

Abstract
Multiple myeloma (MM), a plasma cell malignancy, is the second most prevalent hematologic malignancy in the US. Although much effort has been made trying to understand the etiology and the complexities of this disease with the hope of developing effective therapies, MM remains incurable at this time. Because of their antiproliferative and proapoptotic activities, interferons (IFNs) have been used to treat various malignancies, including MM. Although some success has been observed, the inherent toxicities of IFNs limit their efficacy. To address this problem, we produced anti-CD138 antibody fusion proteins containing either IFNα2 or a mutant IFNα2 (IFNα2YNS) with the goal of targeting IFN to CD138-expressing cells, thereby achieving effective IFN concentrations at the site of the tumor in the absence of toxicity. The fusion proteins inhibited the proliferation and induced apoptosis of U266, ANBL-6, NCI-H929, and MM1-144 MM cell lines. The fusion proteins decreased the expression of IFN regulatory factor 4 (IRF4) in U266. In addition, the fusion proteins were effective against primary cells from MM patients, and treatment with fusion proteins prolonged survival in the U266 murine model of MM. These studies show that IFNα antibody fusion proteins can be effective novel therapeutics for the treatment of MM.
Introduction
Multiple myeloma (MM) is a disease characterized by an excess of malignant plasma cells in the bone marrow. Accumulation and proliferation of malignant myeloma cells result in the disruption of normal hematopoiesis and changes to bone marrow vascularization and bone physiology. Analyses of patient myeloma cells and human myeloma cell lines (HMCLs) have revealed the extensive molecular heterogeneity of this disease (Drexler and Matsuo 2000; Carrasco and others 2006; Lombardi and others 2006; Moreaux and others 2011). The survival rate for MM is 7–8 years when patients are treated with drugs such as the proteasome inhibitor bortezomib, thalidomide, or lenalidomide, which target myeloma cells in the bone marrow microenvironment (Anderson 2012). Currently there is no cure for MM.
Besides their antiviral and immunostimulatory activities, interferons (IFNs) have antiproliferative activity and can induce apoptosis in hematological malignancies and solid tumors (Borden and others 2000; Borden and others 2007). Many studies have shown that type I IFNs, which were the first recombinant proteins used in the treatment of cancer, may be highly effective against a variety of tumor cell targets (Borden and others 2007). However, the effectiveness of type I IFNs for cancer therapy has been limited by their short half-life of only 1 h (Peleg-Shulman and others 2004) and associated side effects when used at high doses (Weiss 1998).
Previously, we have shown that antibody-IFN fusion proteins are an effective strategy for targeting IFNs to cancer cells. Anti-CD20-IFN fusion proteins targeting lymphoma cells showed potent growth inhibitory activity both in vitro and in vivo in murine lymphoma models (Xuan and others 2010; Trinh and others 2013). In addition, fusion of IFNα or IFNβ to IgG increased its half-life to 8 h (Huang and others 2007; Trinh and others 2013). Therefore, we wanted to extend these studies to determine if targeted IFNα would be effective against MM. In this initial study, we used several different HMCLs, but focused our attention on the U266 myeloma cell line. We constructed fusions of anti-CD138 with IFNα2 and IFNα2YNS, a high affinity IFNα2 mutant. We chose CD138, also known as syndecan-1, as the target antigen. CD138 is a heparan sulfate proteoglycan that is highly expressed on HMCLs and malignant plasma cells in peripheral blood and in the bone marrow in patients (Ridley and others 1993; Wijdenes and others 1996; Chilosi and others 1999). Treatment with IFNα2 fusion proteins resulted in decreases in cell viability. The mode of action of the fusion proteins included the induction of apoptosis and in U266 a decrease in expression of IFN regulatory factor 4 (IRF4), a protein which is required for MM cell survival. In addition, the fusion proteins were effective against primary patient cells and in vivo against U266 tumors in a murine model. In some experiments the higher affinity anti-CD138-IFNα2YNS protein showed increased antiproliferative activity over anti-CD138-IFNα2.
Materials and Methods
Cells
HMCLs cells were the generous gift of Dr. W. Michael Kuehl and Dr. Diane Jelinek. Primary cells were obtained after informed consent and approved by the institutional medical ethics committee. Cells were cultured in the RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 5% fetal calf serum (FCS; Atlanta Biologics, Lawrenceville, GA). Growth medium for ANBL-6 cells was supplemented with 2 ng/mL of IL-6. Chinese Hamster Ovary (CHO) cells were cultured in Iscove's Modified Dulbecco's Medium (IMDM; Invitrogen) supplemented with 5% FCS.
Construction of expression vectors, protein production, and purification
The heavy (H) and light (L) chain variable (V) region amino acid sequences of the anti-CD138 antibody B-B4 were obtained from US Patent Application No: 2009/0175863 and used to construct IgG1, κ proteins.
VH sequence: MGWSYIILFLVATATGVHSQVQLQQSGSELMMPGASVKISCKATGYTFSNYWIQRPGHGLEWIGEILPGTGRTIYNEKFKGKATFTADISSNTVQMQLSSLTSEDSAVYYCARRDYYGNFYYAMDYWGQGTSVTVSS.
VL sequence: MKSQTQVFIFLLLCVSGAHGDIQMTQSTSSLSASLGDRVTISCSASQGINNYLNWYQQKPDGTVELLIYYTSTLQSGVPSRFSGSGSGTDYSLTISNLEPEDIGTYYCQQYSKLPRTFGGGTKLEIK.
The DNA sequence encoding a signal peptide was added 5′ of the H chain and L chain V regions (MGWSYIILFLVATATGVHS and MKSQTQVFIFLLLCVSGAHG, respectively) as well as the nucleotide sequence containing a Kozak ribosomal recognition site (5′-GGATATCCACC-3′). To facilitate downstream cloning, the sequence 5′-GCTAGCC-3′ was added 3′ of the H chain V region, and the sequence 5′-CGTAAGTCGACG-3′ was added 3′ of the L chain V region. The DNA sequence was synthesized using codons optimized for CHO expression (DNA2.0, Menlo Park, CA).
The L chain V region flanked by EcoRV and SalI restriction sites was sequence verified before cloning into an expression vector containing the human κ L chain constant region. The H chain V region flanked by EcoRV and NheI restriction sites was sequence verified and cloned into an expression vector containing the human γ1 H chain constant region. Expression of the H and L chains yielded anti-CD138 IgG1. To produce the fusion protein, the anti-CD138 VH was cloned into an expression vector containing the human γ1 H chain, a Gly-Ser linker (SGGGGS), followed by human IFNα2. This expression vector was named pAH6905.
To construct the DNA vector for the expression of anti-CD138-IFNα2YNS, nested PCR was used to introduce 3 amino acid mutations—H57Y, E58N, and Q61S. The first round of PCR was done using the forward primer 5′-CGCGGATCCTGTGATCTGCCTCAAACCCAC-3′ and reverse primer 5′-CCTCTAGAATCATTCCTTACTTCTTAAACT-3′. The nested PCR was done using forward primer 5′-CTCTACAATATGATCTCACAGATC-3′ and reverse primer 5′-GATCTGTGAGATCATATTGTAGAG-3′, which contains the mutations to IFNα2. The insert was cloned into pCR2.1-TOPO vector (Invitrogen) and the DNA sequence was verified. The XbaI/BamHI fragment containing the mutant IFNα2YNS sequence was cloned into an intermediate human γ1 H chain vector and named pAH11015. The BamHI/AvrII fragment from pAH11015 containing the mutations to IFNα2 was then used to replace the wild-type IFNα2 sequence from expression vector pAH6905 (see previous paragraph).
Protein production and purification
Anti-CD138 IgG and fusion proteins were produced in CHO cells by transfection of H and L chain expression vectors. Stably transfected cells were selected with 1 mM histidinol. To produce IgG and the fusion proteins, CHO transfectants were seeded into roller bottles. At confluency, cells were expanded to 100 mL with IMDM+1% Fetal Clone (Thermo Fisher Scientific, Waltham, MA). The supernatant was removed every 2–3 days and replaced with fresh medium. Cell-free culture supernatants were then passed through a protein A-Sepharose 4B fast flow column (Sigma-Aldrich, St. Louis, MO) and the bound protein eluted with 0.1 M citric acid, pH 3.5. Eluted fractions were neutralized immediately with 2 M Tris-HCl pH 8.0. Fractions were run on SDS-PAGE gels and stained with Coomassie blue to verify protein purity and integrity. Protein concentrations were determined using the BCA assay (Pierce, Rockford, IL). Anti-CD20-IFNα2, anti-CD20-IFNα2YNS, and anti-dansyl (DNS)-IFNα2 used as untargeted control proteins were produced as described previously (Xuan and others 2010).
MTS assay to determine metabolic activity
HMCLs were seeded in 96-well plates and incubated with 0.00002 pM–25 nM of IFNα2, anti-CD20-IFNα2, or anti-CD138-IFNα2 at 37°C for 3 or 7 days. Relative metabolic activity was determined using MTS solution (Promega, Madison, WI) by measuring absorbance at 490 nm using a Synergy HT Multi-Detection Microplate Reader (BioTek Instruments, Inc., Winooski, VT) with untreated cells being 100%. The GraphPad Prism (GraphPad Software, Inc., La Jolla, CA) was used to analyze data by nonlinear regression with the log (inhibitor) versus the response with a variable slope. Data are expressed as a percentage of maximum metabolic activity. The experiments were performed in triplicate.
Apoptosis assay
Cells were treated with varying concentrations of IFNα2, anti-CD20-IFNα2, anti-CD138 IgG, anti-CD138-IFNα2 of anti-CD138-IFNα2YNS for 2 or 3 days at 37°C. Cells were stained with Alexa Fluor 488-labeled Annexin V and propidium iodide (PI) using the Vybrant Apoptosis Kit #2 (Molecular Probes, Carlsbad, CA) as per the manufacturer's instructions and analyzed by flow cytometry.
Cells were treated for 2 days at 37°C with 500 pM anti-CD138-IFNα2 in the presence or absence of 1 nM rapamycin or 20 μM LY294002. Apoptosis was monitored by staining with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry as described above.
Caspase inhibition
U266 cells were treated for 2 days with 0.032 pM–2.5 nM of anti-CD138-IFNα2 in the presence or absence of 50 μM zVAD-fmk (Calbiochem, San Diego, CA), a pan-caspase inhibitor. Cell viability was measured using the CellTiter Glo® assay (Promega), as per the manufacturer's instructions.
Detection of IRF4
U266 cells were treated for 48 h with 1 nM of IFNα2, anti-CD138-IFNα2, or anti-CD138-IFNα2YNS. Cells were lysed using the RIPA buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing a protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). The cytosolic fractions were reduced with β-mercaptoethanol and separated by SDS-PAGE. Following transfer to nitrocellulose membrane (Whatman, Piscataway, NJ) and blocking with 3% bovine serum albumin (BSA) in phosphate buffered saline (PBS)+0.1% Tween 20, samples were incubated with rabbit anti-IRF4 (Epitomics, Berlingham, CA) and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Sigma-Aldrich). Secondary anti-rabbit IgG-HRP (GE Healthcare, Billerica, MA) was used and the blots were developed using enhanced chemiluminescence (ECL; Thermo Fisher Scientific). Films were imaged using a MultiImage™ Light Cabinet (Alpha Innotech Corp., San Leandro, CA) and analyzed using the NIH ImageJ. Bands were normalized to a GAPDH loading control and expressed as percent band intensity of untreated cells. The average from 3 different experiments is shown.
Detection of STAT1 and STAT3
U266 cells were treated for 0.5, 24 and 48 h with 100 pM of IFNα2, anti-CD138-IFNα2, or anti-CD138-IFNα2YNS. Cells were lysed using 50 mM Tris–HCl, pH 7.4, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1% glycerol, protease inhibitor cocktail, and PhosSTOP (Roche Applied Science). The cytosolic fractions were reduced with β-mercaptoethanol and separated by SDS-PAGE. Following transfer to PVDF membrane (Boehringer-Mannheim, Germany) and blocking in 3% BSA in PBS+0.1% Tween 20, the blot was incubated with primary antibody overnight at 4°C. Secondary anti-rabbit IgG-HRP (GE Healthcare) was used and the blots were developed using Western Lightning-ECL (PerkinElmer, Waltham, MA). Images were captured using a LAS-1000 from Fujifilm. The following primary antibodies (Cell Signaling Technology, Danvers, MA) were used: rabbit polyclonal serum against phosphorylated STAT1-Y701, STAT1, phosphorylated STAT3-Y705, and STAT3. Rabbit anti-GAPDH was from Sigma-Aldrich. Quantitations of bands are shown in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/jir).
Treatment of primary myeloma cells from patients
Patients with active myeloma were biopsied while off therapy and myeloma cells isolated by negative antibody selection to >95% purity. Cells were incubated with 25 or 100 nM of anti-CD138 IgG, anti-CD138-IFNα2, or anti-CD138-IFNα2YNS for 72 h. Percent viable cell recovery was determined by Trypan Blue staining, with untreated control cells designated as 100%.
In vivo anti-tumor activity against U266 cells
Six to 8 week old female NOD-scid IL2rγnull (NSG) mice were used to establish U266 tumors. Mice were inoculated subcutaneously with 1×107 cells at the base of the tail. Mice were treated intravenously with PBS or 100 μg of anti-CD138, anti-DNS-IFNα2, anti-CD138-IFNα2, anti-CD20-IFNα2YNS, or anti-CD138-IFNα2YNS on days 14, 16, and 18 post tumor challenge. One group of mice received additional treatments with anti-CD138-IFNα2YNS on days 24, 31, and 62 for a total of 6 treatments. Each group consisted of 8 mice. Bidirectional tumor growth was measured throughout the experiment and mice were sacrificed when tumors reached 1.5 cm as per the institutional guidelines. All animal studies were performed in compliance with the US Department of Health and Human Services Guide for the Care and Use of Laboratory Animals and were approved by the UCLA Animal Research Committee.
Results
Production and characterization of anti-CD138-IFNα2 and anti-CD138-IFNα2YNS fusion proteins
The aim of this study was to test the effectiveness of using antibodies specific for CD138 to target IFN to MM cells. The approach was to genetically fuse IFN to the end of the CH3 domain of human IgG1 (Fig. 1A) containing the V regions from the anti-CD138 antibody B-B4 (Wijdenes and others 1996). For the initial experiments we elected to target IFNα2, which has been used successfully in the treatment of MM in the clinic, or a mutant higher affinity IFNα2YNS (see “Increasing the affinity of IFNα2 for its receptor increases its potency in vitro” section).
FIG. 1.

Production of IFNα2 fusion proteins. (A) Schematic diagram of anti-CD138 fusion proteins containing IFNα2 or IFNα2YNS. The fusion proteins contain V regions of murine antibody B-B4, which is specific for CD138, and human IgG1,κ constant regions. Human IFNα2 or IFNα2YNS is fused to the C-terminus of the antibody through a SerGly4Ser peptide linker. (B) SDS-PAGE analysis of reduced and unreduced purified fusion proteins and control IgG. H, heavy; IFN, interferon; L, light.
Anti-CD138 IgG1 either unfused or fused to human IFNα2 was expressed in stable CHO transfectants. Purified proteins were characterized with respect to their size and assembly status. The purified fusion protein possess heavy (H) and light (L) chains of the appropriate molecular weight and assemble into complete H2L2 molecules (Fig. 1B) that bound CD138 expressed on the surface of HMCLs (data not shown).
IFNα2 fusion proteins inhibit the growth of HMCLs with targeting by anti-CD138 further improving efficacy
Several different assays were used to determine whether targeted fusion protein had cytoreductive activity against HMCLs. To determine if fusion of IFNα2 to IgG had any effect on its activity, we compared the activity of IFNα2 alone to untargeted anti-CD20-IFNα2 fusion protein since MM cells do not express CD20. HMCLs were treated with varying concentrations for 3 days (U266, NCI-H929, MM1-144) or 7 days (ANBL-6). Measurement of cellular metabolic activity using the MTS assay showed that the anti-CD20-IFNα2 fusion protein had similar activity as IFNα2 (Fig. 2A). To ascertain the effect of targeting, we compared the activity of the untargeted anti-CD20-IFNα2 fusion protein to that of targeted anti-CD138-IFNα2 in a separate experiment. U266, ANBL-6, NCI-H929, and MM1-144 cells were treated with varying concentrations of untargeted and targeted fusion protein for 3 days. Targeted anti-CD138-IFNα2 was significantly more potent than untargeted IFNα2 (Fig. 2A) with ∼100-fold lower IC50 than untargeted IFNα2 in several different assays.
FIG. 2.

Growth inhibition and induction of apoptosis by untargeted and targeted IFNα2 fusion protein. (A) HMCLs were incubated with varying concentrations (0.00002 pM–1 nM) of IFNα2 or anti-CD20-IFNα2 for 3 days, except ANBL-6 which was treated for 7 days (top panel). In a separate experiment, HMCLs were incubated with (0.03 pM–25 nM) of anti-CD20-IFNα2 or anti-CD138-IFNα2 for 3 days (bottom panel). Cellular metabolic activity was measured using the MTS assay. The experiment was performed in triplicate for each concentration. (B) HMCLs were untreated or treated with 500 pM of anti-CD138-IFNα2 for 3 days. Cells were stained with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry to assess induction of apoptosis. The number in the upper right quadrants indicates the percentage of Annexin V+/PI+ cells. (C) U266 cells were incubated with 0.5, 5, 50, or 500 pM of IFNα2, anti-CD20-IFNα2, anti-CD138-IFNα2, or anti-CD138 for 2 days and analyzed as described above. HMCLs, human myeloma cell lines; PI, propidium iodide.
Since we observed a loss in cell viability in response to fusion protein treatment, we next wanted to determine if apoptosis was being induced. U266, ANBL-6, NCI-H929, and MM1-144 cells were treated for 3 days and stained using Alexa Fluor 488-labeled Annexin V and PI and examined by flow cytometry (Fig. 2B). Anti-CD138-IFNα2 induced apoptosis in all 4 HMCLs. To further examine the induction of apoptosis, U266 cells were incubated for 2 days with varying concentrations of anti-CD138, IFNα2, anti-CD20-IFNα2, and anti-CD138-IFNα2 and then stained with Alexa Fluor 488-labeled Annexin V and PI and examined by flow cytometry (Fig. 2C). Unfused anti-CD138, which did not inhibit cell viability (data not shown), also did not induce apoptosis (Fig. 2C). Anti-CD138-IFNα2, anti-CD20-IFNα2, and IFNα2 all showed a concentration-dependent increase in the percentage of Annexin V+/PI− and Annexin V+/PI+ cells. However, cells treated with the targeted anti-CD138-IFNα2 fusion protein showed the greatest percentage of Annexin V+ cells, consistent with targeted anti-CD138-IFNα2 being more effective than untargeted anti-CD20-IFNα2 in inhibiting cell proliferation (Fig. 2A). Following the shorter 2 day treatment, we observed both Annexin V+/PI− and Annexin V+/PI+ cells whereas by day 3, almost all Annexin V+ cells were also PI+ (Fig. 2B).
Signaling pathways involved in anti-CD138-IFNα2 activity
Many different signaling pathways have been shown to be involved in IFNα-mediated toxicity. One important pathway is the activation of caspases. Previous studies have shown that induction of apoptosis following treatment with IFNα was associated with activation of caspases-1, -2, -3, -8, -9 in U266 cells (Thyrell and others 2002). To determine if caspases play a role in anti-CD138-IFNα2 toxicity against U266, we treated the cells in the presence or absence of the pan-caspase inhibitor, zVAD-fmk. Cells were incubated with increasing concentrations of anti-CD138-IFNα2 with or without 50 μM of zVAD-fmk and cell viability was measured using the CellTiter Glo assay. In the presence of zVAD-fmk, some of the cytotoxic effects of anti-CD138-IFNα2 were decreased, but not eliminated (Fig. 3A). These data confirm previous findings that activation of caspases plays a role in IFNα-mediated cytotoxicity.
FIG. 3.

The antiproliferative effects of IFNα2 fusion proteins include the activation of caspases and the PI3K/mTOR pathways. (A) U266 cells were treated for 2 days with 0.032 pM–2.5 nM of anti-CD138-IFNα2 in the presence or absence of 50 μM of the pan-caspase inhibitor zVAD-fmk. Cell viability was measured using the CellTiter Glo assay. U266 cells were treated for 2 days with 500 pM of anti-CD138-IFNα2 in the presence or absence of 20 μM LY294002 (B) or 1 nM rapamycin (C). Apoptosis was monitored by staining with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry. The number in the upper right quadrants indicates the percentage of Annexin V+/PI+ cells. The number in the lower right quadrants indicates the percentage of Annexin V+/PI− cells. PI3K/mTOR, phosphoinositide 3 kinase/mammalian target of rapamycin.
Thyrell and others (2004) reported that induction of apoptosis by IFNα in U266 cells involves the phosphoinositide 3 kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway. To determine if anti-CD138-IFNα2 acts similarly through this pathway, we analyzed the effects of inhibitors of PI3K/mTOR on the induction of apoptosis. Rapamycin blocks mTOR whereas LY294002 blocks PI3K and to a lesser extent mTOR (Brunn and others 1996). Cells were treated with anti-CD138-IFNα2 for 2 days in the presence or absence of the inhibitors. Apoptosis was monitored by staining with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry. Neither LY294002 nor rapamycin had any effects by itself (Fig. 3B, C, respectively). When the cells were treated with anti-CD138-IFNα2, there was an increase in Annexin V+ cells. However, this induction of apoptosis by the fusion protein was decreased in the presence of LY294002 (Fig. 3B) and rapamycin (Fig. 3C). Therefore, it appears that our fusion protein, like IFNα, utilizes the PI3K/mTOR pathway to induce apoptosis in the U266 cell line.
Increasing the affinity of IFNα2 for its receptor increases its potency in vitro
IFNα and IFNβ are type I IFNs that bind to the same receptor, which is composed of 2 transmembrane proteins, IFNAR1 and IFNAR2. The IFNs differ in that IFNβ has a 20- to 50-fold greater affinity for IFNAR1 than IFNα2 (Lamken and others 2004; Jaitin and others 2006; Jaks and others 2007). This greater affinity has been shown to correlate with a significantly higher antiproliferative activity against some malignancies (Jaitin and others 2006; Kalie and others 2007), making IFNβ an attractive candidate for cancer therapy. However, when human IFNβ was genetically fused to IgG, we found that IFNβ activity was reduced by >100-fold (data not shown). Therefore, we chose an alternative approach to produce a high affinity mutant IFNα2 fusion protein. This mutant contains mutations at 3 positions, H57Y/E58N/Q61S (IFNα2YNS), which confer a 60-fold increased affinity for IFNAR1 and a large increase in antiproliferative activity compared to wild-type IFNα2 (Kalie and others 2007). Using the same approach as described above, we produced anti-CD138-IFNα2YNS in CHO cells (Fig. 1) to compare the activity of the higher affinity fusion protein to that of wild-type fusion protein. The ability of the 2 fusion proteins to induce apoptosis was compared by treating cells with anti-CD138-IFNα2 or anti-CD138-IFNα2YNS for 2 or 3 days. Cells were then stained with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry. For the U266 and NCI-H929 cell lines, treatment with anti-CD138-IFNα2YNS resulted in increased levels of apoptosis compared to cells treated with anti-CD138-IFNα2 (Fig. 4A). In addition, these cell lines showed greater growth inhibition when they were treated with anti-CD138-IFNα2YNS than when they were treated with anti-CD138-IFNα2 and analyzed by the MTS assay (data not shown). Similar results were seen in several other HMCLs (data not shown). Disruption of the PI3K pathway using inhibitors resulted in decreased apoptotic activity against U266 following treatment with anti-CD138-IFNα2YNS (data not shown) similar to what was seen following treatment with anti-CD138-IFNα2 (Fig. 3).
FIG. 4.

Anti-CD138-IFN-α2YNS has greater activity than anti-CD138-IFNα2. (A) Cells were treated with 500 pM of anti-CD138-IFNα2 or anti-CD138-IFNα2YNS for 2 (U266) or 3 (NCI-H929) days. Cells were stained with Alexa Fluor 488-labeled Annexin V and PI and analyzed by flow cytometry to assess induction of apoptosis. The number in the upper right quadrants indicates the percentage of Annexin V+/PI+ cells. The number in the lower right quadrants indicates the percentage of Annexin V+/PI− cells. (B) U266 cells were treated with 100 pM of the indicated proteins for 0.5, 24, or 48 h. Cell lysates were used to determine the levels of pSTAT1, total STAT1 protein, pSTAT3, and total STAT3 protein by western blotting. Protein loading was monitored by probing the same membranes with anti-GAPDH.
To further explore the increased potency of anti-CD138-IFNα2YNS, we tested for the expression and activation of STAT proteins, which are known to be activated by IFNα. U266 cells were treated with 100 pM of IFNα2, anti-CD138, anti-CD138-IFNα2, or anti-CD138-IFNα2YNS for 0.5, 24, and 48 h, and cell lysates were examined for levels of STAT1, pSTAT1, STAT3, and pSTAT3 by western blotting. The housekeeping gene GAPDH was used to control for gel loading. Treatment with anti-CD138 did not activate STAT1 or STAT3. In contrast, pSTAT1 was seen 0.5 h following treatment with IFNα2, anti-CD138-IFNα2, and anti-CD138-IFNα2YNS. However, following treatment with anti-CD138-IFNα2YNS, but not IFNα2 or anti-CD138-IFNα2, STAT1 phosphorylation persisted and strong pSTAT1 signal was observed at 48 h. Similar results were seen for pSTAT3. All treatments except anti-CD138 resulted in increased accumulation of STAT1 over time; increased accumulation of STAT3 was not observed (Fig. 4B and Supplementary Table S1). These data suggest that the increased potency of the higher affinity anti-CD138-IFNα2YNS may be due at least, in part, to the increased strength of the signal and/or the ability to prolong signaling.
IFNα2 induces changes in IRF4 expression in U266 cells
IRF4 is a transcription factor that is important in the development of myeloid, lymphoid, and dendritic cells. IRF4 has also been shown to play a critical role in MM. Some MMs have been shown to contain a chromosomal translocation that juxtaposes the Ig H chain to the IRF4 locus, resulting in the overexpression of IRF4 (Iida and others 1997; Yoshida and others 1999). However, the expression of IRF4 has been shown to be required for survival of MM cells regardless of their genetic etiology even in patient cells and MM cell lines that do not have genetic alterations to the IRF4 locus (Shaffer and others 2008). IRF4 appears to be a master regulator in MM, controlling an aberrant gene-expression program in this disease. Knockdown of IRF4 results in cell death because IRF4 targets numerous genes that have a critical role in proliferation and survival of MM cells (Shaffer and others 2009). To determine if IFNα and fusion protein treatment can cause changes to IRF4 expression, U266 cells were treated for 48 h with IFNα2, anti-CD138-IFNα2, or anti-CD138-IFNα2YNS, cell lysates prepared, and IRF4 levels determined by western blotting. A representative blot is shown in Fig. 5. Protein levels were standardized against GAPDH. The average quantitation from 3 independent experiments is shown. Interestingly, the level of IRF4 protein decreased when U266 cells were treated with IFNα2 or with fusion proteins, suggesting that this decrease may also play a role in the cytotoxic effects of IFNα2 against U266 cells.
FIG. 5.
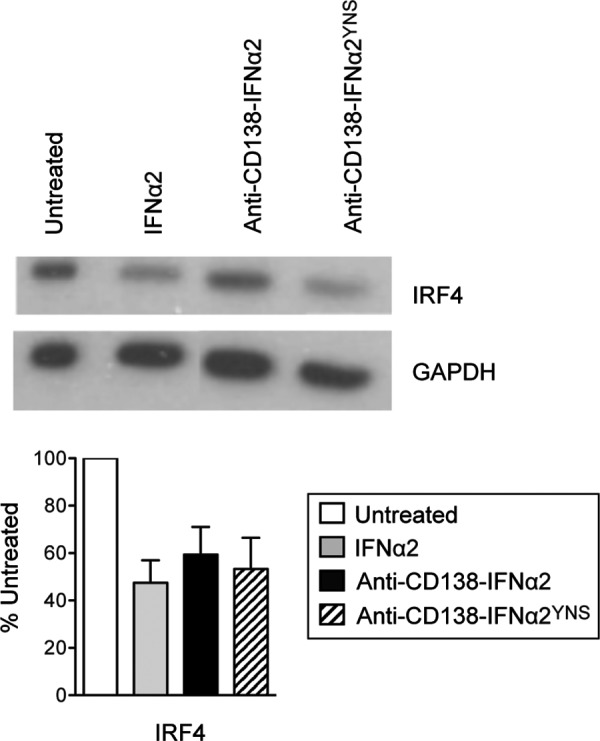
IFNα2 and fusion proteins downregulate IRF4 in U266 cells. Cells were treated for 48 h with 1 nM of IFNα2, anti-CD138-IFNα2, or anti-CD138-IFNα2YNS. Cell lysates were used to determine the levels of IRF4 protein by western blotting. A representative blot is shown. The bands from the blots were quantified using the NIH ImageJ software with levels of GAPDH as a standard. The average of the quantitations from 3 different experiments is shown in the bar graph. IRF4, IFN regulatory factor 4.
Fusion proteins are effective against primary MM cells from patients
To determine if the fusion proteins are effective against primary tumors, patient cells were treated with anti-CD138, anti-CD138-IFNα2, or anti-CD138-IFNα2YNS for 72 h and the percentage of viable recovered cells compared to untreated cells was determined by Trypan Blue exclusion. As expected, untreated primary cells demonstrated no increase in cell number during this time as freshly obtained primary MM cells do not proliferate ex vivo. Not surprisingly, there was some variability in response among the 7 patients' cells. When the proteins were tested at 25 nM (Fig. 6A), anti-CD138, anti-CD138-IFNα2, and IFNα2 (data not shown) had little effect. In contrast, anti-CD138-IFNα2YNS treatment resulted in significant decreases to cell viability when compared to anti-CD138 (P<0.0001) or to anti-CD138-IFNα2 (P=0.0009). Although anti-CD138-IFNα2 had little effect at 25 nM, when the protein was tested at 100 nM, anti-CD138-IFNα2 was able to significantly reduce cell viability when compared to anti-CD138 (P=0.0026; Fig. 6B). These data show that although both fusion proteins can affect cell viability, fusion with the higher affinity IFNα2YNS is more effective at lower concentrations against primary patient cells. In a few primary samples where sufficient cell numbers were available to perform apoptosis assays, fusion proteins appeared to induce apoptotic death (data not shown).
FIG. 6.

Fusion proteins are effective against primary myeloma cells. (A) Purified MM cells from 7 patients were incubated with 25 nM of the indicated proteins for 72 h. Cell viability was determined by Trypan Blue staining by comparing the number of recovered viable cells to that of untreated control cells. (B) Purified MM cells from 7 patients were incubated with 100 nM of the indicated proteins for 72 h. Cell viability was assessed as described above. MM, multiple myeloma.
Fusion proteins are effective in a murine xenograft model of MM
To determine if the fusion proteins are protective against MM in vivo, we used the xenograft model of U266 tumors in NSG mice. The effect of the targeted fusion proteins (anti-CD138-IFNα2 and anti-CD138-IFNα2YNS) was compared to that of several controls. One group was not treated (PBS only), whereas another group was treated with unfused anti-CD138 IgG, which lacks IFNα2 activity. To determine the effects of an untargeted IFNα2 fusion protein, mice were treated with a fusion protein with an irrelevant specificity for the hapten dansyl (anti-DNS-IFNα2). To determine the effects of an untargeted IFNα2YNS fusion protein, mice were treated with anti-CD20-IFNα2YNS. Anti-CD20-IFNα2YNS does not target myeloma cells since it is specific for human CD20 and HMCLs do not express CD20.
U266 tumors were established in NSG mice, which are severely immunocompromised, lacking mature T and B cells, functional NK cells, and are deficient in cytokine signaling. Mice were treated on days 14, 16, and 18 for all treatments. For one of the treatment groups, anti-CD138-IFNα2YNS was administered 3 additional times on days 24, 31, and 62 for a total of 6 treatments. When survival and tumor size were monitored, all treatment groups showed significant improvements in survival when compared with the PBS control group (P≤0.0003; Fig. 7A). Untargeted IFNα2 fusion proteins conferred some level of protection as did anti-CD138 IgG even though it showed no growth inhibitory activity in vitro (Fig. 2C). However, treatment with targeted fusion protein showed the best protection; anti-CD138-IFNα2 was more effective than anti-DNS-IFNα2 (P<0.0001) and anti-CD138-IFNα2YNS was more effective than anti-CD20-IFNα2YNS (P<0.0001). Although anti-CD138-IFNα2YNS was more effective than anti-CD138-IFNα2 in in vitro assays using U266, the 2 proteins had similar protective effects in vivo (P=0.05). The group receiving 6 doses of anti-CD138-IFNα2YNS showed the greatest protection, suggesting that prolonged treatment can enhance survival.
FIG. 7.

Fusion proteins confer protection to mice in a xenograft model of MM. NSG mice were injected subcutaneously with 1×107 U226 cells and treated on days 14, 16, and 18 as indicated by the black arrows with 100 μg of the indicated proteins. One group received additional treatments on days 24, 31, and 62 as indicated by the red arrows. Survival and tumor growth were monitored. Eight mice were treated for each group. P values were calculated between groups. −P≥0.05, *P≤0.0003.
Discussion
IFNα therapy has been used for the treatment of MM, but disagreement exists as to its efficacy. However, meta-analysis of 17 trials, including 2,333 patients who received combination IFNα-chemotherapy or chemotherapy alone showed significantly superior outcomes in IFNα-treated patients for relapse-free and overall survival; similarly, meta-analyses of maintenance treatments also showed significantly better outcomes in the IFNα treatment arms than in untreated controls (Fritz and Ludwig 2000), underscoring the fact that IFNα can be an effective therapeutic against MM. Some of the major problems for IFNα therapy are systemic toxicity and short in vivo half-life. Our approach to circumventing these problems was to fuse IFNα2 to anti-CD138 IgG1 to increase its half-life and, by targeting, deliver an effective dose of IFNα2 to the tumor site without systemic side effects. We have previously successfully used this approach in the treatment of human and murine B cell lymphomas using human IFNα2- and murine IFNβ-antibody fusion proteins, respectively (Xuan and others 2010; Trinh and others 2013).
In this current study, we used the same approach to target CD138 on MM cells. Anti-CD138-IFNα2 was more potent than untargeted anti-CD20-IFNα2 at inhibiting the growth of HMCLs, indicating that targeting enhances efficacy. The activity of the fusion protein was partially dependent on caspase activation as treatment with zVAD-fmk reduced cytotoxicity. Anti-CD138-IFNα2 induced apoptosis of U266 cells in a concentration-dependent manner. The proapoptotic activity of the fusion protein dependent on activation of the PI3K/mTOR pathway as treatment with rapamycin or LY294002 led to decreases in the percentage of apoptotic cells. Previous studies had demonstrated that IFNα-induced apoptosis of U266 was dependent on activation of the PI3K/mTOR pathway (Thyrell and others 2004) and a subset of the genes upregulated by IFNα depends on an active PI3K signaling pathway (Hjortsberg and others 2007). Interestingly, this IFNα-induced apoptosis requiring an active PI3K/mTOR pathway was found to occur in the absence of de novo transcription (Panaretakis and others 2008).
MM is characterized by significant heterogeneity. Studies have shown that not all MM cells are responsive to treatment with IFNα (Crowder and others 2005; Gomez-Benito and others 2005) and responsiveness of HMCLs does not always correlate with the level of IFNAR expression (Gomez-Benito and others 2005). Recently, we showed that anti-CD20-murine IFNβ (mIFNβ) is more effective than untargeted mIFNβ fusion protein and anti-CD20-murine IFNα (mIFNα) against B cell lymphoma in a murine model (Trinh and others 2013). Interestingly, while anti-CD20-mIFNα showed only modest protection against tumors expressing low levels of IFNAR, anti-CD20-mIFNβ was able to significantly prolong survival with some animals remaining tumor-free. These data suggest that IFNs with higher affinity for IFNAR may be better anticancer therapeutics. Therefore, we produced anti-CD138-IFNα2YNS, a mutant designed to mimic IFNβ, which has a higher affinity for IFNAR1 and greater activity than IFNα2. When the efficacy of anti-CD138-IFNα2YNS was compared to that of anti-CD138-IFNα2, we found that anti-CD138-IFNα2YNS was more potent than the wild-type fusion protein in HMCLs and in MM patient cells. Treatment of U266 cells with anti-CD138-IFNα2YNS resulted in increased and prolonged activation of STAT1 as compared with IFNα2 and anti-CD138-IFNα2 treatment, suggesting that this may in part explain the increased potency of anti-CD138-IFNα2YNS.
Expression of IRF4 is associated with many lymphoid malignancies. IRF4 acts as a master regulator of an aberrant, malignancy-specific regulatory network, which influences metabolism, membrane biogenesis, cell cycle progression, cell death, and transcriptional regulation in myeloma cells (Verdelli and others 2009). IRF4 inhibition has been found to be toxic to myeloma cell lines regardless of the transforming oncogenic mechanism and downregulation by as little as 50% can result in MM cell death (Shaffer and others 2008). IRF4 differs from other IRF family members in that it is not induced by IFNs. Therefore, it is of great interest that treatment with IFNα2, anti-CD138-IFNα2, and anti-CD138-IFNα2YNS resulted in decreased expression of IRF4 in U266 cells. However, downregulation of IRF4 may not be a general mechanism for fusion protein efficacy against MM as we did not observe decreased IRF4 levels in 3 other HMCLs treated with anti-CD138-IFNα2 or anti-CD138-IFNα2YNS (unpublished observations). Nevertheless, it would be interesting to expand upon these studies to elucidate the mechanism by which IFNα2 downregulates IRF4 expression in U266. Decreases in IRF4 does not affect cell cycle progression in SKMM1 cells (Shaffer and others 2008), and we found this to be the case for U266 (data not shown). Knockdown of IRF4 expression by RNA interference in MM has been reported to result in nonapoptotic cell death (Shaffer and others 2009). So in addition to proapoptotic activity, IFNα fusion proteins may have other antiproliferative effects.
One of the major challenges in translational research is to determine if in vitro assays are predictive of in vivo outcome. In the case of IFNα2 fusion proteins, targeting improved the efficacy of IFNα treatment both in vitro and in vivo. Anti-CD138-IFNα2 was more effective than untargeted anti-DNS-IFNα2 and anti-CD138-IFNα2YNS was more effective than untargeted anti-CD20-IFNα2YNS against U266 tumors in NSG mice. In addition, increasing the treatment regimen from 3 to 6 doses also improved survival.
Some interesting observations emerged from the in vivo studies. One was that treatment with anti-CD138-IFNα2 and anti-CD138-IFNα2YNS showed similar levels of protection in mice. This is in contrast to what was observed in our in vitro studies in which the higher affinity anti-CD138-IFNα2YNS was more effective than anti-CD138-IFNα2 against U266 cells in the MTS and the apoptosis assays. However, when we tested for the induction of apoptosis at low concentration (1 pM) of fusion proteins, we found that anti-CD138-IFNα2 and anti-CD138-IFNα2YNS had comparable ability to induce apoptosis (data not shown). One explanation for the discrepancy between the in vivo and in vitro data may be that the lower concentration more accurately reflects the in vivo situation. In addition, we found that although anti-CD138 did not display antitumor activity in vitro (Figs. 2 and 6 and data not shown), anti-CD138 provided a significant level of protection in mice and was more protective than untargeted IFNα2 fusion proteins. These data suggest that the antitumor activity may be achieved partly through the effector functions of IgG, which would be observable in vivo, but not in vitro. Although NSG mice are severely immunocompromised, they do contain functional monocytes and neutrophils (Racki and others 2010), which may be involved in tumor killing through antibody-dependent cell-mediated cytotoxicity (ADCC) (Ravetch and Kinet 1991). Therefore, the fusion proteins may prove to be even more effective in the treatment of human patients since the immunomodulatory activities of human IFNα2 and effector functions such as complement-dependent cytotoxcity and ADCC associated with the human IgG Fc region are not fully functioning in mice.
Our studies have shown that targeting of IFNα2 and higher affinity IFNα2YNS through the anti-CD138 moiety can be an effective strategy in the treatment of MM. Fusion of IFNα2 and IFNα2YNS to anti-CD138 should increase their half-life and decrease the systemic side effects of IFNα2, making for an effective therapeutic against MM.
Supplementary Material
Acknowledgments
This work was supported by the Senior Research Award (S.L.M.), the Dean Assink/Multiple Myeloma Research Foundation Senior Research Award (S.L.M.), the National Institutes of Health (grants 2RO1CA111448, 1RO1CA132778 and 1R21CA168491 to A.L.), and grants from the Multiple Myeloma Research Foundation (A.L.), and the Veteran's Administration (A.L.).
Author Disclosure Statement
No competing financial interests exist.
References
- Anderson KC. 2012. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol 30(4):445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden EC, Lindner D, Dreicer R, Hussein M, Peereboom D. 2000. Second-generation interferons for cancer: clinical targets. Semin Cancer Biol 10(2):125–144 [DOI] [PubMed] [Google Scholar]
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 6(12):975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence J, Jr., Abraham RT. 1996. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J 15(19):5256–5267 [PMC free article] [PubMed] [Google Scholar]
- Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, Stewart JP, Zhan F, Khatry D, Protopopova M. 2006. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell 9(4):313–325 [DOI] [PubMed] [Google Scholar]
- Chilosi M, Adami F, Lestani M, Montagna L, Cimarosto L, Semenzato G, Pizzolo G, Menestrina F. 1999. CD138/syndecan-1: a useful immunohistochemical marker of normal and neoplastic plasma cells on routine trephine bone marrow biopsies. Mod Pathol 12(12):1101–1106 [PubMed] [Google Scholar]
- Crowder C, Dahle O, Davis RE, Gabrielsen OS, Rudikoff S. 2005. PML mediates IFN-alpha-induced apoptosis in myeloma by regulating TRAIL induction. Blood 105(3):1280–1287 [DOI] [PubMed] [Google Scholar]
- Drexler H, Matsuo Y. 2000. Malignant hematopoietic cell lines: in vitro models for the study of natural killer cell leukemia-lymphoma. Leukemia 14(5):777–782 [DOI] [PubMed] [Google Scholar]
- Fritz E, Ludwig H. 2000. Interferon-alpha treatment in multiple myeloma: meta-analysis of 30 randomised trials among 3948 patients. Ann Oncol 11(11):1427–1436 [DOI] [PubMed] [Google Scholar]
- Gomez-Benito M, Balsas P, Bosque A, Anel A, Marzo I, Naval J. 2005. Apo2L/TRAIL is an indirect mediator of apoptosis induced by interferon-alpha in human myeloma cells. FEBS Lett 579(27):6217–6222 [DOI] [PubMed] [Google Scholar]
- Hjortsberg L, Lindvall C, Corcoran M, Arulampalam V, Chan D, Thyrell L, Nordenskjold M, Grander D, Pokrovskaja K. 2007. Phosphoinositide 3-kinase regulates a subset of interferon-alpha-stimulated genes. Exp Cell Res 313(2):404–414 [DOI] [PubMed] [Google Scholar]
- Huang TH, Chintalacharuvu KR, Morrison SL. 2007. Targeting IFN-alpha to B cell lymphoma by a tumor-specific antibody elicits potent antitumor activities. J Immunol 179(10):6881–6888 [DOI] [PubMed] [Google Scholar]
- Iida S, Rao PH, Butler M, Corradini P, Boccadoro M, Klein B, Chaganti R, Dalla-Favera R. 1997. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat Genet 17(2):226–230 [DOI] [PubMed] [Google Scholar]
- Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, Uze G, Schreiber G. 2006. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol 26(5):1888–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaks E, Gavutis M, Uze G, Martal J, Piehler J. 2007. Differential receptor subunit affinities of type I interferons govern differential signal activation. J Mol Biol 366(2):525–539 [DOI] [PubMed] [Google Scholar]
- Kalie E, Jaitin DA, Abramovich R, Schreiber G. 2007. An interferon alpha2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J Biol Chem 282(15):11602–11611 [DOI] [PubMed] [Google Scholar]
- Lamken P, Lata S, Gavutis M, Piehler J. 2004. Ligand-induced assembling of the type I interferon receptor on supported lipid bilayers. J Mol Biol 341(1):303–318 [DOI] [PubMed] [Google Scholar]
- Lombardi L, Poretti G, Mattioli M, Fabris S, Agnelli L, Bicciato S, Kwee I, Rinaldi A, Ronchetti D, Verdelli D. 2006. Molecular characterization of human multiple myeloma cell lines by integrative genomics: insights into the biology of the disease. Genes Chromosomes Cancer 46(3):226–238 [DOI] [PubMed] [Google Scholar]
- Moreaux J, Klein B, Bataille R, Descamps G, Maiga S, Hose D, Goldschmidt H, Jauch A, Reme T, Jourdan M. 2011. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica 96(4):574–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaretakis T, Hjortsberg L, Tamm KP, Bjorklund AC, Joseph B, Grander D. 2008. Interferon alpha induces nucleus-independent apoptosis by activating extracellular signal-regulated kinase 1/2 and c-Jun NH2-terminal kinase downstream of phosphatidylinositol 3-kinase and mammalian target of rapamycin. Mol Biol Cell 19(1):41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg-Shulman T, Tsubery H, Mironchik M, Fridkin M, Schreiber G, Shechter Y. 2004. Reversible PEGylation: a novel technology to release native interferon alpha2 over a prolonged time period. J Med Chem 47(20):4897–4904 [DOI] [PubMed] [Google Scholar]
- Racki WJ, Covassin L, Brehm M, Pino S, Ignotz R, Dunn R, Laning J, Graves SK, Rossini AA, Shultz LD. 2010. NOD-scid IL2R gamma-null (NSG) mouse model of human skin transplantation and allograft rejection. Transplantation 89(5):527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravetch JV, Kinet J-P. 1991. Fc receptors. Annu Rev Immunol 9(1):457–492 [DOI] [PubMed] [Google Scholar]
- Ridley R, Xiao H, Hata H, Woodliff J, Epstein J, Sanderson R. 1993. Expression of syndecan regulates human myeloma plasma cell adhesion to type I collagen. Blood 81(3):767–774 [PubMed] [Google Scholar]
- Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H. 2008. IRF4 addiction in multiple myeloma. Nature 454(7201):226–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer AL, Emre NT, Romesser PB, Staudt LM. 2009. IRF4: Immunity. Malignancy! Therapy? Clin Cancer Res 15(9):2954–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, Einhorn S, Grander D. 2002. Mechanisms of interferon-alpha induced apoptosis in malignant cells. Oncogene 21(8):1251–1262 [DOI] [PubMed] [Google Scholar]
- Thyrell L, Hjortsberg L, Arulampalam V, Panaretakis T, Uhles S, Dagnell M, Zhivotovsky B, Leibiger I, Grander D, Pokrovskaja K. 2004. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. J Biol Chem 279(23):24152–24162 [DOI] [PubMed] [Google Scholar]
- Trinh KR, Vasuthasawat A, Steward KK, Yamada RE, Timmerman JM, Morrison SL. 2013. Anti-CD20-interferon-alpha fusion protein therapy of murine B-cell lymphomas. J Immunother 36(5):305–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdelli D, Nobili L, Todoerti K, Intini D, Cosenza M, Civallero M, Bertacchini J, Deliliers GL, Sacchi S, Lombardi L, Neri A. 2009. Molecular targeting of the PKC-beta inhibitor enzastaurin (LY317615) in multiple myeloma involves a coordinated downregulation of MYC and IRF4 expression. Hematol Oncol 27(1):23–30 [DOI] [PubMed] [Google Scholar]
- Weiss K. 1998. Safety profile of interferon-alpha therapy. Semin Oncol 25(1 Suppl 1):9–13 [PubMed] [Google Scholar]
- Wijdenes J, Vooijs WC, Clement C, Post J, Morard F, Vita N, Laurent P, Sun RX, Klein B, Dore JM. 1996. A plasmocyte selective monoclonal antibody (B-B4) recognizes syndecan-1. Br J Haematol 94(2):318–323 [DOI] [PubMed] [Google Scholar]
- Xuan C, Steward KK, Timmerman JM, Morrison SL. 2010. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood 115(14):2864–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Nakazawa N, Iida S, Hayami Y, Sato S, Wakita A, Shimizu S, Taniwaki M, Ueda R. 1999. Detection of MUM1/IRF4-IgH fusion in multiple myeloma. Leukemia 13(11):1812–1816 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.