

Abstract
Endovascular drug-eluting stents have changed the practice of medicine, and yet it is unclear how they so dramatically reduce restenosis and how to distinguish between the different formulations available. Biological drug potency is not the sole determinant of biological effect. Physicochemical drug properties also play important roles. Historically, two classes of therapeutic compounds emerged: hydrophobic drugs, which are retained within tissue and have dramatic effects, and hydrophilic drugs, which are rapidly cleared and ineffective. Researchers are now questioning whether individual properties of different drugs beyond lipid avidity can further distinguish arterial transport and distribution. In bovine internal carotid segments, tissue-loading profiles for hydrophobic paclitaxel and rapamycin are indistinguishable, reaching load steady state after 2 days. Hydrophilic dextran reaches equilibrium in several hours at levels no higher than surrounding solution concentrations. Both paclitaxel and rapamycin bind to the artery at 30–40 times bulk concentration. Competitive binding assays confirm binding to specific tissue elements. Most importantly, transmural drug distribution profiles are markedly different for the two compounds, reflecting, perhaps, different modes of binding. Rapamycin, which binds specifically to FKBP12 binding protein, distributes evenly through the artery, whereas paclitaxel, which binds specifically to microtubules, remains primarily in the subintimal space. The data demonstrate that binding of rapamycin and paclitaxel to specific intracellular proteins plays an essential role in determining arterial transport and distribution and in distinguishing one compound from another. These results offer further insight into the mechanism of local drug delivery and the specific use of existing drug-eluting stent formulations.
It now appears that the success of drug-eluting stents is predicated to as great a degree upon the extent of drug deposition and distribution through the arterial wall as virtually any other factor (1–5). The biological effects of a candidate drug are essential, but, ultimately, tissue residence time will determine effect and toxicity (6, 7). Fueled by clinical relevance (8–11), a number of studies have been carried out to detect, model, and predict the distribution of drugs within arterial segments beneath, adjacent to, and distant from stents (12–15). Drugs that are retained within the blood vessel are far more effective than those that are not. Heparin is a perfect example of a compound whose ubiquitous biological potential is lost by virtue of its physicochemical properties. Heparin regulates virtually every aspect of the vascular response to injury (16). Yet heparin is so soluble and diffusible that it simply cannot stay in the artery for more than a few minutes after release and has no effect on intimal hyperplasia when eluted from a stent (4, 17, 18).
Paclitaxel, in contradistinction, is a far smaller compound, with fewer effects specific to vascular biology, but paclitaxel is so hydrophobic and insoluble that it binds tenaciously to tissue protein elements and remains beneath stent struts long after release (13). The clinical efficacy of paclitaxel at reducing restenosis rates following elution from stents appears incontrovertible (8, 11). In addition to its hydrophobic, nonspecific binding behavior, paclitaxel also binds to its protein target, polymerized microtubules (19), with nanomolar specificity. Analogously, rapamycin, also successful after local vascular delivery (10), acts on a specific target through a series of events that requires binding to the specific binding proteins FKBP12 (FK506 binding protein 12) and FRAP (FKBP12-rapamycin-associated protein) (20, 21). Thus, we hypothesized that the tissue-specific binding proteins can significantly influence arterial-wall drug distribution and local pharmacological effects for compounds such as paclitaxel and rapamycin above and beyond the influence of hydrophobic, nonspecific interactions alone. The distribution of tissue-binding proteins is not uniform in space or time. For example, FKBP12 is found most abundantly in vascular smooth muscle cells at a concentration of ≈10-5 M (22). FKBP12 is up-regulated in neuronal systems with injury (23) and likely after arterial injury as well. Microtubules have a similar cellular concentration of ≈10-5 M (24). The nonuniform distribution of paclitaxel previously observed in the arterial wall may reflect an inhomogeneous distribution (13) of polymerized microtubules or carrier proteins (25).
Although regulated at a coarse level by transport forces and lipid avidity, the distribution of paclitaxel and rapamycin is also regulated at a fine level by the distribution and availability of their protein targets, a level of control not present for drugs such as heparin that are rapidly cleared from arterial tissue. With a more complete understanding of the role of specific binding in arterial drug distribution, stent design, drug composition, and release formulation can be better optimized.
Materials and Methods
Arterial Loading and Elution. We defined the tissue pharmacokinetic profiles of dextran (10,000 Da), paclitaxel (853.9 Da), and rapamycin (914.2 Da) in calf carotid arterial wall segments. Calf internal carotid arteries were harvested and transported in PBS with physiological calcium and magnesium [PBS++/0.01 mol/liter CaCl2/0.01 mol/liter MgCl2 (Sigma)] at 4°C. Arteries were cleaned of excess fascia, opened longitudinally, cut into segments (400–600 mg), and placed in centrifuge tubes with 1.0 ml of [3H]dextran (10-6 M), [3H]paclitaxel (10-6 M), or [14C]rapamycin (10-6 M). All bulk solutions were made fresh immediately before experimentation, and the same initial bulk concentrations were used for both loading and elution experiments.
For loading experiments, segments were removed in triplicate at indicated time points, briefly washed in PBS++, and blot-dried before being dissolved in Solvable (Packard-Canberra). Liquid scintillation mixture (6 ml) was added to dissolved samples before counting with liquid scintillation spectroscopy (2500 TR Liquid Scintillation Analyzer, Packard-Canberra). For elution experiments, segments were allowed to equilibrate for 60 h and were then placed in 50 ml of PBS++ for the indicated time periods before being processed in triplicate as indicated with the loading experiments.
Measurements of Transmural and Planar Diffusion. Planar and transmural diffusivities were measured by using diffusion cells. Planar diffusivities (diffusion in the plane of the elastin sheaths) were measured by mounting opened arterial specimen between two glass slides and contacting the specimen's edge to a drug bath (10-6 M). After 25 min (a time determined to provide an acceptable signal-to-noise ratio while satisfying t << tequilibrium), specimens were processed for liquid scintillation counting. Planar diffusivities were measured in the longitudinal and circumferential directions. For transmural diffusivity, cleaned arteries were opened longitudinally and clamped in a standard Franz diffusion cell consisting of a lower sink compartment containing PBS++, separated from a drug-containing upper compartment by an artery lying en face. The artery was thus exposed only to the drug (10-6 M) on the luminal side and only to the buffer on the other. After 25 min of exposure to the drug, samples were processed for liquid scintillation counting.
The arterial wall is a highly heterogeneous structure composed of different tissue layers that impose individual effective diffusivities. Yet, there is a regularity from the alternating cylindrical bands of connective tissue and smooth muscle cells that permits use of a lumped effective diffusivity parameter to characterize bulk drug-transport properties in the arterial wall (3). Lumped effective diffusivities can be calculated from the measured drug mass M in tissue by using the early time solution to the diffusion equation
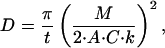 |
[1] |
where t is the time, A is the artery area exposed to drug, k is the binding capacity coefficient, and C is the drug source concentration (3).
Drug Tissue Binding Capacity (TBC) and Distribution. We defined the bulk differential capacities of the arterial wall for dextran, paclitaxel, and rapamycin at equilibrium. Arterial segments were weighed before being placed in drug bath solutions. Segments were allowed to equilibrate for 60 h and were then processed for liquid scintillation counting. The drug concentration of each tissue sample was normalized by tissue mass and then by drug concentration in the bulk fluid during equilibrium incubation to determine the binding capacity. As indicated, tissue was incubated with (i) labeled drug alone to determine TBC; (ii)drugand supersaturated levels of the same unlabeled drug to demonstrate that specific and nonspecific tissue-binding sites can be displaced; and (iii) supersaturated levels of the other nonlabeled hydrophobic drug (i.e., labeled rapamycin was mixed with cold paclitaxel, and vice versa) to displace only nonspecific binding.
Equilibrated transmural drug distribution in the artery was measured through en face cryosectioning. Arterial segments were incubated in the drug bath for 60 h, and then laid flat and snap-frozen in a plastic encasement with Tissue-Tek OCT compound (Sakura Finetechnical, Tokyo). Samples were stored in a -80°C freezer until they were sectioned parallel to the intima with a refrigerated microtome, the Cryotome SME (Shandon, Pittsburgh) (12, 26, 27). Upon sectioning, the segment length and width were measured with a caliper. Sections 0.040 mm thick were cut parallel to the intima, and the drug content of each sample was determined by liquid scintillation spectroscopy. Tissue drug concentration (cT) at each transmural location was calculated as the mass of drug normalized by the measured tissue area and slice thickness. Tissue drug concentration was then normalized by the bulk fluid drug concentration during equilibrium incubation (cbulk) to determine the binding capacity (k) at each transmural location (x).
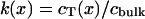 |
[2] |
Results
Tissue-Loading Kinetics. Arterial samples were incubated in [3H]dextran (10-6 M), [3H]paclitaxel (10-6 M), or [14C]rapamycin (10-6 M) and harvested in triplicate over a period of 72 h. Loading data were normalized to an average peak value at 72 h (Fig. 1). Whereas the hydrophilic dextran reached 80% of equilibrium value within several hours, paclitaxel and rapamycin took nearly 1 day to achieve the same level. The loading profiles for these two hydrophobic compounds are indistinguishable and approach steady state only after 60 h.
Fig. 1.

Pharmacokinetic tissue-loading profile of labeled dextran (♦), paclitaxel (□), and rapamycin (⋄) in bovine internal carotid tissue segments normalized to final drug concentrations.
Tissue-Elution Kinetics. Arterial samples were pre-equilibrated in [3H]dextran (10-6 M), [3H]paclitaxel (10-6 M), or [14C]rapamycin (10-6 M) for 60 h and then placed in an elution sink of 50 ml of PBS++. Samples were processed in triplicate over the following 60 h, and data were plotted as percentages of the pre-elution load value (Fig. 2). Dextran elutes most rapidly, losing 90% of its equilibrated load within 2 h and reaching a steady state of ≈10% of original material in <5 h. The hydrophobic drugs take ≈1 day to reach an elution steady-state value. At 48 h, arterial segments loaded with rapamycin retain ≈60% of their initial load, and those loaded with paclitaxel, ≈35%.
Fig. 2.

Pharmacokinetic tissue-elution profile of labeled dextran (♦), paclitaxel (□), and rapamycin (⋄) in bovine internal carotid tissue segments normalized to initial load values.
Bulk Diffusion Measurements. Effective planar and transmural diffusivities in the carotid artery were measured directly from the mass of drug transferred into the arterial wall (n = 3) by using the early-time solution of the diffusion equation (3) (Fig. 3). Diffusivities in the circumferential and longitudinal directions were equivalent (data not shown). Planar coefficients represent measurements in the longitudinal direction. All compounds demonstrated anisotropic diffusivities, with statistically significant (P < 0.05 by the two-tailed Student t test) more rapid planar than transmural diffusivity. Dextran diffusivity was two orders of magnitude greater than that of either of the hydrophobic compounds in both the planar and transmural directions, despite the hydrophobic compounds' 20-fold smaller size. Diffusion is anisotropic and varies nonintuitively with molecular mass, invalidating a common assumption that drug-tissue diffusion problems can be approximated by using a simple molecular conformation in a homogenous media.
Fig. 3.

Planar (□) and transmural (▪) diffusion coefficient of labeled dextran, paclitaxel, and rapamycin in bovine internal carotid tissue segments.
TBC Coefficients. TBC was defined as the tissue concentration (cT) at equilibrium normalized by the bulk concentration at equilibrium (cbulk). Arterial segments were incubated in the drug for 60 h to allow for equilibration. Upon equilibration, bulk solutions were sampled, and tissue samples were processed for liquid scintillation counting. Variation in initial bulk concentrations over an order of magnitude did not affect TBC. Dextran has little binding capacity in arterial tissue with a coefficient of ≈0.60. Because this value is similar to its physically accessible volume fraction in arterial tissue (17), dextran may not leave the extracellular space. The paclitaxel and rapamycin binding coefficients are not statistically different (P > 0.05) by the two-tailed Student t test and are significantly greater than 1, indicating that nonspecific and/or specific binding interactions are sequestering these drugs in the tissue (Fig. 4).
Fig. 4.

TBC of labeled dextran, rapamycin, and paclitaxel in internal carotid tissue segments after 60 h of equilibration.
To assay for the binding specificity, experiments were conducted with mixtures of labeled and nonlabeled drug (Fig. 5). Molar excess of unlabeled paclitaxel or rapamycin displaced the binding of the corresponding labeled drug to <5% of control value. At this level the TBC value fell to nearly 1, indicating displacement of specific and nonspecific binding. When the cold drug was switched, the TBC decreased to ≈35% of control for paclitaxel and ≈50% for rapamycin, suggesting that these compounds were now displacing the unrelated labeled drug, although displacement occurred only from nonspecific sites, leaving specific binding intact. By the two-tailed Student t test, the paclitaxel and rapamycin specific retention fractions are statistically different (P < 0.05).
Fig. 5.

Fractional TBC of labeled paclitaxel (▪) and labeled rapamycin (□) relative to pure labeled drug in internal carotid tissue segments after 60 h of equilibration for the following conditions: (i) Rx, 10-6 M labeled drug plus 10-3 M of the same unlabeled drug and (ii)Rx′,10-6 M labeled paclitaxel plus 10-3 M unlabeled rapamycin and 10-6 M labeled rapamycin plus 10-3 M unlabeled paclitaxel.
Transmural Drug Distribution. Arterial samples were incubated in [3H]dextran, [3H]paclitaxel, or [14C]rapamycin for 60 h and then snap-frozen for en face sectioning (Fig. 6). Previous work with paclitaxel in BSA showed that TBC was maximal in the intima and declined precipitously within the most intimal regions of the arterial media, to less than half the intimal level. At the outer edge of the media, the paclitaxel binding capacity increased gradually and peaked within the adventitia before falling off to unity (13). These data have been recapitulated, but without BSA, for direct comparison with dextran and rapamycin. In this case, paclitaxel shows a very similar medial profile to the BSA case, but rises sharply in the adventitia, suggesting that BSA provides a nonspecific binding sink for adventitial binding. Rapamycin shows a uniform transmural distribution, in stark contrast to the nonuniform distribution of paclitaxel. Dextran again shows little binding capacity throughout the artery.
Fig. 6.

Transmural equilibrium distribution of labeled dextran (♦), paclitaxel (□), and rapamycin (⋄) in 0.040-mm-thick bovine internal carotid tissue segments.
Discussion
Local drug delivery has great theoretical and practical appeal for vascular diseases (28, 29). One important surprise is that biological potency is not the sole determinant of biological effect. Drug-specific physicochemical properties determine, to a great degree, whether concentrations sufficient for therapeutic activity can be sustained. Drugs that bind to tissue elements, for example, are retained within tissue and have dramatic clinical effects; nonbinding hydrophilic drugs are rapidly cleared and ineffective against restenosis. Our data suggest that specific binding also plays a critical role in determining drug distribution. Whereas paclitaxel distributes heterogeneously through arterial tissue, rapamycin distributes more uniformly through the media and adventitia. In addition, the tissue binding and diffusion results suggest that binding site availability and distribution regulate the fine structure of drug deposition beyond the coarse structure imposed by transport forces and lipid avidity. Ultimately, local tissue ultrastructure and the concentrations they enforce on the artery at a microscopic scale, together with specific and nonspecific binding site distribution, become critical considerations in the optimization of vascular drug delivery.
Specific and Nonspecific Binding Determine TBC. Compounds must bind to proteins, intracellular or extracellular, to have a biological effect. This binding can take two forms: nonspecific interactions, such as those influenced by charge or water affinity, and specific binding idiosyncratic to the individual drug. Paclitaxel and rapamycin both can bind nonspecifically to serum proteins and hydrophobic tissue microenvironments. Paclitaxel demonstrates nanomolar specificity to polymerized microtubules, whereas rapamycin shows similar specific binding properties to FKBP12, a ryanodine-receptor-associated protein. Dextran, by virtue of its extreme hydrophilicity, exhibited neither type of binding and, accordingly, its TBC was <1; its potential tissue level can never exceed the concentration in the bathing solution. In contrast, paclitaxel and rapamycin were deposited in the blood vessel at concentrations 30- to 40-fold higher than that in surrounding bulk solutions. Thus, tissue concentrations of paclitaxel and rapamycin can exceed the applied concentration severalfold, establishing an effective volume of distribution within arteries larger than anticipated from surrounding solution concentration.
Because both microtubules and FKBP, which specifically bind paclitaxel and rapamycin, respectively, reside in calf internal carotid segments at ≈10-5 M (24), offering a large specific drug sink, we investigated the specificity and potential of tissue binding by using a competitive binding tissue assay. Both specific and nonspecific binding contributions were observed. When cold rapamycin was substituted for a high concentration molar excess of paclitaxel in the labeled paclitaxel system, the fractional TBC was nearly 35%, indicating that approximately one-third of the TBC was from specific binding. Comparably, when the same procedures were carried out with labeled rapamycin and excess cold paclitaxel, the fractional TBC was 50%, with near equal contributions from specific and nonspecific binding events.
Tissue Pharmacokinetics of Hydrophilic and Hydrophobic Compounds. It is expected that paclitaxel and rapamycin will have similar transport properties, given that both compounds have solubilities of ≈6 μg/ml, molecular masses of <1 kDa, and nanomolar binding constants to their specific protein targets. Indeed, the compounds act quite similar when compared with dextran. Whereas only several hours are required for dextran to reach a steady-state tissue concentration that never exceeds the surrounding media levels, paclitaxel and rapamycin take four to six times longer to reach a steady state. Kinetics, not concentration, accounts for this effect, as the time to reach steady state was independent of the concentration of the same drug applied over a broad range. Although the net binding of paclitaxel and rapamycin were nearly identical, their distribution and tissue elution after binding and uptake were not. At steady state the artery retained only 10% of the applied hydrophilic dextran. Paclitaxel retention was 3.5-fold that of dextran, and rapamycin levels were almost twice those of paclitaxel. These differences in tissue retention closely parallel the percentages attributed to specific binding (Fig. 5), and it is clear that both hydrophobic compounds are retained to a much greater extent than dextran. Moreover, at steady-state loading, whereas paclitaxel remained in the subintimal space and partitioned significantly in the adventitia, rapamycin was evenly distributed throughout the arterial wall (Fig. 6).
Movement of a molecule through a composite structure, such as a blood vessel wall, is driven by a range of forces and phenomena. The influence of effective molecular radius can dominate when all other factors are equal but may recede in importance when other factors are present. For instance, despite being nearly 20 times smaller than dextran, paclitaxel and rapamycin diffuse more slowly in both the transmural and planar directions. This difference may be attributed to the hydrophobicity of the compounds or possibly to the role of binding. Whereas dextran has few binding sites, paclitaxel and rapamycin will repeatedly bind to and dissociate from their respective specific and nonspecific targets as they diffuse through tissue, in effect slowing the leading edge of the diffusion front. We have previously shown that albumin and dextrans diffuse at least an order of magnitude faster in the planar direction than they do in the transmural direction (3). For paclitaxel and rapamycin, the planar diffusivity exceeded transmural diffusivity by at least two orders of magnitude, although both drugs' diffusivities were two orders of magnitude smaller than those of dextran. These phenomena are likely governed by similar forces for all three drugs, despite vastly different lipid avidities. The transport of hydrophilic compounds is enhanced in aqueous regimes of the vessel wall but retarded by hydrophobic elastin layers. For hydrophobic compounds, these layers act in a reverse fashion; the movement of paclitaxel and rapamycin is likely impeded by the more water-rich regions of the blood vessel wall and aided by lipid pools or even the protein-studded elastin lamina. In both cases, however, whereas individual layers might be isotropic, the greater composite of alternating layers of the arterial wall provide for planar diffusion that far exceeds transmural flux (3).
As the number of available drug-eluting devices increases, distinction and choice may reside not only in ease of use but also in the physicochemical functionality of the drug-stent unit. Local drug distribution is modulated by transport and lipid avidity at a coarse level, but for clinically relevant compounds such as rapamycin and paclitaxel, it is also modulated by the distribution of the compounds' specific binding sites. Arterial ultrastructure also influences transport, because alternating tissue layers of varying permeability result in anisotropic transport for both hydrophobic and hydrophilic drugs. Design and evaluation of a drug-eluting device thus requires a unified understanding of the drug, its physicochemical characteristics, and its specific and nonspecific interactions with arterial structures.
Acknowledgments
This work was supported in part by the Hertz Foundation; National Institutes of Health Grants R01 GM/HL 49039, HL 60407, and HL 67246; and a generous gift of [14C]rapamycin and funds for materials from Johnson & Johnson/Cordis (Miami).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: TBC, tissue binding capacity.
References
- 1.Hwang, C. W., Wu, D. & Edelman, E. R. (2001) J. Am. Coll. Cardiol. 37, Suppl. A, 1A-2A.11153722 [Google Scholar]
- 2.Hwang, C. W., Wu, D. & Edelman, E. R. (2001) Circulation 104, 600-605. [DOI] [PubMed] [Google Scholar]
- 3.Hwang, C. W. & Edelman, E. R. (2002) Circ. Res. 90, 826-832. [DOI] [PubMed] [Google Scholar]
- 4.Lovich, M. A., Brown, L. & Edelman, E. R. (1997) J. Am. Coll. Cardiol. 29, 1645-1650. [DOI] [PubMed] [Google Scholar]
- 5.Lovich, M. A. & Edelman, E. R. (1995) Circ. Res. 77, 1143-1150. [DOI] [PubMed] [Google Scholar]
- 6.Edelman, E. R. & Lovich, M. (1998) Nat. Biotechnol. 16, 136-137. [DOI] [PubMed] [Google Scholar]
- 7.Wan, W. K., Lovich, M. A., Hwang, C. W. & Edelman, E. R. (1999) J. Pharm. Sci. 88, 822-829. [DOI] [PubMed] [Google Scholar]
- 8.Grube, E., Silber, S., Hauptmann, K. E., Mueller, R., Buellesfeld, L., Gerckens, U. & Russell, M. E. (2003) Circulation 107, 38-42. [DOI] [PubMed] [Google Scholar]
- 9.Marks, A. R. (2003) N. Engl. J. Med. 349, 1307-1309. [DOI] [PubMed] [Google Scholar]
- 10.Moses, J. W., Leon, M. B., Popma, J. J., Fitzgerald, P. J., Holmes, D. R., O'Shaughnessy, C., Caputo, R. P., Kereiakes, D. J., Williams, D. O., Teirstein, P. S., et al. (2003) N. Engl. J. Med. 349, 1315-1323. [DOI] [PubMed] [Google Scholar]
- 11.Buellesfeld, L., Gerckens, U., Muller, R. & Grube, E. (2003) Z. Kardiol. 92, 825-832. [DOI] [PubMed] [Google Scholar]
- 12.Bratzler, R. L., Chisolm, G. M., Colton, C. K., Smith, K. A., Zilversmit, D. B. & Lees, R. S. (1977) Circ. Res. 40, 182-190. [DOI] [PubMed] [Google Scholar]
- 13.Creel, C. J., Lovich, M. A. & Edelman, E. R. (2000) Circ. Res. 86, 879-884. [DOI] [PubMed] [Google Scholar]
- 14.Edelman, E. R., Nathan, A., Katada, M., Gates, J. & Karnovsky, M. J. (2000) Biomaterials 21, 2279-2286. [DOI] [PubMed] [Google Scholar]
- 15.Edelman, E. R., Nugent, M. A. & Karnovsky, M. J. (1993) Proc. Natl. Acad. Sci. USA 90, 1513-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nugent, M. A., Karnovsky, M. J. & Edelman, E. R. (1993) Circ. Res. 73, 1051-1060. [DOI] [PubMed] [Google Scholar]
- 17.Lovich, M. A. & Edelman, E. R. (1996) Biophys. J. 70, 1553-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lovich, M. A., Philbrook, M., Sawyer, S., Weselcouch, E. & Edelman, E. R. (1998) Am. J. Physiol. 275, H2236-H2242. [DOI] [PubMed] [Google Scholar]
- 19.Manfredi, J. J., Parness, J. & Horwitz, S. B. (1982) J. Cell Biol. 94, 688-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wandless, T. J., Michnick, S. W., Rosen, M. K., Karplus, M. & Schreiber, S. L. (1991) J. Am. Chem. Soc. 113, 2339-2341. [Google Scholar]
- 21.Choi, J. W., Chen, J., Schreiber, S. L. & Clardy, J. (1996) Science 273, 239-242. [DOI] [PubMed] [Google Scholar]
- 22.Siekierka, J. J., Hung, S. H., Poe, M., Lin, C. S. & Sigal, N. H. (1989) Nature 341, 755-757. [DOI] [PubMed] [Google Scholar]
- 23.Sezen, S. F., Blackshaw, S., Steiner, J. P. & Burnett, A. L. (2002) Int. J. Impot. Res. 14, 506-512. [DOI] [PubMed] [Google Scholar]
- 24.Hiller, G. & Weber, K. (1978) Cell 14, 795-804. [DOI] [PubMed] [Google Scholar]
- 25.Lovich, M. A., Creel, C., Hong, K., Hwang, C. W. & Edelman, E. R. (2001) J. Pharm. Sci. 90, 1324-1335. [DOI] [PubMed] [Google Scholar]
- 26.Ramirez, C. A., Colton, C. K., Smith, K. A., Stemerman, M. B. & Lees, R. S. (1984) Arteriosclerosis (Dallas) 4, 283-291. [DOI] [PubMed] [Google Scholar]
- 27.Penn, M. S., Koelle, M. R., Schwartz, S. M. & Chisolm, G. M. (1990) Circ. Res. 67, 11-22. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz, R. S. & Henry, T. D. (2002) Rev. Cardiovasc. Med. 3, Suppl. 5, S4-S9. [PubMed] [Google Scholar]
- 29.Carter, A. J. (2002) Catheter. Cardiovasc. Interv. 57, 69-71. [DOI] [PubMed] [Google Scholar]