is yet to be established. In this study, we begin to investigate the cell signaling pathways involved using a cortical neuron culture model (Kaech and Banker 2006). We examine the roles of type 1 interferon receptor [IFNα receptor (IFNAR)] engagement and the N-methyl-D-asparate receptor (NMDAR). IFNα initiates an antiviral response by binding to the IFNAR and the subsequent activation of the JAK/STAT pathway, and therefore, we expected that IFNAR binding would be required for IFNα-induced neurotoxicity. However, we show that blocking the IFNAR only partially protects neurons from dendritic damage caused by IFNα, indicating a possible role of other pathways. The NMDAR has been linked to IFNα neurotoxicity in clinical studies, where treating patients on IFNα therapy with a mild NMDAR antagonist resulted in decreased side effects (Quarantini and others 2006; Sas and others 2009). The NMDAR is a tetrameric complex where the GluN2 subunit is responsible for receptor function (Paoletti and Neyton 2007). In the adult CNS, the predominant subunits in the cortex and hippocampus are GluN2A and GluN2B, suggesting critical roles in synaptic plasticity and function. Our current studies show that the GluN2A subunit of the NMDAR plays a significant role in IFNα-induced neurotoxicity. Further characterizing signaling molecules (ie, JAK/STAT pathway) involved in IFNα-induced neurotoxicity will help to identify potential therapeutic targets for inflammatory diseases of the CNS, where IFNα is upregulated, such as HAND and the side effects of IFNα therapy.
Materials and Methods
Neuronal cell culture
Neurons were dissected from the frontal cortex of E18 Sprague-Dawley rat embryos (Charles River Laboratory). For the IFNAR KO experiments, Ifnar−/− fetal mice were kindly provided by Kaja Muralie (Emory University). All procedures were approved by the Institutional Animal Care and Use Committee of the Atlanta Veterans Administration Medical Center and were in accordance with the guidelines of the NIH Guide for the Care and Use of Laboratory Animals. High-density cultures for protein and RNA extractions were prepared by plating neurons in poly-L-lysine (PLL; Sigma)-coated dishes at 5×105 cells/60-mm dish and incubated at 37°C with 5% CO2 in a plating medium [MEM supplemented with 10% fetal bovine serum (FBS), glucose 0.6% wt/vol, HEPES]. After 2 h, the plating medium in the high-density cultures was exchanged for a neurobasal medium (neurobasal medium supplemented with B27 and GlutaMax; Invitrogen). Low-density neuronal cultures prepared for coculturing with glia were plated on PLL-coated glass coverslips at 5×105 cells/10-cm dish, as described in Kaech and Banker (2006). Two hours after plating, the low-density neuronal cultures on the glass slides were flipped over onto a dish containing a glial monolayer grown in the neurobasal medium. Coculture with the glial monolayer allows the neurons to grow healthy and robust dendrites even at low density. Cytosine-D-arabinoside (Ara-C, 1 μM; Fluka) was added to both high- and low-density cultures at day 3 after plating to prevent glial cell proliferation. High-density cultures were used to collect large quantities of RNA and protein, and low-density cultures were used to assess dendritic length and branching on individual neurons. Media were exchanged every 7 days. Low-density glass coverslips and high-density neuronal cultures after 14 days are less than 5% glia.
Treatment and reagents
Rat IFNα and IFNβ (PBL Interferon) were diluted in 0.9% saline at a dose range of 300–900 IU/mL based on previous findings (Sas and others 2009), and a single dose was administered to cultures on day 14. Control groups were given equal volumes of saline. For high-density cultures, treatment was left on cultures for different time points varying from 20 min to 48 h before RNA or protein extractions. Low-density neurons cocultured with glia on coverslips were removed from the glia and treated with IFNα or saline for 2 h. The neurons were then returned to the glia for long-term culturing. The type I interferon receptor (IFNAR) mouse antibody (eBioscience), IgG1 K isotype control (eBioscience), or saline was administered at 1 to 50 μg 20 min before IFNα treatment. TCN 201 (Tocris) was given at 0.1, 1, and 10 μM, and ifenprodil (Tocris) was given at 3–10 μM 20 min before IFNα treatment.
Protein extractions and western blot
Proteins were harvested from the high-density neuronal cultures using the RIPA buffer with 10 μL/mL protease inhibitors (Thermo Scientific), incubated on ice for 10 min, and spun down at 14,000 rpm for 10 min. The lysate was removed, and the total protein concentration was determined by a Pierce® BCA Protein Assay Kit. The lysate was aliquoted and stored at −80°C. Five micrograms of each sample was then loaded on a 7.5% polyacrylamide gel. The protein was then transferred to the PVDF membrane run at 120 V for 40 min. The anti-Phospho-STAT1, anti STAT1 (Cell Signaling), and GAPDH (Millipore) antibodies were used as primary antibodies at 1:1,000 each. Western blots were probed according to the manufacturer's protocol. The membrane was then washed and incubated with either the goat anti-rabbit IgG HRP-linked secondary antibody at 1:1,000 (Cell Signaling) or the goat anti-mouse IgG HRP-labeled secondary antibody at 1:1,000 (Perkin Elmer). The membrane was then exposed to X-ray film using SuperSignal® West Pico Chemiluminescent Substrate (Thermo Scientific).
Real-time PCR
High-density neuronal cultures were used to extract RNA according to the RNeasy Mini Kit (Qiagen) protocol. cDNA synthesis was performed using the iScript cDNA Synthesis Kit (Biorad). Levels of rat interferon-stimulated gene 15 (ISG15) were analyzed using real-time polymerase chain reaction (PCR) with the Biorad C1000 Thermocycler, and GAPDH was used as our housekeeping gene and an endogenous control for experiments. ISG15 and GAPDH primers were designed using Assays-by-Design (Applied Biosystems). Levels of the target gene were normalized against the endogenous gene to assess relative quantification after PCR amplification efficiencies were determined according to the MIQE guidelines. The relative changes in gene expression were analyzed using the 2−ΔΔCt method by Livak and Schmittgen (Livak and Schmittgen 2001).
Image acquisition and processing
The low-density neuronal cultures plated on the glass coverslips were removed from the glial coculture and washed in phosphate-buffered saline (PBS) before fixing cells with cold methanol for 20 min at −20°C. Coverslips were rinsed in PBS and TBS50 before permeabilizing the cells in 0.3% Triton X-100/TBS50 for 5 min. Slides stained for IFNAR were not permeabilized. Cells were then rinsed in 0.1% Triton X-100/TBS50 before blocking in the blocking buffer [2% bovine serum albumin (BSA), 2% FBS (Sigma), 0.1% Triton X-100, in TBS50] for 1–2 h. Coverslips were then rinsed in the buffer (2% BSA, 0.1% Triton X-100 in TBS50) and then incubated in the primary anti-microtubule-associated protein 2 (MAP2) antibody (Millipore) at 1:300 at room temperature for 1 h. The secondary fluorescein goat anti-rabbit IgG (Vector) was then added at 1:500 in the dark after rinsing for 30 min.
Coverslips were rinsed and placed on slides with Vectashield fluorescence protection (Vector). Slides were viewed using an Olympus IX71, and photographs were taken using ImagePro Express 6.0. Two 15-mm slides per treatment group were stained, and 20 neurons per treatment group analyzed. ImagePro was also used to measure the dendritic length and branching manually using the trace feature that then totals the measurements of the dendritic length. All dendrites that are not connected to the soma are counted as branches. The researcher was blinded to treatment groups.
Statistics
Analysis of real-time PCR results and dendritic length and branching data was completed using 2-way analysis of variance. Analysis between treatment groups was done with post hoc Tukey tests using SPSS Statistics 19 software. Significance was set at P value <0.05 for all analyses.
Results
IFNα is toxic to neurons cocultured with glia
To determine the timing of IFNα neurotoxicity, we treated low-density rat cortical neurons with 300 IU/mL of IFNα or saline away from the presence of glia. Neurons are removed from glia for treatment to ensure effects are on neurons alone and returned to glia for long-term culturing. After 2 h, IFNα-treated neurons were returned to glia and subsequently fixed at 12, 24, 48, and 72 h after exposure. Dendritic morphology was measured to assess the total length of dendrites per neuron and number of dendritic branches. A comprehensive time course study was done to assess when dendritic morphology damage begins. A significant decrease in dendritic length and branching occurs at 24 h and continues for 72 h, but no effect is seen at 12 h after IFNα exposure (Fig. 1). In addition, neurons treated with IFNα for 72 h showed decreased dendritic arborization, but overall did not show severe abnormalities indicative of neuronal cell death (ie, swollen or abnormally shaped nucleus, dendritic blebbing). Previous studies of cell viability using Trypan Blue staining showed a dose-dependent increase in cell death by IFNα (Sas and others 2009).
FIG. 1.

IFNα is toxic to neurons cocultured with glia. Neurons were treated with a single dose of 300 IU/mL of IFNα over 12, 24, 48, and 72 h. IFNα-induced toxicity is observed at 24 h after a single dose of IFNα and persists for 72 h. No toxicity is seen at 12 h post-treatment. Graphs include data from 3 separate experiments (*P<0.05). IFNα, interferon-alpha.
JAK/STAT pathway activated in neurons treated with IFNα
To assess JAK/STAT pathway activation in neurons, high-density rat cortical neurons were treated with 300 IU/mL of IFNα and protein extracted at 20 min, 2, 12, 24, and 48 h. Neurons show elevated phosphorylated STAT1 at 20 min after a single IFNα treatment compared to untreated neurons. The phosphorylated STAT1 signal persisted for 48 h after a single treatment of IFNα based on western blot analysis (Fig. 2). ISG15 is upregulated after IFNAR activation and used as a marker in this system to assess downstream JAK/STAT pathway activation. ISG15 mRNA was elevated in cultures treated with IFNα 20 min after treatment (Fig. 3A).
FIG. 2.
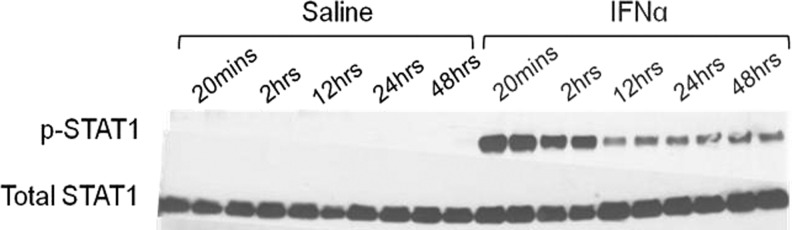
STAT1 is phosphorylated in neurons treated with IFNα. Protein extracts from neurons treated with IFNα at various time points were analyzed by immunoblotting for STAT1 and phospho-STAT1 (p-STAT1) expression. p-STAT1 peaked at 20 min post-treatments and signal persisted for 48 h indicating IFNAR activation. Samples were run in duplicate lanes on western blot. IFNAR, IFNα receptor.
FIG. 3.

IFNAR inhibitor blocks IFNα signaling in IFNα-treated neuronal cultures. Neurons were pretreated with saline or increasing doses of an IFNAR blocker or an IgG1 K isotype control (IFNGR) at 10 and 50 μg. Cultures were then exposed to either saline or IFNα for 20 min. JAK/STAT signaling is inhibited in cultures treated with the IFNAR blocker. ISG15 expression was found to be decreased in cultures pretreated with the IFNAR blocker compared to IFNα-alone-treated cultures (*P<0.05, **P<0.001) (A). p-STAT1 expression was decreased in cultures pretreated with the IFNAR blocker before IFNα treatment compared to cultures treated with IFNα alone STAT1, and p-STAT1 controls were run with the protein extracts as positive controls (B). Experiments were conducted in triplicates. ISG15, interferon stimulating gene 15.
Blocking IFNAR blocks the JAK/STAT pathway
To determine the requirement of the IFNAR for IFNα-induced neurotoxicity, the effectiveness of an IFNAR-blocking antibody was examined in culture. IFNAR engagement induces the JAK/STAT pathway after IFNα binds the receptor. High-density neurons were pretreated with the IFNAR-blocking antibody, IgG1 K isotype control, or saline 20 min before IFNα treatment. To ensure that the pathway was efficiently blocked, ISG15 gene expression was examined. ISG15 expression in IFNAR antibody-pretreated cultures was not significantly different than saline-treated cultures indicating that the JAK/STAT pathway was effectively blocked by the IFNAR antibody (Fig. 3A). The protein was extracted and western blot analysis revealed almost no phosphorylated STAT1 after 20 min from cultures pretreated with the IFNAR-blocking antibody before IFNα exposure compared to neurons treated with an isotype-matched control or IFNα-only-treated cultures (Fig. 3B).
Blocking IFNAR is partially protective against IFNα-induced neurotoxicity
After determining that the IFNAR antibody is efficient in blocking the JAK/STAT pathway in neuronal cultures, we assessed the ability of the IFNAR antibody to block IFNα-induced neurotoxicity. Neuronal cultures were pretreated with the IFNAR antibody, an isotype control, or saline. After 20 min, neurons were exposed to IFNα or saline and cultured for 48 h. Dendritic measurements and analysis of IFNAR antibody-treated cultures showed partial protection against IFNα-induced neurotoxicity based on the total dendritic length and branching (Fig. 4). Cultures treated with varying doses of IFNAR antibody before IFNα exposure were significantly different than cultures treated with IFNα alone. In addition, IFNAR antibody-treated cultures were significantly different from untreated cultures. Previous studies found that IFNα-neutralizing antibodies completely protected neurons from IFNα-induced neurotoxicity (Sas and others 2009). Complete protection was not seen when the IFNAR alone was blocked. Although IFNα is likely to be exerting some neurotoxic effects through its receptor, current studies indicate that receptor associations are complex. In addition, IFNα stimulates a number of substances that could be neurotoxic (Kirkwood and others 2002), therefore, some IFNα effects could be indirect and independent of the IFNAR.
FIG. 4.

Blocking the IFNAR partially protected the neurons from IFNα toxicity. Neurons were pretreated with saline, an IFNAR blocker (1, 10, or 50 μg), or an IgG1 K isotype control (IFNGR) (1, 10, or 50 μg) and then exposed to IFNα for 48 h. Dendritic length and number of branches were significantly improved in cultures pretreated with IFNAR Ab at all doses before IFNα exposure compared to cultures treated with IFNα alone or an isotype control. However, cultures pretreated with IFNAR Ab were not completely protected and were significantly different than saline-treated cultures (*P<0.05, **P<0.001).
Blocking GluN1/GluN2A-containing NMDAR is protective
To determine the role of the NMDAR in IFNα-induced neurotoxicity, the effects of neurons pretreated with TCN 201 (GluN2A antagonist) and ifenprodil (GluN2B antagonist) before IFNα exposure were examined (Fig. 5). Low-density neuronal cultures were treated with doses ranging from 0.1 to 10 μM of TCN 201 or 3 to 10 μM of ifenprodil 20 min before adding 300 IU/mL of IFNα. Cultures were analyzed 48 h later for dendritic length and branching. Neuronal cultures pretreated with higher doses of TCN 201 (1 and 10 μM) were found to be significantly different than cultures treated with IFNα alone based on dendritic length measurements, and the high dose of TCN 201 was found to be significantly different indicating a partial protective effect of TCN 201 against IFNα neurotoxicity. Ifenprodil was found to have no protective effects against IFNα at any dose.
FIG. 5.

Blocking the NR2A-specific subunit of NMDAR was partially protective against IFNα toxicity. Cultures were pretreated with either TCN 201 (0.1, 1, or 10 μM) or ifenprodil (3 or 10 μM) and then exposed to IFNα for 48 h. TCN 201 at 1 and 10 μM had a protective effect against IFNα neurotoxic effects with dendritic length that was not seen in lower doses or with ifenprodil. In addition, the highest dose of TCN 201 was significantly protective against dendritic branching. However, TCN 201 was not completely protective because dendritic length and branching were still significantly different than saline-treated cultures (*P<0.05), suggesting partial involvement of the NR2A subunit of NMDAR in IFNα-induced neurotoxicity. Images are representative of 3 separate experiments that were combined in the graphs. NMDAR, N-methyl-D-asparate receptor.
In addition, neurons extracted from IFNAR KO fetal mice were exposed to IFNα and showed significant decreased dendritic branching and length despite the neurons lacking receptors for IFNα (Fig. 6). However, IFNAR KO fetal mice neurons were completely protected against IFNα-induced neurotoxicity when pretreated with TCN 201 (Fig. 6).
FIG. 6.

IFNα induces neurotoxicity in IFNAR KO mouse neuronal cultures. Significant decreases in dendritic length and branching were evident in neurons extracted from IFNAR KO mouse fetuses that were treated with 300 or 900 U of IFNα for 48 h. Neuron cultures pretreated with TCN 201 before IFNα exposure were not significantly different than saline-treated neurons indicating complete protection from IFNα-induced neurotoxicity when both receptors are inhibited (*P<0.05, **P<0.001, ***P<0.0001).
IFNβ has no toxic effect on neurons
We investigated the effect of IFNβ on neurons cocultured with glia because IFNβ, a type I IFN, along with IFNα, utilizes the IFNAR. Treating neurons with 100, 300, or 900 IU of IFNβ had no effect on the neurons up to 72 h. (Fig. 7).
FIG. 7.
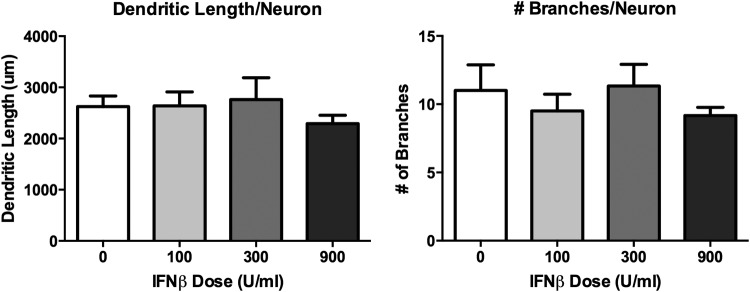
IFNβ causes no significant decrease in dendritic branching or length in rat neuronal cultures. Neurons were treated with IFNβ 100, 300, or 900 IU for 48 h. Neurons displayed no significant decrease in dendritic length or branching, indicating no significant neuronal toxicity of IFNβ compared to IFNα.
Discussion
In this study, we used a primary rat cortical neuronal in vitro model system to determine mechanisms and receptors involved in IFNα-induced neurotoxicity. The neurotoxic effects of IFNα become significant after 24 h of exposure and these effects on dendrites can be seen at 72 h without morphological signs of neuronal cell death (ie, soma swelling, dendritic blebbing). We found that pretreating neurons with either an IFNAR blocker or NMDAR subunit-specific inhibitor was partially protective against IFNα-induced neurotoxicity. This study is the first to look at specific receptor pathways involved in IFNα-induced neurotoxicity. The results imply that IFNα causes toxicity in neurons through indirect and direct pathways and the mechanism for IFNα neurotoxicity is complex.
IFNα neurotoxicity has been studied in several in vivo models (Dunn and Crnic 1993; Dafny 1998; Campbell and others 1999; Makino and others 2000; Mendoza-Fernandez and others 2000; Sas and others 2009). However, very few studies have examined the mechanism of IFNα neurotoxicity in vitro. In the current study using a neuronal in vitro system, we found that a significant decrease in dendritic length and branching is seen between 12 and 24 h after neurons are exposed to IFNα. A significant loss in dendritic arborization was also seen at 48 and 72 h after IFNα exposure. These results confirm and extend previous studies where IFNα was found to cause toxicity after 48 h in a different neuronal culture system (Sas and others 2009).
The current study found that pretreating neurons that are exposed to IFNα with an IFNAR blocker showed only 65% retention of dendritic length compared to 97% dendritic retention, as was seen in a previous study where neurons were treated with neutralizing antibodies to IFNα (Sas and others 2009). Similarly, dendritic branching was more reduced in cultures where the receptor, IFNAR, was targeted compared to using neutralizing antibodies to IFNα. In addition, studies using neurons from IFNAR KO fetal mice showed loss of dendritic branching and length in cultures treated with IFNα (Fig. 6). The results of the present study indicate that IFNα is inducing neurotoxicity through direct engagement of its receptor, but that there are additional important mechanisms involved in IFNα-induced neurotoxicity.
The NMDAR has been previously implicated in IFNα neurotoxicity (Katafuchi and others 1995; Sas and others 2009). The NMDAR is a glutamate-gated cation channel composed of GluN1 and GluN2/3 subunits that play a vital role in synaptic plasticity and cognitive function. The GluN2 subunit is expressed as 4 specific isoforms and this subunit controls the electrophysiological properties of the NMDAR. In addition, there is a GluN2B to GluN2A switch that occurs in the NMDAR as the neuron develops and matures (Wang and others 1995). Our results indicate that neurons pretreated with higher doses of a novel GluN1/GluN2A-specific antagonist, TCN 201, are partially protected against IFNα-induced neurotoxicity. Neurons pretreated with a GluN1/GluN2B antagonist, ifenprodil, were not protected against IFNα exposure. These results suggest that our neuronal cultures represent a more mature phenotype, perhaps more consistent with adult brain neurons. Neurons lacking an IFNAR and treated with TCN 201 were completely protected against IFNα-induced neurotoxicity indicating the involvement of both the IFNAR and NMDAR. In addition, excessive activation of the NMDAR can lead to excitotoxicity, a mechanism of neurotoxicity linked to several neurodegenerative diseases, including HAND (Kaul and others 2001). The ability of TCN 201 to attenuate IFNα-induced neurotoxicity indicates a mechanism of IFNα causing decreased dendritic arborization through overstimulation of the NMDAR and subsequent excitotoxicity. Furthermore and importantly, the data indicate that the GluN2A subunit of the NMDAR is specifically involved and is a possible therapeutic target for IFNα-mediated neurotoxicity. Therefore, our new data extend previous findings by suggesting that NMDAR-mediated neurotoxicity could be dependent on specific NMDAR isotypes. Furthermore, our study emphasizes that the development of effective NMDA antagonists could be applied to multiple diseases.
We also found that despite IFNβ engaging the same receptor as IFNα, treating neurons with IFNβ showed no signs of toxicity based on dendritic arborization assessments (Fig. 7). These data are consistent with numerous studies that have found differential signaling pathways between IFNα and IFNβ after IFNAR binding (MacDonald and others 1990; Platanias and others 1996; Marijanovic and others 2007). It is also consistent with the clinical observation that treatment of patients with IFNβ does not result in cognitive impairment (Jin and others 2007; Lacy and others 2013; Patti and others 2013).
Data shown here indicate that neuronal damage caused by the interaction of IFNα with its receptor is not restricted to the IFNAR and includes NMDAR pathways. TCN 201 only partially restored the neuron's baseline health in the presence of IFNα. It is possible that the cellular cascade initiated by IFNAR activation could affect other ion channels or cytoskeletal pathways in neurons, although no changes were seen in MAP2 or MAP1a expression in neuronal cultures exposed to IFNα (data not shown). Another potential mechanism of IFNα neurotoxicity is through indirect upregulation of other immune factors like nuclear factor-kappa B, a known transcription factor that is upregulated in HAND patients and may play a role in several neurodegenerative diseases (Rostasy and others 2000). Future studies will focus on identifying how IFNAR activation causes changes to dendritic morphology and how IFNα engages the NMDAR.
Identifying potential targets for IFNα neurotoxicity could lead to specific treatments for patients suffering from CNS inflammatory diseases, especially HAND. In addition, IFNα remains the backbone of hepatitis treatment, and complications resulting directly from IFNα treatment involving cognitive dysfunction are common (Okanoue and others 1996; Lieb and others 2006). Blocking IFNα, or other proteins involved in downstream IFNα signaling, could prevent the damage to neurons and the subsequent cognitive dysfunction that is seen both in CNS inflammatory diseases and in patients on IFNα therapy.
Acknowledgments
The authors thank Dr. Gary Bassell for his guidance and his former laboratory members, Kristy Welshans and Sharon Swanger, for training in primary rat neuronal cultures. They thank Dr. Stephen Traynelis for his advice on the NMDAR experiments. Scientific work was supported by the VA Merit Award 1101BX001506-01A2.
Author Contributions
C.F.K. and W.R.T wrote the manuscript. C.F.K. conducted all the experiments. Experiments were designed by C.F.K. and W.R.T.
Author Disclosure Statement
No competing financial interests exist for either author.
References
- Campbell IL, Krucker T, Steffensen S, Akwa Y, Powell HC, Lane T, Carr DJ, Gold LH, Henriksen SJ, Siggins GR. 1999. Structural and functional neuropathology in transgenic mice with CNS expression of IFN-alpha. Brain Res 835(1):46–61 [DOI] [PubMed] [Google Scholar]
- Dafny N. 1998. Is interferon-alpha a neuromodulator? Brain Res Brain Res Rev 26(1):1–15 [DOI] [PubMed] [Google Scholar]
- Dunn AL, Crnic LS. 1993. Repeated injections of interferon-alpha A/D in Balb/c mice: behavioral effects. Brain Behav Immun 7(1):104–111 [DOI] [PubMed] [Google Scholar]
- Fritz-French C, Tyor W. 2012. Interferon-alpha (IFNalpha) neurotoxicity. Cytokine Growth Factor Rev 23(1–2):7–14 [DOI] [PubMed] [Google Scholar]
- Goutieres F, Aicardi J, Barth PG, Lebon P. 1998. Aicardi-Goutieres syndrome: an update and results of interferon-alpha studies. Ann Neurol 44(6):900–907 [DOI] [PubMed] [Google Scholar]
- Jin S, Kawanokuchi J, Mizuno T, Wang J, Sonobe Y, Takeuchi H, Suzumura A. 2007. Interferon-beta is neuroprotective against the toxicity induced by activated microglia. Brain Res 1179:140–146 [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. 2006. Culturing hippocampal neurons. Nat Protoc 1(5):2406–2415 [DOI] [PubMed] [Google Scholar]
- Katafuchi T, Take S, Hori T. 1995. Roles of cytokines in the neural-immune interactions: modulation of NMDA responses by IFN-alpha. Neurobiology (Bp) 3(3–4):319–327 [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. 2001. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410(6831):988–994 [DOI] [PubMed] [Google Scholar]
- Kirkwood JM, Bender C, Agarwala S, Tarhini A, Shipe-Spotloe J, Smelko B, Donnelly S, Stover L. 2002. Mechanisms and management of toxicities associated with high-dose interferon alfa-2b therapy. J Clin Oncol 20(17):3703–3718 [DOI] [PubMed] [Google Scholar]
- Lacy M, Hauser M, Pliskin N, Assuras S, Valentine MO, Reder A. 2013. The effects of long-term interferon-beta-1b treatment on cognitive functioning in multiple sclerosis: a 16-year longitudinal study. Mult Scler 19(13):1765–1772 [DOI] [PubMed] [Google Scholar]
- Lieb K, Engelbrecht MA, Gut O, Fiebich BL, Bauer J, Janssen G, Schaefer M. 2006. Cognitive impairment in patients with chronic hepatitis treated with interferon alpha (IFNalpha): results from a prospective study. Eur Psychiatry 21(3):204–210 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25(4):402–408 [DOI] [PubMed] [Google Scholar]
- MacDonald NJ, Kuhl D, Maguire D, Naf D, Gallant P, Goswamy A, Hug H, Bueler H, Chaturvedi M, de la Fuente J, and others. 1990. Different pathways mediate virus inducibility of the human IFN-alpha 1 and IFN-beta genes. Cell 60(5):767–779 [DOI] [PubMed] [Google Scholar]
- Makino M, Kitano Y, Komiyama C, Hirohashi M, Kohno M, Moriyama M, Takasuna K. 2000. Human interferon-alpha induces immobility in the mouse forced swimming test: involvement of the opioid system. Brain Res 852(2):482–484 [DOI] [PubMed] [Google Scholar]
- Marijanovic Z, Ragimbeau J, van der Heyden J, Uze G, Pellegrini S. 2007. Comparable potency of IFNalpha2 and IFNbeta on immediate JAK/STAT activation but differential down-regulation of IFNAR2. Biochem J 407(1):141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza-Fernandez V, Andrew RD, Barajas-Lopez C. 2000. Interferon-alpha inhibits long-term potentiation and unmasks a long-term depression in the rat hippocampus. Brain Res 885(1):14–24 [DOI] [PubMed] [Google Scholar]
- Okanoue T, Sakamoto S, Itoh Y, Minami M, Yasui K, Sakamoto M, Nishioji K, Katagishi T, Nakagawa Y, Tada H, Sawa Y, Mizuno M, Kagawa K, Kashima K. 1996. Side effects of high-dose interferon therapy for chronic hepatitis C. J Hepatol 25(3):283–291 [DOI] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. 2007. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol 7(1):39–47 [DOI] [PubMed] [Google Scholar]
- Patti F, Morra VB, Amato MP, Trojano M, Bastianello S, Tola MR, Cottone S, Plant A, Picconi O, Group CS. 2013. Subcutaneous interferon beta-1a may protect against cognitive impairment in patients with relapsing-remitting multiple sclerosis: 5-year follow-up of the COGIMUS study. PLoS One 8(8):e74111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC, Uddin S, Domanski P, Colamonici OR. 1996. Differences in interferon alpha and beta signaling. Interferon beta selectively induces the interaction of the alpha and betaL subunits of the type I interferon receptor. J Biol Chem 271(39):23630–23633 [DOI] [PubMed] [Google Scholar]
- Quarantini LC, Miranda-Scippa A, Schinoni MI, Sampaio AS, Santos-Jesus R, Bressan RA, Tatsch F, de Oliveira I, Parana R. 2006. Effect of amantadine on depressive symptoms in chronic hepatitis C patients treated with pegylated interferon: a randomized, controlled pilot study. Clin Neuropharmacol 29(3):138–143 [DOI] [PubMed] [Google Scholar]
- Rho MB, Wesselingh S, Glass JD, McArthur JC, Choi S, Griffin J, Tyor WR. 1995. A potential role for interferon-alpha in the pathogenesis of HIV-associated dementia. Brain Behav Immun 9(4):366–377 [DOI] [PubMed] [Google Scholar]
- Rostasy K, Monti L, Yiannoutsos C, Wu J, Bell J, Hedreen J, Navia BA. 2000. NFkappaB activation, TNF-alpha expression, and apoptosis in the AIDS-Dementia-Complex. J Neurovirol 6(6):537–543 [DOI] [PubMed] [Google Scholar]
- Sas AR, Bimonte-Nelson H, Smothers CT, Woodward J, Tyor WR. 2009. Interferon-alpha causes neuronal dysfunction in encephalitis. J Neurosci 29(12):3948–3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sas AR, Bimonte-Nelson HA, Tyor WR. 2007. Cognitive dysfunction in HIV encephalitic SCID mice correlates with levels of Interferon-alpha in the brain. AIDS 21(16):2151–2159 [DOI] [PubMed] [Google Scholar]
- Schaefer M, Engelbrecht MA, Gut O, Fiebich BL, Bauer J, Schmidt F, Grunze H, Lieb K. 2002. Interferon alpha (IFNalpha) and psychiatric syndromes: a review. Prog Neuropsychopharmacol Biol Psychiatry 26(4):731–746 [DOI] [PubMed] [Google Scholar]
- Shiozawa S, Kuroki Y, Kim M, Hirohata S, Ogino T. 1992. Interferon-alpha in lupus psychosis. Arthritis Rheum 35(4):417–422 [DOI] [PubMed] [Google Scholar]
- Traugott U, Lebon P. 1988. Multiple sclerosis: involvement of interferons in lesion pathogenesis. Ann Neurol 24(2):243–251 [DOI] [PubMed] [Google Scholar]
- Tzartos JS, Khan G, Vossenkamper A, Cruz-Sadaba M, Lonardi S, Sefia E, Meager A, Elia A, Middeldorp JM, Clemens M, Farrell PJ, Giovannoni G, Meier UC. (2012). Association of innate immune activation with latent Epstein-Barr virus in active MS lesions. Neurology 78(1):15–23 [DOI] [PubMed] [Google Scholar]
- Valentine AD, Meyers CA, Kling MA, Richelson E, Hauser P. 1998. Mood and cognitive side effects of interferon-alpha therapy. Semin Oncol 25(1 Suppl 1):39–47 [PubMed] [Google Scholar]
- Wang YH, Bosy TZ, Yasuda RP, Grayson DR, Vicini S, Pizzorusso T, Wolfe BB. 1995. Characterization of NMDA receptor subunit-specific antibodies: distribution of NR2A and NR2B receptor subunits in rat brain and ontogenic profile in the cerebellum. J Neurochem 65(1):176–183 [DOI] [PubMed] [Google Scholar]