

Abstract
Background. Neoangiogenesis is crucial in plaque progression and instability. Previous data from our group showed that Nestin-positive intraplaque neovessels correlated with histological complications. The aim of the present work is to evaluate the relationship between neoangiogenesis, plaque morphology, and clinical instability of the plaque. Materials and Methods. Seventy-three patients (53 males and 20 females, mean age 71 years) were consecutively enrolled. Clinical data and 14 histological variables, including intraplaque hemorrhage and calcifications, were collected. Immunohistochemistry for CD34 and Nestin was performed. RT-PCR was performed to evaluate Nestin mRNA (including 5 healthy arteries as controls). Results. Diffusely calcified plaques (13/73) were found predominantly in females (P = 0.017), with a significantly lower incidence of symptoms (TIA/stroke (P = 0.019) than noncalcified plaques but with the same incidence of histological complications (P = 0.156)). Accordingly, calcified and noncalcified plaques showed similar mean densities of positivity for CD34 and Nestin. Nestin density, but not CD34, correlated with the occurrence of intraplaque hemorrhage. Conclusions. Plaques with massive calcifications show the same incidence of histological complications but without influencing symptomatology, especially in female patients, and regardless of the amount of neoangiogenesis. These results can be applied in a future presurgical identification of patients at major risk of developing symptoms.
1. Introduction
The vulnerable atheromatous plaques have been originally described as characterized by a large lipid core, a thin fibrous cap, a rich infiltrate of macrophages, and little smooth muscle cell component [1]. Neoangiogenesis began to stand out as one of the most important pathological processes involved in the plaque progression only in the recent years [2], when neovessel formation was related to an increased plaque vulnerability and to the onset of clinical symptoms [3, 4]. In particular, the intraplaque hemorrhage and the incidence of symptomatic plaques were directly related to the neovessel density, simply measured by means of immunohistochemistry (IHC) for CD34 [3]. If the density of neovessels is likely to be directly related with plaque growth and progression [5], the morphology of the neoangiogenetic structures plays a key role in the onset of the plaque instability. In fact plaque neovessels are reported to lack extracellular junctions [6]; moreover, symptomatic plaques show larger and more irregular neoangiogenetic structures compared to the neovessels of asymptomatic plaques [7]. Recently we described the IHC and immunofluorescence positivity for Nestin and WT1 in vasa vasorum from healthy arteries, hypothesizing that they might represent the starting point of the neoangiogenesis during atherosclerosis [8]. Afterwards we actually confirmed the expression of Nestin and WT1 in diseased arteries but together with the observation that nearly 36% of the neovessels showed positivity for Nestin and negativity for WT1 (Nestin+/WT1−). As a matter of fact, hemorrhagic plaques showed significantly more Nestin+/WT1− neoangiogenesis than uncomplicated plaques at both IHC and RT-PCR [9].
The aims of the present study were (i) to evaluate the relationship between the intraplaque neoangiogenesis (as quantified by CD34 and Nestin IHC) and the main histopathological plaque characteristics (especially complications and calcifications) and (ii) to evaluate the relationship between the immunohistochemical and histopathological characteristics and the clinical plaque instability.
2. Materials and Methods
2.1. Patients and Clinical Data
This study was carried out in conformity to the ethical guidelines of the 1975 Declaration of Helsinki (and following modifications); informed consent was obtained from all patients before surgery. We evaluated all consecutive cases of carotid endarterectomy specimens that came to our Surgery Unit in 2 years, from January 2011. According to the European Society for Vascular Surgery (ESVS) and the Society of Vascular Surgeons (SVS) recommendations [11, 12], the patients were submitted to carotid endarterectomy (CEA) for either symptomatic carotid plaques ≥50% or asymptomatic carotid stenosis ≥70%. For each patient the following clinical data were collected: occurrence of symptoms related to the carotid disease, that is, transient ischemic attack (TIA) and stroke, association with chronic ischemic cardiopathy, chronic obstructive bronchopneumonia and/or chronic renal failure, smoke, diabetes, dyslipidemia, and therapy with acetylsalicylic acid or statins.
2.2. Histopathological Analysis
Endarterectomy specimens were sent to our Pathology Unit, fixed in formalin, routinely processed, and paraffin embedded. Two μm thick slices were cut from the paraffin blocks for haematoxylin-eosin and trichrome stains. For each case, the following histopathological features were collected as single variables, as previously reported [9]: the occurrence of intraplaque complications (hemorrhage, thrombosis, and/or surface defects) which put the plaque into the AHA type VI [10], maximum and minimum size of the fibrous cap, extension of the lipid core, and extension of the inflammatory infiltrate. For the purposes of the study, the amount of calcifications was assessed and graded from 0 to 4+ in relation to their extension along the vessel circumference; the intraplaque calcification was therefore simplified in low-grade (if 0 to 2+) and high-grade (if 3+ or 4+).
2.3. Immunohistochemistry
The monoclonal antibodies used in this study are listed in Table 1. Immunohistochemistry (IHC) for Nestin was performed manually, as previously described [8, 9]. IHC for CD34 was performed automatically, by means of Benchmark XT (Ventana Medical System), using the XT ultraView DAB v3 program.
Table 1.
Technical characteristics of the antibodies used for immunohistochemistry.
| Antibody | Clone | Manufacturer |
|---|---|---|
| Nestin | 10C2 (mouse IgG) | Millipore |
| CD34 | QBEnd/10 (mouse IgG) | Roche Ventana |
The microvessel “density of positivity” [9] for Nestin was evaluated after the identification of specific Regions of Interest (ROI). ROI were defined as areas with CD34-positive neoangiogenesis, and at 20x magnification each ROI was divided into 1 mm2 fields using an Olympus ocular micrometer (1 length unit = 5 μm, which means that an area of 100 × 100 units is equal to 0.25 mm2). Firstly, CD34 and Nestin microvessel positivity were counted separately; the Nestin-positive vessels were counted in the same ROI where CD34 was evaluated. Afterwards we calculated the “density of positivity” for CD34 and Nestin by dividing the sum of all the positive vascular structures observed by the number of the counted fields in each section. Finally we calculated the ratio between the densities of positivity of Nestin and CD34 in each case: the Nestin/CD34 ratio represents how many CD34-positive neovessels concomitantly express Nestin in the intraplaque neoangiogenesis.
2.4. RT-PCR
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) for the product of the Nestin gene was carried out on 5 type V and on 5 type VI plaques, in order to evaluate the different expression in complicated and uncomplicated carotid plaques. A pool composed of 5 healthy carotid arteries was used as controls. Healthy carotids were kindly provided by the Cardiovascular Tissue Bank of S. Orsola-Malpighi University Hospital of Bologna from 5 multiorgan donors (3 males and 2 females, mean age 33.8 ± 13.2 years, range 18–53 years), without known comorbidities.
Tissues were homogenized with an Ultraturax and incubated with 800 ul of Trizol reagent (TRIzol Life Technologies, Carlsbad, CA, USA) for 5 min at RT. RNA extraction with Trizol was performed following manufacturer instructions. RNA quality and concentration were measured by using an ND-1000 spectrophotometer (NanoDrop, Thermo Fisher Scientific, Wilmington, DE, USA). Reverse transcription assay was performed using 2.0 μg of starting total RNA quantity per 25 μL of mix, following the manufacturer's protocol (High capacity cDNA Archive kit, Life Technologies). The cDNA was stored at −20°C until RT-PCR was performed. RT-PCR was carried out following MasterMix TaqMan Protocol (TaqMan Univ PCR MasterMix, Life Technologies). Four μL of neat cDNA was amplified using specific probes for Nestin (NC_000001.10) and GUSB (NM_000181.3) in the RT-PCR mix (TaqMan Gene Expression Assay, Life Technologies, respective ID assay: Hs04187831_g1, Hs00939627_m1). Reactions were run on ABI PRISM 7900HT Sequence Detection System (Life Technologies). Cycling conditions were as follows: 10 min at 95°C, 50 cycles at 95°C for 15 s, and 60°C for 60 sec. Each assay was carried out in triplicate and the transcription level was normalized using GUSB as a reference gene.
2.5. Statistical Analysis
All statistical analyses were carried out with SPSS software for Windows, version 20. All continuous variables are expressed as means, standard deviations, and ranges; all categorical variables (both nominal and ordinal) are expressed as number of cases and percentages. The Spearman test, the chi-square test, the Mann-Whitney U test, and the Kruskal-Wallis test were used when appropriate. The mRNA expression values for atheromatous type V and type VI plaques are presented as fold expression in relation to healthy arteries; the actual values were calculated using the 2−ΔΔCT equation, where ΔΔCT = [CT Target − CT GUSB] (atheromatous sample V or VI) − [CT Target − CT GUSB] (healthy sample). RT-PCR data were analyzed assuming the null hypothesis that the CT differences between target and reference genes will be the same in type V tissue versus type VI tissue. If the null hypothesis is not rejected, then the ΔΔCT would not be significantly different from 0. Analyses of differences between the three groups (healthy, class V, and class VI plaques) were performed with one-way ANOVA test, followed by Tukey's test. All the P values are derived from testing the null hypothesis that ΔΔCT are equal to 0 (at P = 0.05). SEM, SD, and the confidence interval (CI) of 2−(ΔΔCT) are all derived from the SEM, SD, and CI of ΔΔCT as explained by Yuan et al. [13].
3. Results
3.1. Patients and Histopathological Analysis
Seventy-three patients were finally enrolled, 53 (72.6%) males and 20 (27.4%) females, with a mean age at the time of endarterectomy of 70.8 ± 8.7 years (range 42–86 years). The clinical characteristics of the patients, including ongoing therapy, are summarized in Table 2. Notably, 29 (39.7%) plaques were symptomatic, since 11 (15.1%) patients had a stroke as clinical presentation and 18 (24.6%) had a transient ischemic attack (TIA).
Table 2.
Summary of the baseline clinical characteristics of the 73 patients.
| Number of patients | Percentage | |
|---|---|---|
| Stroke | 11 | 15.1% |
| Transient ischemic attack | 18 | 24.6% |
| Hypertension | 65 | 89.0% |
| Dyslipidemia | 51 | 69.9% |
| Diabetes | 20 | 27.4% |
| Smoke | 6 | 8.5% |
| Chronic ischemic cardiopathy | 25 | 34.2% |
| Chronic obstructive pulmonary Disease | 4 | 5.5% |
| Chronic renal failure | 4 | 5.5% |
| Acetylsalicylic acid use | 64 | 87.7% |
| Statins use | 42 | 57.5% |
At histopathological analysis, mean maximum cap size was 1132.8 ± 485.6 μm (range 120–2500 μm) and mean minimum cap size was 284.3 ± 199.7 μm (range 40–1125 μm); the lipid core was not evident in 8 (11.0%) cases, 1/4 in 16 (21.9%), 2/4 in 18 (24.7%), 3/4 in 23 (31.4%), and 4/4 in 8 (11.0%); the inflammatory infiltrate was mild/focal or absent in 14 (19.2%) cases, moderate in 20 (27.4%) cases, and severe/diffuse in 39 (53.4%) cases. Fifty-two (71.2%) plaques were classified as AHA type VI [10]: the most common complication observed was intraplaque hemorrhage, present in 41 cases, followed by endothelial erosion in 22 cases and thrombosis in 4 cases. Eight (11.0%) further noncomplicated plaques were classified as type VII due to the prevalently calcified core in 8 (11.0%) cases, while the remaining 13 (17.8%) cases were classified as type VIII (prevalently fibrotic core) or type V (fibroatheroma) [10].
Intraplaque calcifications were graded 0 in 11 (15.1%) cases, 1+ in 21 (28.8%), 2+ in 20 (27.4%), 3+ in 16 (21.9%), and 4+ in 5 (6.8%). According to this semiquantitative assessment of the calcification extent, low-grade calcifications (up to 2+) were recorded in 52 (71.2%) plaques, and high-grade calcification (3+ and 4+) was recorded in 21 (28.8%) plaques (Figure 1).
Figure 1.
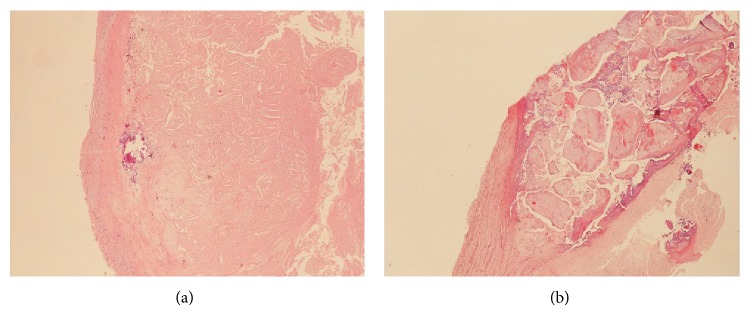
Details of two cases of carotid plaque with low-grade calcification (a) and high-grade calcification (b), respectively. Haematoxylin-eosin stain, magnification 10x.
Finally, according to the occurrence of intraplaque complications and/or calcifications, our cases were sorted in noncalcified complicated plaques (type VI-nc, N = 39), calcified and complicated plaques (type VI-c N = 13), calcified noncomplicated plaques (type VII, N = 8), and noncalcified noncomplicated plaques (types V–VIII, N = 13). No correlations were found between plaque morphological criteria used in the present study (i.e., calcifications and histological complications) and the occurrence of chronic ischemic cardiopathy, chronic obstructive bronchopneumonia, chronic renal failure, smoke, diabetes, hypertension, dyslipidemia, or therapy with acetylsalicylic acid or statins (data not shown, chi-square test).
3.2. Nestin-Positive Neoangiogenesis
Two cases had no appreciable intraplaque neoangiogenesis after IHC for CD34. In the remaining 71 cases, the mean density of positivity for CD34 was 10.1 ± 3.9 structures/field (range 3.5–22.1/field). The mean density of positivity for Nestin, evaluated in the same ROI, was 6.8 ± 3.7 structures/field (range 1.4–18.5/field). The mean Nestin/CD34 ratio was 0.7 ± 0.2, which means that 70% of the CD34-positive neovessels examined coexpressed Nestin. This result is in line with what is previously described [9].
The total amount of neoangiogenesis, expressed as CD34-positive vessels, was not significantly different between type VI plaques and uncomplicated plaques (P = 0.111, Mann-Whitney U test), while the density of Nestin-positive neovessels was significantly higher in type VI (P = 0.015, Table 3).
Table 3.
Densities of positivity of intraplaque neovessels for CD34 and Nestin in our series of plaques, sorted by calcification and by the occurrence of complications.
| Noncalcified plaques | Calcified plaques | Complicated plaques | Uncomplicated plaques | |
|---|---|---|---|---|
| CD34 density | 11.19 ± 3.84 | 7.52 ± 3.09 | 10.68 ± 3.79 | 8.99 ± 4.13 |
| Mann-Whitney | P < 0.001 | P = 0.111 | ||
|
| ||||
| Nestin density | 7.69 ± 3.77 | 4.59 ± 2.31 | 7.45 ± 3.71 | 5.31 ± 3.09 |
| Mann-Whitney | P < 0.001 | P = 0.015 | ||
The calcified plaques (including complicated and uncomplicated) showed overall less neoangiogenesis, measured with both CD34 and Nestin, than noncalcified plaques (P < 0.001, Table 3; Figure 2).
Figure 2.

Immunohistochemical staining for CD34 (a), (c), and (e) and Nestin (b), (d), and (f) in an uncomplicated noncalcified plaque (a) and (b), a calcified plaque (c) and (d), and a complicated noncalcified plaque (e) and (f). Neoangiogenesis in uncomplicated plaques is generally Nestin-negative. The overall neoangiogenesis in calcified plaques (complicated or not) is generally lower (both CD34 and Nestin) than in complicated plaques. Magnification 20x.
At RT-PCR, the total mean extracted mRNA from healthy tissue and type V and type VI plaques was 8764 ng, 5069.4 ng, and 2172 ng, respectively. The mean CT values of endogenous control GUSB were 36.28 ± 0.21 in healthy tissue, 31.36 ± 0.32 in type V plaques, and 34.20 ± 0.22 in type VI plaques. Mean CT for tested gene Nestin were 33.04 ± 0.06 in healthy tissue, 32.37 ± 0.12 in type V plaques, and 34.21 ± 0.30 in type VI plaques. ΔΔCT Nestin was significantly different from 0 (P = 0.0001); thus the null hypothesis was rejected, which indicated a change in Nestin gene expression among healthy, type V and type VI plaques. In type V and type VI plaques, the mean ΔΔCT Nestin was, respectively, 4.25 and 3.25; this corresponds to 2−(ΔΔCT) of 0.05 for Nestin gene expression in type V plaques and 0.11 in type VI plaques. The type VI plaques showed a 2-fold expression increase for Nestin gene compared to type V plaques, as a confirmation of the IHC results (Figure 3).
Figure 3.
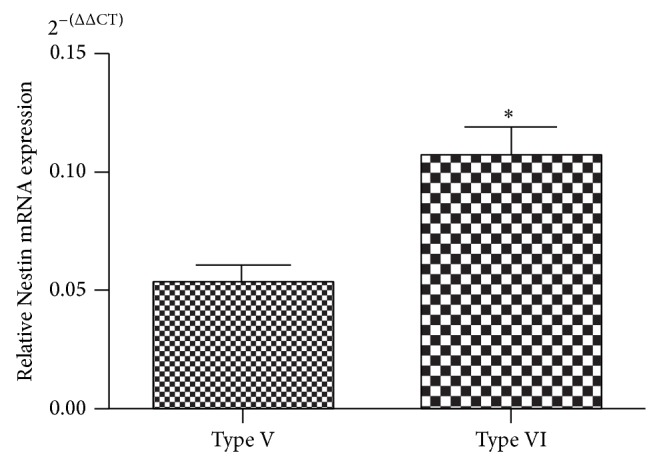
Difference in Nestin mRNA expression between type V (uncomplicated) and type VI (complicated) plaques (* P < 0.001).
These data confirm that Nestin-positive neoangiogenesis, studied both on the protein (IHC) and the mRNA level (RT-PCR), plays a key role in the development of intraplaque complications and that plaques with massive calcifications generally have a minor density of neoangiogenesis compared to other plaques.
3.3. Intraplaque Calcifications and Clinical Stability
Despite the differences in neoangiogenesis, the incidence of histological complications did not differ significantly between calcified and noncalcified plaques (P = 0.167, chi-square test; Figure 4).
Figure 4.
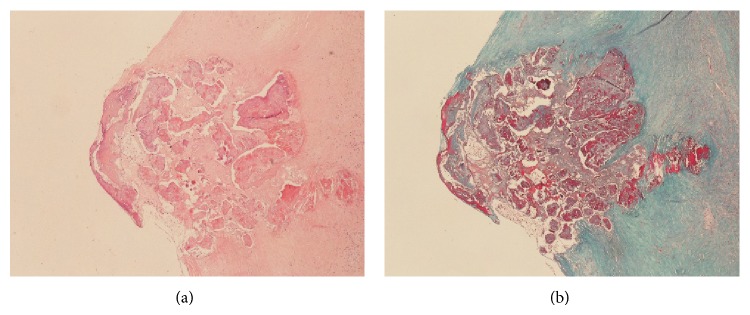
An example of calcified plaque with histopathological complications (i.e., hemorrhage) with haematoxylin-eosin stain (a). The trichrome stain highlights the hemorrhagic foci (in red (b)). Magnification 10x.
As for the clinical presentation, unsurprisingly, 25 out of 29 (86.2%) symptomatic patients (i.e., with stroke or TIA) had a type VI plaque, versus 27 out of 44 (61.4%) asymptomatic patients (P = 0.026, chi-square test). Interestingly, among the 21 patients with calcified plaques, only 4 (19.0%) had symptoms at the onset, regardless of the occurrence of histological complications; conversely 25 out of 52 (48.1%) patients with noncalcified plaques were symptomatic (P = 0.019, chi-square test).
The patients' gender was correlated with the plaque morphology and instability as well; first of all only 3 (15.0%) of the 20 female patients in our study were symptomatic versus 26 (49.1%) out of 53 males (P = 0.007, chi-square test). All three symptomatic female patients had a type VI-nc atheromatous lesion. Notably, a higher incidence of calcified plaques was observed in female patients (P = 0.017): 10 (50.0%) females had calcified plaques (5 type VII and 5 type VI-c) versus only 11 (20.8%) males (3 type VII and 8 type VI-c).
4. Discussion
The severity and extent of calcification reflect the atherosclerotic plaque burden and strongly predict cardiovascular morbidity and mortality [14, 15]; in a relatively recent study, no patients were found to have calcifications confined only to the coronary or carotid beds [16]. The extent of calcification is associated with a worse prognosis, albeit the real impact of calcifications within a specific vascular pathological district remains unclear [17]. For example, in the coronary vessels small calcium depositions increase the probability of atherosclerotic plaque rupture, especially on their edges, while massive calcification seems to be associated with a decreased risk [17, 18]. Anyway, vascular calcification is considered a worsening factor, probably due to its coexistence with the general risk factors; a study by Iribarren et al. [19] found that aortic arch calcification was associated with coronary heart disease risk both in men and in women. Thus aortic arch calcification may reflect the general burden of disease or be a marker of more aggressive disease. At any chance, the clinical impact of a plaque in which both calcification and histological complications coexist is far from being clarified.
Our aims were to evaluate the relationship between the intraplaque neoangiogenesis, the main histopathological characteristics (histological complications and calcifications), and the clinical plaque instability. For these purposes, neoangiogenesis was evaluated and semiquantified by CD34 and Nestin IHC, followed by RT-PCR. The majority of tissue obtained from endarterectomy was used for the routine histological diagnosis of surgical specimens. The remaining tissue had to be fully processed for RT-PCR and Western blot analysis was not included in our protocol. However, in our experience [8, 9] we observed that Nestin staining in IHC is very reliable, allowing us to evaluate its cell location and its expression at a protein level as well, without the need of the more sensible immunoblot techniques. The correlation between IHC and RT-PCR confirmed that there is a direct relationship between Nestin protein and mRNA.
According to our data calcified plaques show less inflammatory infiltrate, a smaller lipid core, and less neoangiogenesis than other plaques, but with the same incidence of hemorrhage, thrombosis, and surface defects, which define the plaque as histologically complicated [10]. Yet, interestingly, in these calcified plaques complications are not correlated with clinical plaque instability (evaluated as symptomatology); indeed, the incidence of TIA/stroke in patients with calcified plaques was sensibly lower than patients with noncalcified plaques, despite the same incidence of intraplaque complications. This is noteworthy, since at least in the carotid district the presence of calcifications seems to imply a sort of clinical “protection” to complications, making the histological complications play second fiddle. For these reasons, in our opinion, these plaques can be classified among the type VII plaques, instead of type VI, at least on clinical grounds. Alternatively, they can be classified as type VI, but the extension of the calcifications should be stated in the pathological report, to highlight their protective nature.
The reason why in the massively calcified plaques the neoangiogenesis and the histological complications do not affect symptomatology remains unclear, but it is possible that the hemorrhages and erosions found in these plaques might have a different pathogenesis. For example, it is possible that they can be due directly by the mechanical stresses of the calcified mass and not by immature neoangiogenesis and endothelial damage. Another possible explanation is that the “calcified type VI” plaques can represent an early stage of type VII plaques, in which the regressive process is more recent, and the complications have not disappeared yet (Figure 5).
Figure 5.

Flowchart illustrating the hypothetical plaque progression in relation to the Nestin-positive neoangiogenesis and calcification features. According to AHA classification [10], type V plaque is the uncomplicated fibroatheroma, type VI is the complicated plaque, type VII is the calcified plaque, and type VIII is the fibrotic plaque.
The last result that emerged from our data is that 50% of the female patients had calcified plaques, showing a significantly lower incidence of symptoms and type VI plaques than the male patients. Ten years ago, Allison et al. have found 53% and 30% prevalence of “zero calcification” in female and male patients, respectively, before age of 50; after that age the prevalence of a diffuse vessel calcification increases, in a linear fashion in males and exponentially in females [16]. Actually, female sex hormones play an important role in bone tissue metabolism, increasing bone density and inhibiting osteoclast activity [20–22]. Nevertheless, it should be kept in mind that most women in our series were postmenopausal and their age at the moment of surgery did not differ from males (70.2 ± 9.5 versus 71.0 ± 8.5 years), so the question whether the postmenopausal hormone therapy might play a role in the pathophysiology of the atherosclerosis is still open.
Study Limitations. A limitation of our study is represented by heterogeneity in the sample size of each plaque group (e.g., 8 cases of type VII plaques were available). However, this is a monocentric perspective study, and our series reflects the incidence of the different plaque types in the general population. Moreover, the patients submitted to surgery generally have advanced plaques, and complicated type VI plaques are the most represented. Furthermore, there is a discrepancy between the age of the patients with plaques and the controls, due to obvious differences in the populations of multiorgan donors and atheromatous patients.
5. Conclusions
In conclusion, this study confirms that the Nestin+/WT1− phenotype characterizes the plaques with morphological features of instability, regardless of the actual amount of the neoangiogenesis (expressed as CD34-positive vessels). The plaques with massive calcifications show the same incidence of histological complications but with a lower incidence of neurological symptoms. Female patients show a much higher incidence of noncomplicated or calcified plaques, receiving de facto a sort of protection compared to male patients.
A possible indication emerging from these findings could be a comparison between the plaque dynamic imaging and the histological assessment of calcification, to evaluate the possibility of a presurgical risk stratification of patients, based on their sex, risk factors, and intraplaque calcification. The presurgical identification of those patients at major risk of developing stroke or brain lesions is likely to make the priority for endarterectomy more rational.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Kullo I. J., Edwards W. D., Schwartz R. S. Vulnerable plaque: pathobiology and clinical implications. Annals of Internal Medicine. 1998;129(12):1050–1060. doi: 10.7326/0003-4819-129-12-199812150-00010. [DOI] [PubMed] [Google Scholar]
- 2.Hermus L., Tielliu I. F. J., de Wallis Vries B. M., van den Dungen J. J. A. M., Zeebregts C. J. Imaging the vulnerable carotid artery plaque. Acta Chirurgica Belgica. 2010;110(2):159–164. doi: 10.1080/00015458.2010.11680590. [DOI] [PubMed] [Google Scholar]
- 3.Mofidi R., Crotty T. B., McCarthy P., Sheehan S. J., Mehigan D., Keaveny T. V. Association between plaque instability, angiogenesis and symptomatic carotid occlusive disease. British Journal of Surgery. 2001;88(7):945–950. doi: 10.1046/j.0007-1323.2001.01823.x. [DOI] [PubMed] [Google Scholar]
- 4.Faggioli G. L., Pini R., Mauro R., et al. Identification of carotid ‘Vulnerable Plaque’ by contrast-enhanced ultrasonography: correlation with plaque histology, symptoms and cerebral computed tomography. European Journal of Vascular and Endovascular Surgery. 2011;41(2):238–248. doi: 10.1016/j.ejvs.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Moreno P. R., Purushothaman K.-R., Sirol M., Levy A. P., Fuster V. Neovascularization in human atherosclerosis. Circulation. 2006;113(18):2245–2252. doi: 10.1161/CIRCULATIONAHA.105.578955. [DOI] [PubMed] [Google Scholar]
- 6.Sluimer J. C., Kolodgie F. D., Bijnens A. P. J. J., et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. Journal of the American College of Cardiology. 2009;53(17):1517–1527. doi: 10.1016/j.jacc.2008.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarthy M. J., Loftus I. M., Thompson M. M., et al. Angiogenesis and the atherosclerotic carotid plaque: an association between symptomatology and plaque morphology. Journal of Vascular Surgery. 1999;30(2):261–268. doi: 10.1016/s0741-5214(99)70136-9. [DOI] [PubMed] [Google Scholar]
- 8.Vasuri F., Fittipaldi S., Buzzi M., et al. Nestin and WT1 expression in small-sized vasa vasorum from human normal arteries. Histology & Histopathology. 2012;27(9):1195–1202. doi: 10.14670/HH-27.1195. [DOI] [PubMed] [Google Scholar]
- 9.Fittipaldi S., Vasuri F., Degiovanni A., et al. Nestin and WT1 expression in atheromatous plaque neovessels: association with vulnerability. Histology and Histopathology. 2014;29:1565–1573. doi: 10.14670/HH-29.1565. [DOI] [PubMed] [Google Scholar]
- 10.Stary H. C. Natural history and histological classification of atherosclerotic lesions an update. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(5):1177–1178. doi: 10.1161/01.ATV.20.5.1177. [DOI] [PubMed] [Google Scholar]
- 11.Hobson R. W., II, Mackey W. C., Ascher E., et al. Management of atherosclerotic carotid artery disease: clinical practice guidelines of the Society for Vascular Surgery. Journal of Vascular Surgery. 2008;48(2):480–486. doi: 10.1016/j.jvs.2008.05.036. [DOI] [PubMed] [Google Scholar]
- 12.Liapis C. D., Bell P. R., Mikhailidis D., et al. ESVS guidelines. Invasive treatment for carotid stenosis: indications, techniques. European Journal of Vascular and Endovascular Surgery. 2009;37(4, supplement):1–19. doi: 10.1016/j.ejvs.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Yuan J. S., Reed A., Chen F., Stewart C. N., Jr. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7, article 85 doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sangiorgi G., Rumberger J. A., Severson A., et al. Arterial calcification and not lumen stenosis is highly correlated with atherosclerotic plaque burden in humans: a histologic study of 723 coronary artery segments using nondecalcifying methodology. Journal of the American College of Cardiology. 1998;31(1):126–133. doi: 10.1016/s0735-1097(97)00443-9. [DOI] [PubMed] [Google Scholar]
- 15.Vliegenthart R., Oudkerk M., Hofman A., et al. Coronary calcification improves cardiovascular risk prediction in the elderly. Circulation. 2005;112(4):572–577. doi: 10.1161/CIRCULATIONAHA.104.488916. [DOI] [PubMed] [Google Scholar]
- 16.Allison M. A., Criqui M. H., Wright C. M. Patterns and risk factors for systemic calcified atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(2):331–336. doi: 10.1161/01.atv.0000110786.02097.0c. [DOI] [PubMed] [Google Scholar]
- 17.Huang H., Virmani R., Younis H., Burke A. P., Kamm R. D., Lee R. T. The impact of calcification on the biomechanical stability of atherosclerotic plaques. Circulation. 2001;103(8):1051–1056. doi: 10.1161/01.CIR.103.8.1051. [DOI] [PubMed] [Google Scholar]
- 18.Vengrenyuk Y., Carlier S., Xanthos S., et al. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(40):14678–14683. doi: 10.1073/pnas.0606310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iribarren C., Sidney S., Sternfeld B., Browner W. S. Calcification of the aortic arch: risk factors and association with coronary heart disease, stroke, and peripheral vascular disease. The Journal of the American Medical Association. 2000;283(21):2810–2815. doi: 10.1001/jama.283.21.2810. [DOI] [PubMed] [Google Scholar]
- 20.Mackey R. H., Kuller L. H., Sutton-Tyrrell K., Evans R. W., Holubkov R., Matthews K. A. Hormone therapy, lipoprotein subclasses, and coronary calcification: the healthy women study. Archives of Internal Medicine. 2005;165(5):510–515. doi: 10.1001/archinte.165.5.510. [DOI] [PubMed] [Google Scholar]
- 21.Manson J. E., Allison M. A., Rossouw J. E., et al. Estrogen therapy and coronary-artery calcification. The New England Journal of Medicine. 2007;356(25):2591–2602. doi: 10.1056/NEJMoa071513. [DOI] [PubMed] [Google Scholar]
- 22.Jeon G.-H., Kim S. H., Yun S.-C., Chae H. D., Kim C.-H., Kang B. M. Association between serum estradiol level and coronary artery calcification in postmenopausal women. Menopause. 2010;17(5):902–907. doi: 10.1097/gme.0b013e3181d76768. [DOI] [PubMed] [Google Scholar]