

Abstract
Tuberculosis (TB), a chronic infectious disease mainly caused by the tubercle bacillus Mycobacterium tuberculosis, is one of the world's deadliest diseases that has afflicted humanity since ancient times. Although the number of people falling ill with TB each year is declining, its incidence in many developing countries is still a major cause of concern. Upon invading host cells by phagocytosis, M. tuberculosis can replicate within infected cells by arresting the maturation of the phagosome whose function is to target the pathogen for elimination. Host cells have mechanisms of controlling this evasion by inducing autophagy, an elaborate cellular process that targets bacteria for progressive elimination, decreasing bacterial loads within infected cells. In addition, autophagy activation also aids in the control of inflammation, contributing to a more efficient innate immune response against M. tuberculosis. Several innovative TB therapies have been envisaged based on autophagy manipulation, with some of them revealing high potential for future clinical trials and eventual implementation in healthcare systems. Thus, this review highlights the recent advances on the innate immune response regulation by autophagy upon M. tuberculosis infection and the promising new autophagy-based therapies for TB.
Mycobacterium tuberculosis: Biology and Infection
Mycobacterium tuberculosis, the major causative agent of tuberculosis (TB), is estimated to latently infect one-third of the world's population and continues to claim more lives than any single other bacterial pathogen, despite the availability of drugs since 1944 when streptomycin was first administered to a TB patient. Although only a small percentage of the latently infected individuals will ever develop active disease, this translates into a huge number of 8 million new TB cases and nearly 1.5 million deaths per year.
TB is often regarded as a developing world disease where debilitated healthcare systems, together with the HIV epidemic, allow it to remain rampant. In addition, the increasing incidence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) TB, which in some countries accounts for over 20% of new infections, has the potential to exert a heavy toll also in developed countries (WHO Global tuberculosis report 2014).
M. tuberculosis and all other species of mycobacteria, many of which are opportunistic intracellular pathogens, owe much of their resilience to a distinctive lipid-rich cell envelope that not only protects the cells against harsh environments but also contains many molecules that are immune effectors crucial in evading the host immune response (Briken et al., 2004; Ishikawa et al., 2009; Schafer et al., 2009; Court et al., 2010; Ehlers, 2010; Philips and Ernst, 2012; Lang, 2013; Nobre et al., 2014). Besides glycolipids and glucans that constitute most of the M. tuberculosis cell envelope, several secreted proteins have been identified and shown to also play fundamental roles in M. tuberculosis survival and proliferation within the host (Abdallah et al., 2007; Philips, 2008; Philips and Ernst, 2012).
This extremely successful human pathogen enters our bodies through inhalation of aerosols containing M. tuberculosis cells. This organism then employs an array of immune modulators to invade and thrive in the host professional phagocytic cells, such as macrophages, neutrophils, monocytes, and dendritic cells (DCs), by arresting phagosome maturation and fusion with lysosomes (Cooper, 2009; Ernst, 2012). While for many other pathogens the recruitment of phagocytic cells to the infection site halts and eradicates invading organisms, in mycobacterial infections it actually helps the pathogen to proliferate by providing further cells for infection (Ernst, 2012; Philips and Ernst, 2012). As recruited cells get infected, the host immune system coordinates the edification of specific protective structures named granulomas, the histological hallmark of this disease (Ernst, 2012; Philips and Ernst, 2012). Granulomas are traditionally viewed as an attempt by the host to control the infection that is achieved with variable degrees of success (Davis and Ramakrishnan, 2009; Philips and Ernst, 2012; Lin et al., 2014). The dogma claims that M. tuberculosis resides inside phagosomes; however, some reports have described that this pathogen can also grow in the cytoplasm environment (van der Wel et al., 2007). The bacteria that replicate inside phagocytes control cell death pathways toward necrosis and recruit more noninfected macrophages, expanding the infection (Lee et al., 2011; Philips and Ernst, 2012).
Understanding how this organism evades and exploits our immune defense mechanisms has the potential to significantly change how we tackle this disease, improving the lives and health of millions worldwide.
Autophagy: A General Antibacterial Host Mechanism
Macroautophagy (hereafter referred as autophagy) is a mechanism that relies on the formation of a double-membrane vesicle, the autophagosome, which engulfs components of the cytoplasm and delivers them to degradation in the lysosome. Autophagy is crucial for the maintenance of cellular homeostasis by continuously degrading damaged organelles, long-lived proteins, protein aggregates, and intracellular pathogenic microorganisms (Mizushima, 2011). It also provides a way of recycling nutrients, which then participate in de novo protein synthesis and energy production. Autophagy can be induced by a variety of stimuli and/or environmental stresses such as nutrient starvation, low oxygen levels, oxidative stress, pathogen infection, and certain drugs treatments (Bento et al., 2013; Gomes and Dikic, 2014).
Autophagy-related (ATG) proteins are the key players in the regulation of autophagy, being hierarchically organized in functional complexes that control all the autophagy steps, from the initiation signaling point to the autophagosome fusion with the lysosome. The formation of new autophagosomes is triggered and assisted by a core of ATG proteins that can be subdivided in some groups: (1) the unc-51-like kinase (ULK) complex, comprising the mammalian Atg1 orthologs ULK1 and ULK2, ATG13, and the focal adhesion kinase family-interacting protein of 200 kDa (FIP200), which is controlled by AMP-activated protein kinase (AMPK) and mechanistic target of rapamycin (mTOR), both being responsive to signals, such as amino acids and glucose availability, growth factors stimulation, stress conditions, and AMP/ATP energetic status of the cell; (2) class III phosphatidylinositol 3-kinase (PI3K) complex (controlled through phosphorylation by the ULK complex) comprising VPS34, ATG6 (also known as Beclin-1), ATG14 (also known as Barkor), and VPS15 (also known as p150), which is involved in the synthesis of phosphatidylinositol 3-phosphate (PI3P), whose function in autophagy is not very clear, but seems to favor the recruitment of WD repeat domain phosphoinositide-interacting proteins (WIPIs in mammals and Atg18 in yeast) to the phagophore membrane, marking membranes for autophagosome nucleation; (3) ATG9, which is involved in the supply of lipid bilayers to the formation of autophagosomes; (4) ATG12-ATG5-ATG16L1 complex formed by an ubiquitination-like reaction where ATG12 is conjugated to ATG5 (by a mechanism dependent on ATG7 and ATG10, which act similar to an E1ubiquitin-activating enzyme and an E2 ubiquitin-conjugating enzyme, respectively), which is then associated with ATG16L1 and subsequently with the nascent phagophore; and (5) the ubiquitin-like microtubule-associated protein 1 light chain 3 (LC3 in mammals or Atg8 in yeast) family system where pro-LC3 is cleaved by ATG4B, resulting in LC3-I, which is then conjugated to phosphatidylethanolamine (PE) by ATG7 and ATG3 to form LC3-II, the autophagosome-associated form of LC3. The ATG12-ATG5-ATG16L1 complex enhances the recruitment of LC3 to the site of lipidation and conjugation of LC3 to PE (Fig. 1). LC3-II is thought to be involved in the elongation and closure of the autophagosome and it is also important in the recruitment of cargo by a mechanism dependent on proteins similar to P62 (also known as sequestosome 1 or SQSTM1) and the neighbor of the BRCA gene 1 (NBR1), containing LC3-interacting region (LIR) and ubiquitin-associated (UBA) domains. These types of proteins recognize ubiquitin-tagged substrates through their UBA domains and interact with LC3-II through the LIR domain, acting as cargo adapters for ubiquitinated proteins that can be degraded by autophagy (Bento et al., 2013; Gomes and Dikic, 2014).
FIG. 1.

The autophagy pathway and its main regulators. Autophagy is typically subdivided in different steps: (i) vesicle nucleation/initiation, (ii) phagophore elongation, (iii) autophagosome maturation, (iv) autophagosome/lysosome fusion, and (v) cargo degradation. Following AMPK activation and/or mTORC1 inhibition (by factors such as nutrient depletion and energetic stress), the complex formed by ULK1/2, FIP200, and ATG13 is activated, which in turn activates the VPS34 complex by phosphorylation. Both complexes regulate the nucleation/initiation step of autophagy, with VPS34 providing PI3P to the phagophore, which is likely to assist the recruitment of WIPI to the phagophore. On the other hand, membrane expansion depends on ATG9, which is postulated to supply lipid bilayers to the phagophore, and on two ubiquitin-like conjugation systems that conjugate LC3 and ATG12 to PE and ATG5, respectively. The ATG12-ATG5 complex further interacts with ATG16L1, presumably at the surface of the autophagosome membrane. LC3-II seems to be involved in the elongation and closure of the autophagosome membrane, as well as in the recruitment of cargo to the phagophore. Subsequently, the autophagosome fuses with the lysosome, forming the autolysosome, where degradation of the autophagosomal contents occurs. AMPK, AMP-activated protein kinase; FIP200, focal adhesion kinase family-interacting protein of 200 kDa; mTORC1, mechanistic target of rapamycin complex 1; PE, phosphatidylethanolamine; ULK, unc-51-like kinase.
The formation of autophagosomes can be regulated by a wide variety of signals that are usually categorized into mTOR dependent and mTOR independent (Sarkar, 2013). mTOR is a classic negative regulator of autophagy and its activity is canonically inhibited by starvation or rapamycin treatment, which leads to activation of the ATG13-ULK1/ULK2-FIP200 complex, thereby inducing autophagy. ULK1 phosphorylation/activation by AMPK and upregulation of the phosphatase and tensin homologue (PTEN) through inhibition of AKT kinase, both induced by P53, also inhibit mTOR. AMPK can also phosphorylate the tuberous sclerosis complex 2 (TSC2), which impacts on the activity of the TSC-Ras homology enriched in the brain (RHEB) axis, ultimately leading to mTOR inactivation and autophagy induction. Among the mTOR-independent mechanisms, inhibition of inositol monophosphatase (IMPase), which reduces the levels of free inositol and inositol (1,4,5)-triphosphate, and activation of AMPK through Ca2+-transfer from the endoplasmic reticulum to the mitochondria are some of the most well-characterized mechanisms regulating autophagy (Ravikumar et al., 2009; Bento et al., 2013).
With particular interest for the context of this review, autophagy constitutes a cell-autonomous defense mechanism against a wide range of intracellular pathogens from bacteria (i.e., M. tuberculosis, Streptococcus pyogenes, Shigella flexneri, Salmonella enterica) to protozoa and viruses (Gomes and Dikic, 2014).
In the specific case of bacteria, after invading host cells they reside within vacuoles or phagosomes, whose maturation tends to be blocked. Eventually, some bacteria damage the membrane of the phagosome and escape into the cytosol. Bacteria in damaged phagosomes or in the cytosol can then be targeted to autophagy and degradation in the lysosome by a mechanism that relies on the binding of ubiquitin or galectin to bacteria and/or to the membrane of phagosome-containing bacteria, which are recognized by the autophagic adaptors P62, NBR1, optineurin or calcium binding and coiled-coil domain 2 (CALCOCO2 or NDP52). However, some bacteria have the ability to manipulate autophagy for survival by secreting effectors that inhibit the pathway (Gomes and Dikic, 2014; Huang and Brumell, 2014). In the next sections, we will revise the main findings that have implicated autophagy in the clearance of mycobacteria as well as some of the mycobacterial defense mechanisms against elimination by host cells and potential TB therapeutics based on autophagy antimicrobial responses.
Autophagy: A Host Defense Mechanism Against M. tuberculosis
M. tuberculosis is an intracellular pathogen that can replicate within infected macrophages by arresting the maturation of the phagosome where the bacteria reside. This is, at least in part, attributed to the failure of phagosomes to undergo fusion with lysosomes by selective exclusion of RAB7 GTPase and lysosomal-associated membrane protein 1 (LAMP1) (markers of late endosome and lysosome) coupled with the retention of RAB5 (an early endosome marker) on the phagosome, which allows M. tuberculosis to avert the usual physiological destination of phagocytosed material (Via et al., 1997). In addition, M. tuberculosis also interferes with the delivery of V-ATPase subunits and lysosomal hydrolases from the trans-Golgi network (TGN) to the phagocytic compartment, which impacts on its biogenesis and function. This is connected to the fact that M. tuberculosis produces an array of lipids and lipoglycans (i.e., lipoarabinomannan) that mimic certain mammalian phosphatidylinositols important for the synthesis of PI3P through VPS34, which not only inhibits autophagy but also blocks PI3P-dependent trafficking pathways, such as the one between the TGN and the phagosome (Fratti et al., 2003; Vergne et al., 2004; Shui et al., 2011). M. tuberculosis also secretes a tyrosine phosphatase (MptpA) that further reduces the phagosomal levels of PI3P and blocks phagosome/lysosome fusion by interacting with vacuolar protein sorting 33b (VPS33b), a host protein typically associated with vesicle trafficking steps in the endosome/lysosome pathway (Vergne et al., 2005; Bach et al., 2008) (Fig. 2).
FIG. 2.

M. tuberculosis clearance by autophagy. M. tuberculosis invades macrophages by phagocytosis and arrests the maturation of the phagosome by excluding late endosome and lysosome markers (i.e., RAB7, V-ATPase, VPS33b, LAMP1) from the phagosome and by promoting the retention of early endosome markers (i.e., RAB5) in the phagocytic compartment. Host cells have developed ways of overcoming the evasion of M. tuberculosis from the phagocytic pathway by taking advantage of some intrinsic M. tuberculosis mechanisms. For instance, phagosomal permeabilization induced by the bacterial ESX-1/ESAT-6 system allows the host protein STING to recognize extracellular bacterial DNA, which then promotes ubiquitin marking of bacteria (mostly through K63-linkage chain formation by the E3 ligase Parkin). Ubiquitin is then recognized by autophagy adaptors, such as P62, which deliver the bacilli to autophagosomes. TBK-1-induced phosphorylation of Ser403 of P62 increases the affinity of P62 to ubiquitin. Autophagosomes are subsequently fused to lysosomes, where degradation of mycobacteria occurs. LAMP1, lysosomal-associated membrane protein 1; STING, stimulator of the interferon gene; TBK-1, TANK-binding kinase 1.
Nutrient starvation, a conventional inducer of the VPS34 kinase complex, has been shown to act as an effective promoter of biogenesis, acidification, and maturation of mycobacterial phagosomes by increasing the delivery of late endosome/lysosome markers (e.g., vacuolar-type H+-ATPase or V-ATPase, LAMP1, and cathepsin D) and the recruitment of membrane-associated LC3 to the phagosome, which directly impacts on the survival of mycobacteria in infected cells (Gutierrez et al., 2004). Interferon-γ (IFN-γ), a cytokine associated with protective immunity against M. tuberculosis, and the immunity-related GTPase family, M (IRGM or LRG-47), a downstream effector of IFN-γ, show similar effects to starvation, while the PI3K inhibitors, 3-methyladenine (3-MA) and wortmannin, abrogate this response (Gutierrez et al., 2004). Depletion of Beclin-1 and ATG7, critical autophagy regulators, also inhibits IFN-γ- or IRGM1-induced phagosomal maturation (Singh et al., 2006). Therefore, immunological or pharmacological VPS34- or autophagy-targeted manipulation can render infected cells more resistant to mycobacterial infection.
Besides inhibiting VPS34, mycobacteria have other ways of protecting themselves against autophagy-mediated clearance. For instance, the enhanced intracellular survival (eis) gene enhances the survival of M. tuberculosis and other mycobacteria inside cells by diminishing autophagy and proinflammation (Wei et al., 2000; Shin et al., 2010a). Infection of macrophages with an eis-deleted M. tuberculosis strain (Mtb-Δeis) was indeed shown to augment the formation of LC3-positive and double-membrane vesicles (autophagosomes) compared with the Mtb-WT strain. These vesicles enclose the bacilli and subsequently fuse with multivesicular structures, leading to the formation of late or degradative autophagic vacuoles, which correspond to autolysosomes. In addition, Mtb-Δeis also upregulates the production of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), and the generation of reactive oxygen species (ROS) by a c-Jun N-terminal kinase (JNK)-dependent mechanism in host macrophages, being the last event triggering autophagy and proinflammation (Shin et al., 2010a). Interestingly, the Eis protein secreted by M. tuberculosis appears to enhance survival of other mycobacteria, namely Mycobacterium smegmatis, in macrophages. M. tuberculosis-Eis is an efficient Nɛ-acetyltransferase rapidly acetylating Lys55 of the dual-specificity protein phosphatase 16 (DUSP16; also known as MKP-7), a JNK-specific phosphatase, whereas M. smegmatis-Eis is more efficient as an Nα-acetyltransferase and preferentially acetylates the terminal amino group of peptides (Kim et al., 2012b). This difference between both proteins is likely to be explained by a structural dissimilarity in the peptide recognition pocket of the enzymes; M. tuberculosis-Eis is characterized by the presence of a narrow channel, while M. smegmatis-Eis has a deep, round-shaped substrate-binding pocket, which seems more suitable for accommodating the terminal amino group of peptides than specific sequences within proteins (Kim et al., 2012b). This structural difference seems, indeed, to have a physiological impact on the survival of both species. While M. tuberculosis-Eis significantly downregulates lipopolysaccharide-induced JNK phosphorylation, M. smegmatis-Eis does not reveal this function. Therefore, acetylation of DUSP16 by M. tuberculosis-Eis seems to be the key initial event in the JNK-dependent inhibition of autophagy, phagosome maturation, and ROS generation, which ultimately contribute to enhanced survival of M. tuberculosis within the macrophage cells (Kim et al., 2012b). This also suggests that M. tuberculosis developed adaptive evolutionary strategies to potentiate the suppression of the host innate immune system.
M. tuberculosis has indeed been suggested to pervert the function of some intrinsic host mechanisms favoring the survival of the bacteria in macrophages. This is the case observed for coronin 1a (CORO1A), a host F-actin-binding protein, which seems to be activated by M. tuberculosis and inhibit autophagosome formation around the bacillus-containing phagosomes, most likely by most likely by inhibiting the activation of p38 mitogen-activated protein kinase (p38 MAPK) necessary for autophagy induction through Toll-like receptor (TLR) signaling pathways in innate immunity (Seto et al., 2012).
However, host adaptive responses have also evolved to take advantage of some intrinsic mycobacteria mechanisms such as the type VII secretion system ESX-1 (Watson et al., 2012). Recruitment of LC3 to Mycobacterium bovis bacilli Calmette Guerin (BCG)-containing vacuoles depends on exogenous stimulation of autophagy (Gutierrez et al., 2004; Singh et al., 2006; Watson et al., 2012). On the contrary, targeting of LC3 to M. tuberculosis-containing vacuoles seems to be triggered without any extrinsic stimulatory signal. One of the major differences between BCG and virulent mycobacteria is the lack of ESX-1 from the BCG strain, which may explain their different effectiveness in replicating within macrophages and in activating innate immune responses by the host (Harboe et al., 1996).
Mycobacterium marinum has been used in many studies as a surrogate model of M. tuberculosis due to its relative safety for humans (fish and amphibians are the preferred hosts) and its shared mechanisms of pathogenesis (Tobin and Ramakrishnan, 2008). Although ESX-1 is present in both pathogenic species, it seems to play different roles in targeting bacteria to degradation. For instance, ESX-1 is required for the total vacuolar escape of M. marinum (contributing to an intracellular phase where it resides in the cytosol without being enclosed by any membrane organelle), which becomes ubiquitinated and sequestered by LAMP1-positive host vesicles by a mechanism that seems to be ATG5 independent and to occur in the absence of LC3-membrane association (Collins et al., 2009). In contrast, ESX-1 has been suggested to induce M. tuberculosis-containing phagosome permeabilization/perforation, which allows cytosolic components of the ubiquitin-mediated autophagy pathway access to the enclosed bacillus. Stimulator of the interferon gene (STING), which works both as a direct cytosolic DNA sensor and an adaptor protein that functions upstream of TANK-binding kinase 1 (TBK-1) in type 1 interferon signaling, recognizes extracellular bacterial DNA (which has been shown to be exposed during macrophage infection) and promotes ubiquitin tagging of the bacilli (mostly through K63-linkage) and subsequent LC3 recruitment to the phagosome by a mechanism dependent on the ubiquitin-autophagy adaptors P62 and NDP52 (Watson et al., 2012). This leads to the formation of bacilli-containing autophagosomes that mature through fusion with lysosomes to create autolysosomes (Fig. 2). In opposition to M. marinum, this mechanism was shown to require the activity of ATG5 as macrophages with ATG5 genomic deletion were unable to recruit LC3 to phagosomes containing M. tuberculosis and induce their maturation into autolysosomes, as assessed by a strong decrease of LAMP1 colocalization (Collins et al., 2009; Watson et al., 2012). This whole mechanism is clearly dependent on ESX-1 since an M. tuberculosis mutant defective in early secreted Ag of 6 kDa (ESAT-6) (the major ESX-1-secreted substrate with membrane-damaging activity) fails to recruit LC3 to the phagosome. Therefore, M. tuberculosis clearance by the autophagy-lysosomal pathway provides a way of cell-autonomous control of bacterial replication within macrophages and appears to be fundamental for the host survival upon M. tuberculosis infection. Indeed, Atg5−/− deletion renders mice extremely sensitive to M. tuberculosis as all mice succumb to infection by 4 weeks postinfection and a 1000-fold increase in bacilli replication is observed within the lungs compared with Atg5+/+-infected mice (Watson et al., 2012). However, it is interesting to note that only one-third of intracellular bacteria are targeted by the ubiquitin-autophagy pathway, which suggests that infection control may be potentiated by strategies that augment autophagy activity in the host cells and avoid autophagy evasion by the remaining bacilli.
TBK-1 also plays a critical role in regulating mycobacteria clearance by autophagy and cell defense against mycobacteria-triggered infection (Pilli et al., 2012). TBK-1 was previously shown to control the type I interferon response elicited by intracellular DNA and, more recently, to orchestrate autophagy clearance of mycobacteria by a mechanism dependent on RAB8b, optineurin, and P62. TBK-1 depletion does not suppress formation of autophagosomes, but suppresses their maturation into autolysosomes due to inhibited delivery of lysosomal hydrolases to the autophagosomal compartment. In addition, TBK-1 also triggers phosphorylation of serine 403 in the UBA domain of P62, strongly increasing its affinity for ubiquitin-tagged substrates, such as mycobacteria tagged with K63-linked ubiquitin chains, which induces their elimination by autophagy (Fig. 2). Of interest is also the fact that TBK-1 is required for IL-1β-induced clearance of M. tuberculosis by autophagy since the TBK-1 inhibitor BX795 or TBK-1 depletion reduces mycobacterial killing when autophagy is induced by IL-1β (Pilli et al., 2012).
Elimination of M. tuberculosis through autophagy has been clearly elucidated as an ubiquitin-dependent mechanism where phagosomes enclosing bacteria are tagged with ubiquitin chains, which are subsequently recognized by the autophagy adaptors, P62 and NDP52, that recruit all the autophagy machinery necessary for their degradation (Gomes and Dikic, 2014; Huang and Brumell, 2014). However, the E3 ligase (Parkin) that triggers K63-ubiquitination of mycobacteria was only recently identified. Parkin (also known as PARK2) mutations and polymorphisms, apart from being well-known Parkinson's disease risk factors, are also associated with increased susceptibility to Mycobacterium leprae infection (Mira et al., 2004). In fact, Parkin is important for the host defense against M. tuberculosis by promoting xenophagy, by a mechanism similar to the one that implicates the protein in mitophagy induction (Geisler et al., 2010; Youle and Narendra, 2011). This resemblance is likely to be explained by the fact that endosymbiotic bacteria are the most probable evolutionary origin of mitochondria, which suggests an evolutionarily conserved role for Parkin (Manzanillo et al., 2013).
The major evidences implicating Parkin in M. tuberculosis xenophagy showed that infected Park2−/− macrophages present a significant reduction in ubiquitin-positive mycobacteria compared with normal cells (Manzanillo et al., 2013). While expression of wild-type Park2 in Park2−/− macrophages restored ubiquitin localization around mycobacterial cells, two parkin pathogenic RING domain mutants with no E3 ligase activity (T240R and P437L) failed to do so (Manzanillo et al., 2013). Infected Park2−/− macrophages also revealed decreased recruitment of the ubiquitin adaptors, P62, NDP52, and NBR1, and the autophagy proteins, LC3 and ATG12, to mycobacterial cells, compromising their efficient elimination. Parkin deficiency indeed increased bacterial viability and replication within infected macrophages, being Park2−/− mice extremely susceptible to M. tuberculosis; all infected Park2-deficient mice succumbed to M. tuberculosis infection by 85 days postinfection, whereas all infected wild-type mice remained alive and with no overt signs of stress (Manzanillo et al., 2013).
Cross Talk Between Innate Immunity and Autophagy in Tuberculosis
Induction of cytokine expression is a key host defense mechanism against M. tuberculosis infection and can be triggered by activation of TLR- and non-TLR-dependent signaling cascades (Jo, 2013). Apart from regulating other defense responses that are beyond the scope of this review, IFN-γ was shown to be an important cytokine in the regulation of mycobacteria clearance by autophagy, while the cytokines, IL-4 and IL-13, seem to inhibit this effect (Harris et al., 2007). On the other hand, TNF-α appears to synergize the antimicrobial and autophagic responses triggered by IFN-γ (Harris et al., 2008), while many other cytokines positively aid in the autophagic response against mycobacteria (i.e., TNF-β, IL-2, IL-6, CCL2) (Harris, 2011). Interestingly, maturation of M. tuberculosis-containing phagosomes induced by IFN-γ was shown to be abrogated by the TNF blockers adalimumab, etanercept, and infliximab (Harris et al., 2008). Overall, T helper-1 (Th1) cytokines appear to induce autophagy, whereas the Th2 cytokines, IL-4 and IL-13, seem to inhibit it. Apart from inhibiting IFN-γ-induced autophagy, IL-4 and IL-13 also inhibit starvation-induced autophagy in a way dependent on protein kinase B (AKT) (Harris et al., 2007). Therefore, a precise balance of different cytokines is critical for an efficient host response against M. tuberculosis.
TLR-dependent signaling pathways account for the maintenance of this balance. For instance, the receptors TLR2, TLR4, and TLR9, appear to be the main TLRs implicated in the recognition of mycobacteria and production of antimicrobial effectors and cytokines upon M. tuberculosis infection (Kleinnijenhuis et al., 2011). TLR2, TLR4, and TLR9 stimulation was interestingly shown to induce maturation of bacterium-containing phagosomes, activate autophagy, and increase degradation of bacteria (Sanjuan et al., 2007; Xu et al., 2007; Delgado et al., 2008) by mechanisms dependent on the myeloid differentiation primary response gene 88 (MyD88), TIR-domain-containing adapter-inducing interferon-β (TRIF), and MAPK (Xu et al., 2007; Delgado et al., 2008; Shi and Kehrl, 2008; Jo, 2013). Non-TLR pathways have also been implicated in regulation of autophagy upon M. tuberculosis infection. One of the examples is the signaling cascade triggered by NOD-like receptor 2 (NOD2), which is an intracellular receptor that recognizes bacterial molecules (i.e., peptidoglycan) and induces expression of proteins that upregulate autophagy, such as IRGM, LC3, and ATG16L1, contributing to decreased M. tuberculosis virulence (Juarez et al., 2012).
Regulation of cytokine production and autophagy activation seem to be mutually regulated by each other; as mentioned before, cytokines regulate autophagy, but the opposite is also true. For instance, autophagy seems to positively regulate the expression and secretion of TNF-α (Crisan et al., 2011; Jo, 2013) and to negatively regulate the secretion of several other proinflammatory cytokines, including IL-1α, IL-1β, and IL-18 (Saitoh et al., 2008; Crisan et al., 2011; Harris et al., 2011; Nakahira et al., 2011; Zhou et al., 2011; Jo, 2013). For the specific case of IL-1β, autophagy was shown to control its expression by different ways: increasing degradation of pro-IL-1β and inhibiting AIM2 and NLRP3 inflammasomes, which decrease IL-1β processing and secretion (Harris et al., 2011; Nakahira et al., 2011; Zhou et al., 2011; Bradfute et al., 2013; Shah et al., 2013). Although IL-1 is necessary for protection against mycobacteria, negative regulation of IL-1 by autophagy is likely to have beneficial effects to the infected cells as high levels of IL-1 are associated with excessive inflammation and pathology, suggesting that a precise control of IL-1 expression and release is needed for a successful response against infection (Bradfute et al., 2013). ATG5 deficiency in mice indeed causes an excessive pulmonary inflammatory response characterized by neutrophil infiltration and IL-17 response with increased IL-1α secretion (Castillo et al., 2012) (Fig. 3).
FIG. 3.
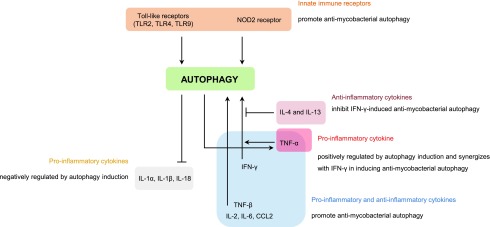
Cross talk between autophagy and inflammation during M. tuberculosis infection. Autophagy activation in macrophages is controlled by membrane and intracellular innate immune receptors, as well as by several inflammatory cytokines released by Th1 and Th2 cells upon M. tuberculosis infection. The receptors, TLR2, TLR4, TLR9, and NOD2, and the proinflammatory cytokines, TNF-α, IFN-γ, IL-2, IL-6, and CCL2, promote autophagy activation. On the other hand, the anti-inflammatory cytokines, IL-4 and IL-13, appear to inhibit IFN-γ-induced autophagy activation. However, cytokine expression and secretion are also regulated by autophagy. For instance, IL-1α, IL-1β, and IL-18 are negatively regulated by autophagy, while TNF-α is upregulated by this mechanism. IFN-γ, interferon-γ; IL, interleukin; NOD2, NOD-like receptor 2; Th1, T helper 1; Th2, T helper 2; TLR, Toll-like receptor; TNF-α, tumor necrosis factor-α.
In addition to acting as a modulator of proinflammatory cytokine secretion, autophagy also plays a role in antigen processing and presentation. In fact, autophagy was shown to be directly associated with enhanced delivery of intracellular material to the major histocompatibility complex class II pathway under mycobacteria infection (Jagannath et al., 2009). Rapamycin-induced autophagy indeed enhanced mycobacterial Ag85B presentation by antigen-presenting cells infected with M. tuberculosis, while suppression of autophagy by 3-MA or knockdown of Beclin-1 attenuated this effect (Jagannath et al., 2009).
Association Between Genetic Variants of Autophagy-Related Genes and Susceptibility to TB
Although Crohn's disease is considered to have an autoimmune origin, increasing evidence points to an infectious etiology involving mycobacteria (Greenstein, 2003). Some genome-wide association studies have indeed been suggesting an overlap between genetic susceptibility for inflammatory bowel disease, such as Chron's disease, and TB (Jostins et al., 2012). Therefore, it is not surprising that both diseases share similar profiles of genetic variants and risk factors. IRGM polymorphisms are an example as they were initially identified as autophagy risk loci for Chron's disease and more recently as TB risk factors in different populations (Wellcome Trust Case Control Consortium, 2007; King et al., 2011). In fact, genetic variants of the IRGM gene are the most consensually associated with TB infection, with at least five different genetic variants identified so far. Although the detailed mechanism by which this gene regulates autophagy is not clear, IRGM was shown to induce clearance of mycobacteria in infected macrophages by inducing phagosomal maturation and autophagy. Most of the variants are associated with increased protection against TB (Intemann et al., 2009; Che et al., 2010; Bahari et al., 2012). However, carriers of the Chron's disease-related T allele of rs10065172 reveal increased susceptibility to TB (King et al., 2011), while the −1208G/−1161T/−947T haplotype is also positively associated with the disease (Che et al., 2010).
TLR2 is another gene that has been associated with TB and, in opposition to IRGM variants, all the TLR2 variants identified so far appear to be risk factors for developing TB (Ben-Ali et al., 2004; Ogus et al., 2004; Etokebe et al., 2010). A variety of cell wall components of mycobacteria are known to activate macrophages through TLR2, suggesting that this innate immune receptor plays a role in the host response to M. tuberculosis infection (Drennan et al., 2004; Bowdish et al., 2009). The direct role of TLR2 activation in the regulation of autophagy is not well ascribed; however, several evidences have been suggesting that TLR2 activation is capable of inducing autophagy by a mechanism dependent on the activation of p38 MAPK (Seto et al., 2012). Interestingly, the R753Q TLR2 polymorphism was shown to render TLR2 incapable of inducing tyrosine phosphorylation and heterodimerization with TLR6 upon agonist binding, ultimately leading to impaired capacity of p38 and autophagy activation (Xiong et al., 2012).
Genetic polymorphisms in the vitamin D receptor (VDR) are also associated with predisposition to TB when combined with 25-hydroxycholecalciferol (calcidiol) deficiency (Wilkinson et al., 2000). Calcidiol is a precursor of calcitriol (1,25-dihydroxyvitamin D3), the active form of vitamin D, which has been shown to protect against M. tuberculosis infection through upregulation of autophagy (Yuk et al., 2009; Fabri et al., 2011; Campbell and Spector, 2012). Interestingly, regulation of autophagy by TLR2 seems to occur through the activation of VDR by binding to calcitriol, triggering the expression of the antimicrobial peptide, cathelicidin, which induces autophagy and promotes autophagosome/lysosome fusion (Shin et al., 2010b).
These and other autophagy-related genetic variants associated with TB are listed and summarized in Table 1.
Table 1.
Autophagy-Related Genetic Variants Associated with Tuberculosis
| Gene | Variant | TB-association | Reference* |
|---|---|---|---|
| VDR | Combination of TT or Tt genotype (Taq1) with 25-hydroxycholecalciferol deficiency; ff (Fok1) genotype | The combination of genotype TT/Tt and 25-hydroxycholecalciferol deficiency, and the genotype ff are positively associated with TB (individuals of Hindu and Gujarati origins). | Wilkinson et al. (2000) |
| TLR2 | Arg753Gln (AA genotype) | Carriers of this polymorphism present higher risk of developing TB (Turkish individuals). | Ogus et al. (2004) |
| TLR2 | Arg677Trp (C2029T) | This polymorphism is associated with increased susceptibility to TB (Tunisian individuals). | Ben-Ali et al. (2004) |
| IL-1β | −31position genotypes | A significant difference of IL-1β −31 genotypes was found between 98 TB patients and healthy controls. | Sun et al. (2007) |
| IRGM | −261TT | It confers protection against TB (Ghanaian individuals). | Intemann et al. (2009) |
| TLR2 | P631H | It is significantly overrepresented in TB when TB patients were compared with controls, indicating a possible low-risk predisposition (Croatian individuals). | Etokebe et al. (2010) |
| IRGM | −1208AA genotype and −1208G/−1161T/−947T haplotype | The −1208AA genotype is associated with decreased susceptibility to TB, while the −1208G/−1161T/−947T haplotype is positively associated with TB (Chinese individuals). | Che et al. (2010) |
| IRGM | rs10065172C/T | The carriers of the Chron's disease-related rs10065172C/T SNP present increased susceptibility to TB (African American individuals). | King et al. (2011) |
| LAMP1 and MTOR | rs9577229 (LAMP1) rs6701524 (MTOR) |
Associations were observed between SNPs in LAMP1, MTOR, and infection with M. tuberculosis Beijing genotype, but statistical significance was lost after correction for multiple testing (Indonesian individuals). | Songane et al. (2012) |
| IRGM | −1161C/T and −947C/T | Both polymorphisms are associated with decreased susceptibility to TB (Iranian individuals). | Bahari et al. (2012) |
Note: Genes are listed according to the respective date of publication (from earliest to latest).
IL, interleukin; IRGM, immunity-related GTPase family M; LAMP1, lysosomal-associated membrane protein 1; TB, tuberculosis; TLR2, toll-like receptor 2; VDR, vitamin D receptor.
TB Therapeutics Based on Autophagy Antimycobacteria Responses
The current TB numbers associated with the HIV epidemic and the growing number of the immunocompromised population, under medication of immunosuppressive drugs or due to aging, are a serious cause of concern for public health. When we further combine the portfolio of dated anti-TB drugs in use and the emergence of MDR and XDR strains, the potential threat to public health urges for the development of new and more effective strategies.
As described before, autophagy plays a critical role in the host immune response against M. tuberculosis and therefore the development of autophagy-based therapies to combat TB represents an appealing strategy. In fact, it has been established that prolonged use of autophagy inhibitors, such as azitromycin, inhibits intracellular killing of mycobacteria and predisposes cystic fibrosis patients to mycobacterial disease (Renna et al., 2011). Furthermore, autophagy was shown to be the determinant in the intracellular killing effect of the first-line TB drugs, isoniazid and pyrazinamide, through a mechanism based on the release of ROS (Kim et al., 2012a). Therefore, autophagy inducers can and are being explored as potential new TB therapies.
Rapamycin (sirolimus) and everolimus are potent mTOR inhibitors and autophagy inducers currently approved for clinical use to prevent transplant rejection (Gutierrez et al., 2004; Ni Cheallaigh et al., 2011). Although these drugs are strong autophagy inducers, they are also immunosuppressing drugs and therefore their direct use in TB therapies is counterproductive (Ni Cheallaigh et al., 2011; Yu et al., 2013). Nevertheless, direct delivery of these drugs to the lungs using a nanoparticle system to enable specific particle uptake by professional phagocytic cells has been proposed in an attempt to minimize the systemic side effects (Ni Cheallaigh et al., 2011). An in vitro study, recently published, showed the potential of investing further in this approach since rapamycin-carrying nanoparticles were efficiently taken up by macrophages and exhibited substantial activity against intracellular M. tuberculosis (Gupta et al., 2014). Another possible approach is to enhance rapamycin potency toward autophagy induction and therefore to reduce the amount of rapamycin that needs to be administered, reducing adverse side effects. To this end, small-molecule enhancers of rapamycin (SMERs) in combination with rapamycin have been shown to increase the killing of mycobacteria by primary human macrophages, suggesting a possible application in TB therapy (Floto et al., 2007). Nevertheless, these SMERs were shown to act either independently or downstream of mTOR (Sarkar et al., 2007).
Niclosamide, an approved drug currently used to treat worm infections in the intestinal tract, was also found to be an inhibitor of mechanistic target of rapamycin complex 1 (mTORC1) signaling and a potent stimulator of autophagy (Balgi et al., 2009). Despite its effectiveness in the gastrointestinal tract, its poor absorption precludes any use in TB therapy. However, the niclosamide derivative drug, nitazoxanide, already in use as an antiprotozoal agent and with good intestinal absorption, was also shown to inhibit mTORC1 signaling and potently induce autophagy (Lam et al., 2012). It was demonstrated that nitazoxanide and its active form, tizoxanide, inhibit intracellular M. tuberculosis proliferation at the concentration normally found in the blood after oral administration (Lam et al., 2012). Interestingly, this drug was previously shown to kill both replicating and nonreplicating M. tuberculosis in vitro by an unknown mechanism (de Carvalho et al., 2009). It is possible that these promising results in TB therapy are not only the consequence of a dual mode of action involving direct targeting and killing of the bacteria but also autophagy induction in phagocytic cells, which promotes intracellular clearance of M. tuberculosis (Lam et al., 2012). It was also found that this effect was not compromised in the presence of first-line anti-TB drugs, such as isoniazid, pyrazinamide, ethambutol, rifampicin, and streptomycin (Lam et al., 2012). Furthermore, this drug has been proven safe without significant side effects, in very long treatment regimens (up to 4 years), in AIDS-related cryptosporidiosis (Fox and Saravolatz, 2005; Rossignol et al., 2006). All of these characteristics make nitazoxanide a drug with very promising applications in TB therapy.
Recently, it was also found that treatment of M. tuberculosis-infected macrophages with the AMP-mimetic 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) robustly activates autophagy and inhibits the survival of intracellular bacilli (Yang et al., 2014). This was shown to be dependent on AMPK-induced mTOR inhibition and AMPK-induced peroxisome proliferator-activated receptor-γ coactivator 1α (PPARGC1A) upregulation, accounting for increased expression of autophagy-related genes through CCAAT/enhancer-binding protein β (CEBPB) and autophagy induction (Yang et al., 2014). Therefore, the development of drugs that activate AMPK may have potential application in TB therapy (Yang et al., 2014).
There are, however, other methods of inducing autophagy in an mTOR-independent manner. Immunoadjuvants that induce autophagy have likely applications in TB treatment. INF-γ is a cytokine that has been demonstrated to be absolutely essential to control M. tuberculosis infection in both animal models and humans (Jouanguy et al., 1999). Besides activating macrophages to kill bacteria through the production of reactive nitrogen intermediates (Chan et al., 1992), IFN-γ stimulates delivery of mycobacteria to lysosomes by activating autophagy (MacMicking et al., 2003; Singh et al., 2006). A pilot study involving the administration of IFN-γ as an immune adjuvant for drug-resistant TB therapy obtained promising results, suggesting the efficacy of IFN-γ in drug-resistant TB (Suarez-Mendez et al., 2004). It is possible that by blocking the Th2 cytokines, IL-4 and IL-13, known to restrict autophagy, similar promising results may be achieved since a high-throughput RNA interference screen in a human monocytic cell line (THP-1) found that these autophagy-negative regulators are absolutely essential for intracellular mycobacterial survival and growth (Kumar et al., 2010). In fact, it has been demonstrated in vitro that lactic acid bacteria enhance the bacterial killing ability of mononuclear phagocytes by increasing autophagy-inducing cytokine IFN-γ levels and by reducing IL-4 and IL-13 (Ghadimi et al., 2010). In addition, oral treatment with lactic acid bacteria was sufficient to downregulate the lung Th2 response (Forsythe et al., 2007; Ghadimi et al., 2010).
Calcitriol is the hormonally active form of vitamin D. It is not only produced by the kidney but also in human macrophages from its precursor 25-hydroxy vitamin D by a specific 1-α hydroxylase, and it has been demonstrated to be required for IFN-γ-induced autophagy (Fabri et al., 2011). Vitamin D and IFN-γ-induced autophagy have been shown to promote lysosomal fusion with phagosomes containing M. tuberculosis and to consequently inhibit mycobacterial expansion in the host (Campbell and Spector, 2012; Bradfute et al., 2013). It was therefore tempting to test whether vitamin D could be used as a dietary supplement in TB treatment since, historically, vitamin D sources such as sunbathing and cod liver oil were used to treat TB. Many clinical trials have been performed to test this hypothesis; the results have, however, been inconclusive with several studies showing positive results, especially in patients with vitamin D deficiency (Salahuddin et al., 2013; Kearns et al., 2014), but many other major clinical trials showing no benefits overall (Wejse et al., 2009; Martineau et al., 2011; Ralph et al., 2013; Kearns et al., 2014). It is still uncertain whether vitamin D will have any use in TB therapy, with some researchers advocating that better knowledge is needed about vitamin D concentrations for optimal immune response to perform adequate clinical trials (Ralph et al., 2013).
Lithium, carbamazepine, and sodium valproate are currently approved drugs used to treat mood disorders and epilepsy. They target the d-myo-inositol-1,4,5-triphosphate (IP3)-regulated pathway, depleting intracellular inositol, and therefore induce autophagy (Sarkar et al., 2005; Rubinsztein et al., 2007). Tamoxifen is a drug currently used to treat a wide variety of diseases, from breast cancer to mood disorders and infertility, among others. It was shown to be an inducer of autophagy in a Beclin-1-dependent manner (Wienecke et al., 2006; Rubinsztein et al., 2007). Ridaifen-B, a tamoxifen derivative, was also shown to be an inducer of autophagy, but through a currently unknown Beclin-1-independent mechanism (Nagahara et al., 2013). Gefitinib, an epidermal growth factor receptor (EGFR) inhibitor used to treat breast and other types of cancers, has also been shown to induce autophagy and to be effective in M. tuberculosis killing in a mouse model (Stanley et al., 2014). Nortriptyline and fluoxetine, currently used to treat depression, were also reported to promote autophagy and enhance mycobacteria clearance (Sundaramurthy et al., 2013; Stanley et al., 2014). While the mechanisms behind autophagy enhancement by these two drugs are currently unknown, the fluoxetine effect is related to increased TNF-α secretion (Stanley et al., 2014). Since all these FDA-approved drugs have been reported as autophagy inducers, they have the potential to be used as complementary treatment(s) to current TB therapies.
Statins are widely used HMG-CoA reductase inhibitors currently approved for clinical use to lower cholesterol levels (Ray et al., 2010). It has been recently documented that statins also have immunomodulatory and anti-inflammatory effects with reports of reduced mortality in patients with bacteremia (Kwak et al., 2000; Liao and Laufs, 2005; Tleyjeh et al., 2009; Parihar et al., 2014). In TB mice models, treatment with statins significantly reduced the bacterial load and the pulmonary pathological effects of TB infection (Parihar et al., 2014). It was further shown in TB-infected macrophages and in mice models that treatment with statins improves the efficacy of first-line TB drug regimens and of rifampicin alone (Lobato et al., 2014; Skerry et al., 2014). It was demonstrated that statins improve bacterial clearance by the host and improve the efficacy of TB drugs by promoting autophagy through inhibition of the HMG-CoA reductase pathway (Parihar et al., 2014). Furthermore, there are some reports claiming that statins also enhance autophagy in an mTOR-dependent way by inhibiting the RAC1-mTOR signaling pathway (Wei et al., 2013). The full mechanisms behind the autophagy-inducing effects of statins are still not fully elucidated; however, the effects observed from statin administration are promising and may provide another possible complement to TB therapy.
Several TLRs have been shown to be involved in autophagy induction and play a critical role in the formation of the immune response (Sanjuan et al., 2007; Xu et al., 2007; Delgado et al., 2008). However, prolonged stimulation of TLRs (by abundant TLR-interacting mycobacterial compounds) results in the production of immunosuppressive cytokines, decreased antigen presentation, and survival of bacteria inside macrophages (Saraav et al., 2014). Nevertheless, TLR4-mediated autophagy was found to promote mycobacteria containment in macrophages (Xu et al., 2007). Finding the right equilibrium of different TLR activations using either drug or vaccine approaches might lead to increased immunogenic response and improved TB therapies.
The existing prophylactic approach to TB, the BCG vaccine, was first tested in humans over 90 years ago and has been used extensively, despite its unreliability in terms of averting TB in adults (Colditz et al., 1995; Fine, 1995). A new vaccine or an innovative strategy to improve the efficacy of the current BCG vaccine would have a profound impact on the current situation of the TB epidemic. Stimulation of autophagy was found to increase the efficacy of attenuated H37rv and BCG vaccines through enhancement of the ability of the macrophage and DCs to present mycobacterial antigens (Jagannath et al., 2009). Rapamycin-treated macrophages exhibited a substantial increase in antigen presentation when infected with the tested TB vaccine strains and the wild-type H37rv strain (Jagannath et al., 2009). Furthermore, the results showed that DCs also had enhanced antigen presentation when treated with rapamycin (Jagannath et al., 2009). The increased in vitro antigen presentation was observed to translate into in vivo protection in a TB mouse model (Jagannath et al., 2009). It was further demonstrated that M. smegmatis and M. bovis BCG strains that were modified to overexpress immunogenic antigens targeted by the autophagy-lysosome pathway (such as Ag85B) led to an increase in antigen presentation (Jagannath et al., 2009). This is in agreement with a different study showing that a live BCG strain overexpressing Ag85B is a more efficient vaccine when compared with the wild-type BCG strain (Horwitz et al., 2006).
DNA vaccines used directly or as prime-boost are alternative promising approaches to either improve the efficacy of the current BCG vaccine or to create a new more effective one (Rivas-Santiago and Cervantes-Villagrana, 2014). Plasmids containing M. tuberculosis DNA (from Ag85, Hsp65, and the 23 members of Esx gene family) used in experimental DNA vaccines have been found to lead to higher INF-γ production and consequent induction of autophagy (Meerak et al., 2013; Zarate-Blades et al., 2013; Villarreal et al., 2014).
These promising TB therapies are listed and summarized in Table 2.
Table 2.
Potential Tuberculosis Therapies Based on Autophagy Manipulation
| Therapy | Effects | Reference |
|---|---|---|
| Sirolimus (rapamycin), everolimus | mTORC1 inhibitors and autophagy inducers; increase BCG vaccine efficacy | Gutierrez et al. (2004), Jagannath et al. (2009), Gupta et al. (2014) |
| SMERs | Synergistic effect with rapamycin on autophagy, acting independently or downstream of mTOR; autophagy inducer | Floto et al. (2007) |
| Nitazoxanide (tizoxanide) | Inhibitor of mTORC1 signaling and autophagy inducer; kills mycobacteria directly by an unknown mechanism | de Carvalho et al. (2009), Lam et al. (2012) |
| IFN-γ | Increases production of reactive nitrogen species in macrophages and induces autophagy | Chan et al. (1992), MacMicking et al. (2003), Singh et al. (2006) |
| Lactic acid bacteria | Increase production of IFN-γ and decrease the autophagy negative regulators, IL-4 and IL-13; induce autophagy | Ghadimi et al. (2010) |
| Vitamin D | Required for IFN-γ-induced autophagy | Campbell and Spector (2012) |
| Lithium, carbamazepine, sodium valproate | Target the d-myo-inosiltol-1,4,5 triphosphate (IP3)-regulated pathway, deplete intracellular inositol, and induce autophagy in an mTOR-independent way | Sarkar et al. (2005) |
| Tamoxifen | Inducer of autophagy in a Beclin-1-dependent manner | Wienecke et al. (2006) |
| Ridaifen-B | Tamoxifen derivative that induces autophagy through an unknown Beclin-1-independent mechanism | Nagahara et al. (2013) |
| Gefitinib | Induces autophagy (although it is not clear, the authors postulate that gefitinib-induction of autophagy relies on p38 MAPK inhibition) | Stanley et al. (2014) |
| Nortriptyline | Induces autophagy by an unknown mechanism | Sundaramurthy et al. (2013) |
| Fluoxetine | Induces autophagy possibly due to enhanced TNF-α secretion | Stanley et al. (2014) |
| Statins | Promote autophagy by inhibiting the HMG-CoA reductase pathway and in an mTOR-dependent way; improve the efficacy of first-line TB drugs | Wei et al. (2013), Lobato et al. (2014), Parihar et al. (2014), Skerry et al. (2014) |
| Ag85-overexpressing strains for vaccines | Ag85 is targeted by the autophagy-lysosome pathway and increases antigen presentation | Horwitz et al. (2006), Jagannath et al. (2009) |
| DNA vaccines (Ag85, Hsp65, Esx genes) | Potentiate INF-γ production and induce autophagy | Meerak et al. (2013), Zarate-Blades et al. (2013), Villarreal et al. (2014) |
BCG, bacilli Calmette Guerin; IFN-γ, interferon-γ; mTOR, mechanistic target of rapamycin; mTORC1, mechanistic target of rapamycin complex 1; SMERs, small-molecule enhancers of rapamycin; TNF, tumor necrosis factor.
Final Remarks
Autophagy is a key mechanism in eukaryotic cell resistance to M. tuberculosis infection as it plays a vital role in the intracellular clearance of this pathogen. It potentiates the effect of some of the current first-line TB drugs, influences antigen presentation, and modulates the release of cytokines that are determinants for the infection outcome. Although the knowledge of how autophagy influences immunity is still far from complete, there is a clear potential for autophagy-based therapies in advanced TB treatment strategies. Promotion of autophagy through pharmacological means by administrating autophagy-inducing drugs and cytokines has produced positive results in vitro in TB mice models and even in a human pilot study. Enhancing autophagy also increases the efficacy of the only TB-prophylactic method available, the BCG vaccine. The results of using autophagy-inducing approaches to combat TB are very promising and an autophagy-based therapy for TB may soon be a reality.
Acknowledgments
This work was funded by the Bill and Melinda Gates Foundation (subcontract by the Foundation for the National Institutes of Health—NIH) (OPP1024021) and the Fundação para a Ciência e a Tecnologia and EU-FEDER-COMPETE funding (PTDC/BIA-MIC/2779/2012; FCOMP-01-0124-FEDER-028359). V.M. would like to acknowledge Fundação para a Ciência e a Tecnologia for a postdoctoral fellowship (SFRH/BPD/79531/2011).
Disclosure Statement
The authors do not have financial or commercial competing interests.
References
- Abdallah A.M., Gey van Pittius N.C., Champion P.A., Cox J., Luirink J., Vandenbroucke-Grauls C.M., et al. (2007). Type VII secretion—mycobacteria show the way. Nat Rev Microbiol 5,883–891 [DOI] [PubMed] [Google Scholar]
- Bach H., Papavinasasundaram K.G., Wong D., Hmama Z., and Av-Gay Y. (2008). Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe 3,316–322 [DOI] [PubMed] [Google Scholar]
- Bahari G., Hashemi M., Taheri M., Naderi M., Eskandari-Nasab E., and Atabaki M. (2012). Association of IRGM polymorphisms and susceptibility to pulmonary tuberculosis in Zahedan, Southeast Iran. Sci World J 2012,950801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balgi A.D., Fonseca B.D., Donohue E., Tsang T.C., Lajoie P., Proud C.G., et al. (2009). Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 4,e7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ali M., Barbouche M.R., Bousnina S., Chabbou A., and Dellagi K. (2004). Toll-like receptor 2 Arg677Trp polymorphism is associated with susceptibility to tuberculosis in Tunisian patients. Clin Diagn Lab Immunol 11,625–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento C.F., Puri C., Moreau K., and Rubinsztein D.C. (2013). The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci 126,1059–1069 [DOI] [PubMed] [Google Scholar]
- Bowdish D.M., Sakamoto K., Kim M.J., Kroos M., Mukhopadhyay S., Leifer C.A., et al. (2009). MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathog 5,e1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradfute S.B., Castillo E.F., Arko-Mensah J., Chauhan S., Jiang S., Mandell M., et al. (2013). Autophagy as an immune effector against tuberculosis. Curr Opin Microbiol 16,355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briken V., Porcelli S.A., Besra G.S., and Kremer L. (2004). Mycobacterial lipoarabinomannan and related lipoglycans: from biogenesis to modulation of the immune response. Mol Microbiol 53,391–403 [DOI] [PubMed] [Google Scholar]
- Campbell G.R., and Spector S.A. (2012). Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog 8,e1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo E.F., Dekonenko A., Arko-Mensah J., Mandell M.A., Dupont N., Jiang S., et al. (2012). Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA 109,E3168–E3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J., Xing Y., Magliozzo R.S., and Bloom B.R. (1992). Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med 175,1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che N., Li S., Gao T., Zhang Z., Han Y., Zhang X., et al. (2010). Identification of a novel IRGM promoter single nucleotide polymorphism associated with tuberculosis. Clin Chim Acta 411,1645–1649 [DOI] [PubMed] [Google Scholar]
- Colditz G.A., Berkey C.S., Mosteller F., Brewer T.F., Wilson M.E., Burdick E., et al. (1995). The efficacy of bacillus Calmette-Guerin vaccination of newborns and infants in the prevention of tuberculosis: meta-analyses of the published literature. Pediatrics 96,29–35 [PubMed] [Google Scholar]
- Collins C.A., De Maziere A., van Dijk S., Carlsson F., Klumperman J., and Brown E.J. (2009). Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog 5,e1000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.M. (2009). Cell-mediated immune responses in tuberculosis. Annu Rev Immunol 27,393–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court N., Vasseur V., Vacher R., Fremond C., Shebzukhov Y., Yeremeev V.V., et al. (2010). Partial redundancy of the pattern recognition receptors, scavenger receptors, and C-type lectins for the long-term control of Mycobacterium tuberculosis infection. J Immunol 184,7057–7070 [DOI] [PubMed] [Google Scholar]
- Crisan T.O., Plantinga T.S., van de Veerdonk F.L., Farcas M.F., Stoffels M., Kullberg B.J., et al. (2011). Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One 6,e18666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.M., and Ramakrishnan L. (2009). The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136,37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho L.P., Lin G., Jiang X., and Nathan C. (2009). Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J Med Chem 52,5789–5792 [DOI] [PubMed] [Google Scholar]
- Delgado M.A., Elmaoued R.A., Davis A.S., Kyei G., and Deretic V. (2008). Toll-like receptors control autophagy. EMBO J 27,1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drennan M.B., Nicolle D., Quesniaux V.J., Jacobs M., Allie N., Mpagi J., et al. (2004). Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol 164,49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers S. (2010). DC-SIGN and mannosylated surface structures of Mycobacterium tuberculosis: a deceptive liaison. Eur J Cell Biol 89,95–101 [DOI] [PubMed] [Google Scholar]
- Ernst J.D. (2012). The immunological life cycle of tuberculosis. Nat Rev Immunol 12,581–591 [DOI] [PubMed] [Google Scholar]
- Etokebe G.E., Skjeldal F., Nilsen N., Rodionov D., Knezevic J., Bulat-Kardum L., et al. (2010). Toll-like receptor 2 (P631H) mutant impairs membrane internalization and is a dominant negative allele. Scand J Immunol 71,369–381 [DOI] [PubMed] [Google Scholar]
- Fabri M., Stenger S., Shin D.M., Yuk J.M., Liu P.T., Realegeno S., et al. (2011). Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med 3,104ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine P.E. (1995). Variation in protection by BCG: implications of and for heterologous immunity. Lancet 346,1339–1345 [DOI] [PubMed] [Google Scholar]
- Floto R.A., Sarkar S., Perlstein E.O., Kampmann B., Schreiber S.L., and Rubinsztein D.C. (2007). Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington's disease models and enhance killing of mycobacteria by macrophages. Autophagy 3,620–622 [DOI] [PubMed] [Google Scholar]
- Forsythe P., Inman M.D., and Bienenstock J. (2007). Oral treatment with live Lactobacillus reuteri inhibits the allergic airway response in mice. Am J Respir Crit Care Med 175,561–569 [DOI] [PubMed] [Google Scholar]
- Fox L.M., and Saravolatz L.D. (2005). Nitazoxanide: a new thiazolide antiparasitic agent. Clin Infect Dis 40,1173–1180 [DOI] [PubMed] [Google Scholar]
- Fratti R.A., Chua J., Vergne I., and Deretic V. (2003). Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci USA 100,5437–5442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S., Holmstrom K.M., Skujat D., Fiesel F.C., Rothfuss O.C., Kahle P.J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12,119–131 [DOI] [PubMed] [Google Scholar]
- Ghadimi D., de Vrese M., Heller K.J., and Schrezenmeir J. (2010). Lactic acid bacteria enhance autophagic ability of mononuclear phagocytes by increasing Th1 autophagy-promoting cytokine (IFN-gamma) and nitric oxide (NO) levels and reducing Th2 autophagy-restraining cytokines (IL-4 and IL-13) in response to Mycobacterium tuberculosis antigen. Int Immunopharmacol 10,694–706 [DOI] [PubMed] [Google Scholar]
- Gomes L.C., and Dikic I. (2014). Autophagy in antimicrobial immunity. Mol Cell 54,224–233 [DOI] [PubMed] [Google Scholar]
- Greenstein R.J. (2003). Is Crohn's disease caused by a mycobacterium? Comparisons with leprosy, tuberculosis, and Johne's disease. Lancet Infect Dis 3,507–514 [DOI] [PubMed] [Google Scholar]
- Gupta A., Pant G., Mitra K., Madan J., Chourasia M.K., and Misra A. (2014). Inhalable particles containing rapamycin for induction of autophagy in macrophages infected with Mycobacterium tuberculosis. Mol Pharm 11,1201–1207 [DOI] [PubMed] [Google Scholar]
- Gutierrez M.G., Master S.S., Singh S.B., Taylor G.A., Colombo M.I., and Deretic V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119,753–766 [DOI] [PubMed] [Google Scholar]
- Harboe M., Oettinger T., Wiker H.G., Rosenkrands I., and Andersen P. (1996). Evidence for occurrence of the ESAT-6 protein in Mycobacterium tuberculosis and virulent Mycobacterium bovis and for its absence in Mycobacterium bovis BCG. Infect Immun 64,16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. (2011). Autophagy and cytokines. Cytokine 56,140–144 [DOI] [PubMed] [Google Scholar]
- Harris J., De Haro S.A., Master S.S., Keane J., Roberts E.A., Delgado M., et al. (2007). T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 27,505–517 [DOI] [PubMed] [Google Scholar]
- Harris J., Hartman M., Roche C., Zeng S.G., O'Shea A., Sharp F.A., et al. (2011). Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 286,9587–9597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J., Hope J.C., and Keane J. (2008). Tumor necrosis factor blockers influence macrophage responses to Mycobacterium tuberculosis. J Infect Dis 198,1842–1850 [DOI] [PubMed] [Google Scholar]
- Horwitz M.A., Harth G., Dillon B.J., and Maslesa-Galic S. (2006). A novel live recombinant mycobacterial vaccine against bovine tuberculosis more potent than BCG. Vaccine 24,1593–1600 [DOI] [PubMed] [Google Scholar]
- Huang J., and Brumell J.H. (2014). Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 12,101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intemann C.D., Thye T., Niemann S., Browne E.N., Amanua Chinbuah M., Enimil A., et al. (2009). Autophagy gene variant IRGM −261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5,e1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa E., Ishikawa T., Morita Y.S., Toyonaga K., Yamada H., Takeuchi O., et al. (2009). Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med 206,2879–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannath C., Lindsey D.R., Dhandayuthapani S., Xu Y., Hunter R.L., Jr., and Eissa N.T. (2009). Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med 15,267–276 [DOI] [PubMed] [Google Scholar]
- Jo E.K. (2013). Autophagy as an innate defense against mycobacteria. Pathog Dis 67,108–118 [DOI] [PubMed] [Google Scholar]
- Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491,119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouanguy E., Lamhamedi-Cherradi S., Lammas D., Dorman S.E., Fondaneche M.C., Dupuis S., et al. (1999). A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet 21,370–378 [DOI] [PubMed] [Google Scholar]
- Juarez E., Carranza C., Hernandez-Sanchez F., Leon-Contreras J.C., Hernandez-Pando R., Escobedo D., et al. (2012). NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur J Immunol 42,880–889 [DOI] [PubMed] [Google Scholar]
- Kearns M.D., Alvarez J.A., Seidel N., and Tangpricha V. (2014). Impact of vitamin D on infectious disease: a systematic review of controlled trials. Am J Med Sci [Epub ahead of print]; DOI: 10.1097/MAJ.0000000000000360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.J., Lee H.M., Shin D.M., Kim W., Yuk J.M., Jin H.S., et al. (2012a). Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 11,457–468 [DOI] [PubMed] [Google Scholar]
- Kim K.H., An D.R., Song J., Yoon J.Y., Kim H.S., Yoon H.J., et al. (2012b). Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci USA 109,7729–7734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K.Y., Lew J.D., Ha N.P., Lin J.S., Ma X., Graviss E.A., et al. (2011). Polymorphic allele of human IRGM1 is associated with susceptibility to tuberculosis in African Americans. PLoS One 6,e16317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinnijenhuis J., Oosting M., Joosten L.A., Netea M.G., and Van Crevel R. (2011). Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol 2011,405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D., Nath L., Kamal M.A., Varshney A., Jain A., Singh S., et al. (2010). Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 140,731–743 [DOI] [PubMed] [Google Scholar]
- Kwak B., Mulhaupt F., Myit S., and Mach F. (2000). Statins as a newly recognized type of immunomodulator. Nat Med 6,1399–1402 [DOI] [PubMed] [Google Scholar]
- Lam K.K., Zheng X., Forestieri R., Balgi A.D., Nodwell M., Vollett S., et al. (2012). Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog 8,e1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R. (2013). Recognition of the mycobacterial cord factor by Mincle: relevance for granuloma formation and resistance to tuberculosis. Front Immunol 4,5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Repasy T., Papavinasasundaram K., Sassetti C., and Kornfeld H. (2011). Mycobacterium tuberculosis induces an atypical cell death mode to escape from infected macrophages. PLoS One 6,e18367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J.K., and Laufs U. (2005). Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 45,89–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P.L., Ford C.B., Coleman M.T., Myers A.J., Gawande R., Ioerger T., et al. (2014). Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med 20,75–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato L.S., Rosa P.S., Ferreira Jda S., Neumann Ada S., da Silva M.G., do Nascimento D.C., et al. (2014). Statins increase rifampin mycobactericidal effect. Antimicrob Agents Chemother 58,5766–5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J.D., Taylor G.A., and McKinney J.D. (2003). Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302,654–659 [DOI] [PubMed] [Google Scholar]
- Manzanillo P.S., Ayres J.S., Watson R.O., Collins A.C., Souza G., Rae C.S., et al. (2013). The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501,512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau A.R., Timms P.M., Bothamley G.H., Hanifa Y., Islam K., Claxton A.P., et al. (2011). High-dose vitamin D(3) during intensive-phase antimicrobial treatment of pulmonary tuberculosis: a double-blind randomised controlled trial. Lancet 377,242–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerak J., Wanichwecharungruang S.P., and Palaga T. (2013). Enhancement of immune response to a DNA vaccine against Mycobacterium tuberculosis Ag85B by incorporation of an autophagy inducing system. Vaccine 31,784–790 [DOI] [PubMed] [Google Scholar]
- Mira M.T., Alcais A., Nguyen V.T., Moraes M.O., Di Flumeri C., Vu H.T., et al. (2004). Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427,636–640 [DOI] [PubMed] [Google Scholar]
- Mizushima N. (2011). Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol 76,397–402 [DOI] [PubMed] [Google Scholar]
- Nagahara Y., Takeyoshi M., Sakemoto S., Shiina I., Nakata K., Fujimori K., et al. (2013). Novel tamoxifen derivative Ridaifen-B induces Bcl-2 independent autophagy without estrogen receptor involvement. Biochem Biophys Res Commun 435,657–663 [DOI] [PubMed] [Google Scholar]
- Nakahira K., Haspel J.A., Rathinam V.A., Lee S.J., Dolinay T., Lam H.C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12,222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Cheallaigh C., Keane J., Lavelle E.C., Hope J.C., and Harris J. (2011). Autophagy in the immune response to tuberculosis: clinical perspectives. Clin Exp Immunol 164,291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobre A., Alarico S., Maranha A., Mendes V., and Empadinhas N. (2014). The molecular biology of mycobacterial trehalose in the quest for advanced tuberculosis therapies. Microbiology 160,1547–1570 [DOI] [PubMed] [Google Scholar]
- Ogus A.C., Yoldas B., Ozdemir T., Uguz A., Olcen S., Keser I., et al. (2004). The Arg753GLn polymorphism of the human toll-like receptor 2 gene in tuberculosis disease. Eur Respir J 23,219–223 [DOI] [PubMed] [Google Scholar]
- Parihar S.P., Guler R., Khutlang R., Lang D.M., Hurdayal R., Mhlanga M.M., et al. (2014). Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis 209,754–763 [DOI] [PubMed] [Google Scholar]
- Philips J.A. (2008). Mycobacterial manipulation of vacuolar sorting. Cell Microbiol 10,2408–2415 [DOI] [PubMed] [Google Scholar]
- Philips J.A., and Ernst J.D. (2012). Tuberculosis pathogenesis and immunity. Annu Rev Pathol 7,353–384 [DOI] [PubMed] [Google Scholar]
- Pilli M., Arko-Mensah J., Ponpuak M., Roberts E., Master S., Mandell M.A., et al. (2012). TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37,223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph A.P., Lucas R.M., and Norval M. (2013). Vitamin D and solar ultraviolet radiation in the risk and treatment of tuberculosis. Lancet Infect Dis 13,77–88 [DOI] [PubMed] [Google Scholar]
- Ravikumar B., Futter M., Jahreiss L., Korolchuk V.I., Lichtenberg M., Luo S., et al. (2009). Mammalian macroautophagy at a glance. J Cell Sci 122,1707–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray K.K., Seshasai S.R., Erqou S., Sever P., Jukema J.W., Ford I., et al. (2010). Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med 170,1024–1031 [DOI] [PubMed] [Google Scholar]
- Renna M., Schaffner C., Brown K., Shang S., Tamayo M.H., Hegyi K., et al. (2011). Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest 121,3554–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas-Santiago B., and Cervantes-Villagrana A.R. (2014). Novel approaches to tuberculosis prevention: DNA vaccines. Scand J Infect Dis 46,161–168 [DOI] [PubMed] [Google Scholar]
- Rossignol J.F., Kabil S.M., el-Gohary Y., and Younis A.M. (2006). Effect of nitazoxanide in diarrhea and enteritis caused by Cryptosporidium species. Clin Gastroenterol Hepatol 4,320–324 [DOI] [PubMed] [Google Scholar]
- Rubinsztein D.C., Gestwicki J.E., Murphy L.O., and Klionsky D.J. (2007). Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6,304–312 [DOI] [PubMed] [Google Scholar]
- Saitoh T., Fujita N., Jang M.H., Uematsu S., Yang B.G., Satoh T., et al. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456,264–268 [DOI] [PubMed] [Google Scholar]
- Salahuddin N., Ali F., Hasan Z., Rao N., Aqeel M., and Mahmood F. (2013). Vitamin D accelerates clinical recovery from tuberculosis: results of the SUCCINCT Study [Supplementary Cholecalciferol in recovery from tuberculosis]. A randomized, placebo-controlled, clinical trial of vitamin D supplementation in patients with pulmonary tuberculosis'. BMC Infect Dis 13,22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan M.A., Dillon C.P., Tait S.W., Moshiach S., Dorsey F., Connell S., et al. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450,1253–1257 [DOI] [PubMed] [Google Scholar]
- Saraav I., Singh S., and Sharma S. (2014). Outcome of Mycobacterium tuberculosis and Toll-like receptor interaction: immune response or immune evasion? Immunol Cell Biol 92,741–746 [DOI] [PubMed] [Google Scholar]
- Sarkar S. (2013). Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 41,1103–1130 [DOI] [PubMed] [Google Scholar]
- Sarkar S., Floto R.A., Berger Z., Imarisio S., Cordenier A., Pasco M., et al. (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170,1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S., Perlstein E.O., Imarisio S., Pineau S., Cordenier A., Maglathlin R.L., et al. (2007). Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol 3,331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer G., Jacobs M., Wilkinson R.J., and Brown G.D. (2009). Non-opsonic recognition of Mycobacterium tuberculosis by phagocytes. J Innate Immun 1,231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto S., Tsujimura K., and Koide Y. (2012). Coronin-1a inhibits autophagosome formation around Mycobacterium tuberculosis-containing phagosomes and assists mycobacterial survival in macrophages. Cell Microbiol 14,710–727 [DOI] [PubMed] [Google Scholar]
- Shah S., Bohsali A., Ahlbrand S.E., Srinivasan L., Rathinam V.A., Vogel S.N., et al. (2013). Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-beta and AIM2 inflammasome-dependent IL-1beta production via its ESX-1 secretion system. J Immunol 191,3514–3518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.S., and Kehrl J.H. (2008). MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 283,33175–33182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D.M., Jeon B.Y., Lee H.M., Jin H.S., Yuk J.M., Song C.H., et al. (2010a). Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog 6,e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D.M., Yuk J.M., Lee H.M., Lee S.H., Son J.W., Harding C.V., et al. (2010b). Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol 12,1648–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui W., Petzold C.J., Redding A., Liu J., Pitcher A., Sheu L., et al. (2011). Organelle membrane proteomics reveals differential influence of mycobacterial lipoglycans on macrophage phagosome maturation and autophagosome accumulation. J Proteome Res 10,339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.B., Davis A.S., Taylor G.A., and Deretic V. (2006). Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313,1438–1441 [DOI] [PubMed] [Google Scholar]
- Skerry C., Pinn M.L., Bruiners N., Pine R., Gennaro M.L., and Karakousis P.C. (2014). Simvastatin increases the in vivo activity of the first-line tuberculosis regimen. J Antimicrob Chemother 69,2453–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songane M., Kleinnijenhuis J., Alisjahbana B., Sahiratmadja E., Parwati I., Oosting M., et al. (2012). Polymorphisms in autophagy genes and susceptibility to tuberculosis. Plos one 7,e41618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S.A., Barczak A.K., Silvis M.R., Luo S.S., Sogi K., Vokes M., et al. (2014). Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog 10,e1003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Mendez R., Garcia-Garcia I., Fernandez-Olivera N., Valdes-Quintana M., Milanes-Virelles M.T., Carbonell D., et al. (2004). Adjuvant interferon gamma in patients with drug - resistant pulmonary tuberculosis: a pilot study. BMC Infect Dis 4,44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Wang Y., Ma X., Pei F., Sun H., Zhang Y., Yu B. (2007). A method of oligochip for single nucleotide polymorphism genotyping in the promoter region of the interleukin-1 beta gene and its clinical application. Oligonucleotides 17,336–344 [DOI] [PubMed] [Google Scholar]
- Sundaramurthy V., Barsacchi R., Samusik N., Marsico G., Gilleron J., Kalaidzidis I., et al. (2013). Integration of chemical and RNAi multiparametric profiles identifies triggers of intracellular mycobacterial killing. Cell Host Microbe 13,129–142 [DOI] [PubMed] [Google Scholar]
- Tleyjeh I.M., Kashour T., Hakim F.A., Zimmerman V.A., Erwin P.J., Sutton A.J., et al. (2009). Statins for the prevention and treatment of infections: a systematic review and meta-analysis. Arch Intern Med 169,1658–1667 [DOI] [PubMed] [Google Scholar]
- Tobin D.M., and Ramakrishnan L. (2008). Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell Microbiol 10,1027–1039 [DOI] [PubMed] [Google Scholar]
- van der Wel N., Hava D., Houben D., Fluitsma D., van Zon M., Pierson J., et al. (2007). M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129,1287–1298 [DOI] [PubMed] [Google Scholar]
- Vergne I., Chua J., Lee H.H., Lucas M., Belisle J., and Deretic V. (2005). Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci USA 102,4033–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergne I., Fratti R.A., Hill P.J., Chua J., Belisle J., and Deretic V. (2004). Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol Biol Cell 15,751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via L.E., Deretic D., Ulmer R.J., Hibler N.S., Huber L.A., and Deretic V. (1997). Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem 272,13326–13331 [DOI] [PubMed] [Google Scholar]
- Villarreal D.O., Walters J., Laddy D.J., Yan J., and Weiner D.B. (2014). Multivalent TB vaccines targeting the esx gene family generate potent and broad cell-mediated immune responses superior to BCG. Hum Vaccin Immunother 10,2188–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson R.O., Manzanillo P.S., and Cox J.S. (2012). Extracellular, M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150,803–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Dahl J.L., Moulder J.W., Roberts E.A., O'Gaora P., Young D.B., et al. (2000). Identification of a Mycobacterium tuberculosis gene that enhances mycobacterial survival in macrophages. J Bacteriol 182,377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y.M., Li X., Xu M., Abais J.M., Chen Y., Riebling C.R., et al. (2013). Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell Physiol Biochem 31,925–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wejse C., Gomes V.F., Rabna P., Gustafson P., Aaby P., Lisse I.M., et al. (2009). Vitamin D as supplementary treatment for tuberculosis: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 179,843–850 [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. (2007). Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447,661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienecke R., Fackler I., Linsenmaier U., Mayer K., Licht T., and Kretzler M. (2006). Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis 48,e27–e29 [DOI] [PubMed] [Google Scholar]
- Wilkinson R.J., Llewelyn M., Toossi Z., Patel P., Pasvol G., Lalvani A., et al. (2000). Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: a case-control study. Lancet 355,618–621 [DOI] [PubMed] [Google Scholar]
- Xiong Y., Song C., Snyder G.A., Sundberg E.J., and Medvedev A.E. (2012). R753Q polymorphism inhibits Toll-like receptor (TLR) 2 tyrosine phosphorylation, dimerization with TLR6, and recruitment of myeloid differentiation primary response protein 88. J Biol Chem 287,38327–38337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Jagannath C., Liu X.D., Sharafkhaneh A., Kolodziejska K.E., and Eissa N.T. (2007). Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27,135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.S., Kim J.J., Lee H.M., Jin H.S., Lee S.H., Park J.H., et al. (2014). The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy 10,785–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle R.J., and Narendra D.P. (2011). Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12,9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Li C., Hong W., Pan W., and Xie J. (2013). Autophagy during Mycobacterium tuberculosis infection and implications for future tuberculosis medications. Cell Signal 25,1272–1278 [DOI] [PubMed] [Google Scholar]
- Yuk J.M., Shin D.M., Lee H.M., Yang C.S., Jin H.S., Kim K.K., et al. (2009). Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6,231–243 [DOI] [PubMed] [Google Scholar]
- Zarate-Blades C.R., Rodrigues R.F., Souza P.R., Rios W.M., Soares L.S., Rosada R.S., et al. (2013). Evaluation of the overall IFN-gamma and IL-17 pro-inflammatory responses after DNA therapy of tuberculosis. Hum Vaccin Immunother 9,1093–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R., Yazdi A.S., Menu P., and Tschopp J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469,221–225 [DOI] [PubMed] [Google Scholar]