

Abstract
Initiation of Na+-glucose cotransport in intestinal epithelial cells leads to activation of the apical Na+-H+ exchanger NHE3 and subsequent increases in cytoplasmic pH (pHi). This process requires activation of p38 mitogen-activated protein (MAP) kinase, but additional signaling intermediates have not been identified. One candidate is the cytoskeletal linker protein ezrin, which interacts with NHE3 via specific regulatory proteins. The data show that initiation of Na+-glucose cotransport resulted in rapid increases in both apical membrane-associated NHE3 and cytoskeletal-associated ezrin and occurred in parallel with ezrin phosphorylation at threonine 567. Phosphorylation at this site is known to activate ezrin and increase its association with actin. Consistent with a central role for ezrin activation in this NHE3 regulation, an N-terminal dominant negative ezrin construct inhibited both NHE3 recruitment and pHi increases after Na+-glucose cotransport. Ezrin phosphorylation occurred in parallel with p38 MAP kinase activation, and the latter proceeded normally in cells expressing dominant negative ezrin. In contrast, inhibition of p38 MAP kinase prevented increases in ezrin phosphorylation after initiation of Na+-glucose cotransport. Thus, ezrin phosphorylation after Na+-glucose cotransport requires p38 MAP kinase activity, but p38 MAP kinase activation does not require ezrin function. These data describe a specific role for ezrin in the coordinate regulation of Na+-glucose cotransport and Na+-H+ exchange. Intact ezrin function is necessary for NHE3 recruitment to the apical membrane and NHE3-dependent pHi increases triggered by Na+-glucose cotransport. The data also define a pathway of p38 MAP kinase-dependent ezrin activation.
Vectorial nutrient and ion transport is of particular importance in small intestinal and renal tubular epithelium. Transcellular transport of each ion or nutrient is typically accomplished by one or more specific transporter proteins. Although early studies assumed that each of these processes occurred in isolation, transactivation of one transporter by another is now recognized as a mechanism by which the coordinated regulation of numerous transport processes occurs. For example, we have previously shown that initiation of glucose transport by the apical Na+-glucose cotransporter SGLT1 leads to activation of the apical Na+-H+ exchanger NHE3 (1). Thwaites et al. (2) have shown that, in turn, NHE3 can regulate the activity of the dipeptide transporter hPepT1. Thus, SGLT1-mediated Na+-glucose cotransport activates both NHE3 and hPepT1, suggesting that the presence of Na+ and glucose in the lumen may activate transporters globally, shifting the cell from a quiescent to an active state with regard to nutrient and ion absorption.
The mechanisms by which Na+-glucose cotransport triggers activation of NHE3-mediated Na+-H+ exchange are largely unknown. Although we showed that this process requires activation of p38 mitogen-activated protein (MAP) kinase, the targets of p38 MAP kinase are not yet identified (1). To better understand the mechanisms by which SGLT1 and p38 MAP kinase regulate NHE3, we examined the role of the cytoskeletal crosslinking protein ezrin in this process. Ezrin has been implicated in NHE3 regulation via a signaling complex (3) that includes NHE3, ezrin, and the regulatory factors NHERF-1/EBP50 or NHERF-2/E3KARP (4, 5). The interaction of ezrin with NHERF proteins occurs via an N-terminal membrane-targeting site that binds cargo molecules, whereas the C terminus contains an F-actin-binding site (6). These ezrin domains interact strongly with each other, masking the membrane and F-actin-binding sites and maintaining ezrin as an inactive monomer in the cytoplasm (7). Phosphorylation at threonine 567 releases the intramolecular interaction, thereby activating ezrin and allowing it to bind to cargo molecules and F-actin (8, 9).
We began by assessing the association of ezrin with the cytoskeleton and ezrin phosphorylation at threonine 567. Both were increased after initiation of Na+-glucose cotransport. These increases correlated with increased NHE3 at the apical membrane and cytoplasmic alkalinization. Stable expression of dominant negative ezrin prevented both NHE3 recruitment to the apical membrane and cytoplasmic alkalinization after initiation of Na+-glucose cotransport, showing that ezrin activation is required for this NHE3 transactivation. Inhibition of p38 MAP kinase prevented both cytoplasmic alkalinization and ezrin phosphorylation at threonine 567. Thus, p38 MAP kinase is required for ezrin activation, which directs translocation of NHE3 to the apical membrane and cytoplasmic alkalinization. In addition to defining a specific role for ezrin in the coordinate regulation of Na+-glucose cotransport and Na+-H+ exchange, these data describe a previously unrecognized pathway of p38 MAP kinase-dependent ezrin activation.
Methods
Generation of Ezrin-Transfected Cell Lines. Vesicular stomatitis virus G protein epitope-tagged full length wild-type Y353F and N-terminal (1–309) ezrin cDNA constructs (6, 10) were ligated into pcDNA3.1 Zeo (Invitrogen) and used to stably transfect Caco-2 cells expressing wild-type SGLT1 (11), as described (12). Isolated clones were screened by immunoblot and immunofluorescence microscopy with anti-vesicular stomatitis virus G epitope tag antibody P5D4 (Sigma). Maintenance of SGLT1 expression and SGLT1-dependent Na+-glucose cotransport were confirmed by using 14C-α-d-methyl glucoside, as described (12).
Measurement of Cytoplasmic pH (pHi). Confluent monolayers were washed with nominally 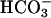 -free Hanks' balanced salt solution (HBSS) with 25 mM mannose and incubated for 15 min at room temperature in 3.5 μM of the acetomethoxyl ester of 2′-7′-bis(2-carboxyethyl)-5 (6)carboxyfluorescein (BCECF-AM, Molecular Probes) as described (1). BCECF-loaded cells were analyzed by using a Model RC-M fluorometer (Photon Technology International, Monmouth Junction, NJ). Fluorescence was measured at excitations of 439 and 502 nm and emission at 535 nm. Flourometric ratios corresponding to pHs of 7.00, 7.25, 7.50, and 7.75 were determined by clamping pHi by using medium (at the designated pH) containing 110 mM KCl (in place of NaCl) and 10 μg/ml nigericin. Standard curves were generated, and experimental data were analyzed by using felix software (version 1.42, Photon Technology International). Initial pHi values of monolayers studied were 7.47 ± 0.01. Given the relatively small change in steady-state pHi seen upon initiation of Na+-glucose cotransport (≈0.1 pH unit), data for each monolayer were standardized to the initial pHi of that monolayer to facilitate comparison between samples. For comparison of pHi responses, the pHi 2 min after initiation of Na+-glucose cotransport, at which time the new steady-state pHi had been achieved, was compared to the pHi before activation of Na+-glucose cotransport. The difference in pHi between these values was considered as the final pHi response. In comparisons between clones, this final pHi response was used. HOE694 and S3226 were generously provided by Hans-Jochen Lang (Hoechst-Marion Roussel, Frankfurt, Germany). PD169316 was from Calbiochem. Pharmacological inhibitors were added in nominally
-free Hanks' balanced salt solution (HBSS) with 25 mM mannose and incubated for 15 min at room temperature in 3.5 μM of the acetomethoxyl ester of 2′-7′-bis(2-carboxyethyl)-5 (6)carboxyfluorescein (BCECF-AM, Molecular Probes) as described (1). BCECF-loaded cells were analyzed by using a Model RC-M fluorometer (Photon Technology International, Monmouth Junction, NJ). Fluorescence was measured at excitations of 439 and 502 nm and emission at 535 nm. Flourometric ratios corresponding to pHs of 7.00, 7.25, 7.50, and 7.75 were determined by clamping pHi by using medium (at the designated pH) containing 110 mM KCl (in place of NaCl) and 10 μg/ml nigericin. Standard curves were generated, and experimental data were analyzed by using felix software (version 1.42, Photon Technology International). Initial pHi values of monolayers studied were 7.47 ± 0.01. Given the relatively small change in steady-state pHi seen upon initiation of Na+-glucose cotransport (≈0.1 pH unit), data for each monolayer were standardized to the initial pHi of that monolayer to facilitate comparison between samples. For comparison of pHi responses, the pHi 2 min after initiation of Na+-glucose cotransport, at which time the new steady-state pHi had been achieved, was compared to the pHi before activation of Na+-glucose cotransport. The difference in pHi between these values was considered as the final pHi response. In comparisons between clones, this final pHi response was used. HOE694 and S3226 were generously provided by Hans-Jochen Lang (Hoechst-Marion Roussel, Frankfurt, Germany). PD169316 was from Calbiochem. Pharmacological inhibitors were added in nominally 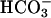 -free HBSS containing 25 mM mannose 15 min before isoosmotic exchange for nominally
-free HBSS containing 25 mM mannose 15 min before isoosmotic exchange for nominally 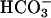 -free HBSS containing 25 mM of glucose and the indicated drug.
-free HBSS containing 25 mM of glucose and the indicated drug.
To assess NHE3-dependent pHi recovery, BCECF-loaded monolayers were incubated with isoosmotic HBSS containing 25 mM glucose, 50 mM NH4Cl, and 50 μM HOE694 (to inhibit NHE1 and NHE2) for 15 min. The buffer was then exchanged isoosmotically for buffer without NH4Cl, but with HOE694.
Detergent Fractionation and Immunoblots. Confluent monolayers were preincubated in nominally 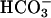 -free HBSS with 25 mM mannose and 0.5 mM phloridzin for 20 min at 37°C. The buffer was then exchanged isoosmotically for media with 25 mM glucose at 37°C. Drugs were included in both buffers. For analysis of detergent-soluble and -insoluble fractions, cells were lysed in 500 μl (per 10 cm2) of csk buffer [50 mM 2-(N-morpholino)ethanesulfonic acid, pH 7.4/3 mM EGTA/5 mM MgCl2/0.5% Triton X-100/2 mM Na2P2O7/1 mM Na3VO4/0.5 μM calyculin A/1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride/0.8 μM aprotinin/0.5 mM bestatin/15 μM E-64/20 μM leupeptin/10 μM pepstatin A), as described (9), and monolayers extracted for 2 min at room temperature with gentle rocking. The csk buffer, representing the detergent-soluble fraction, was removed and residual detergent-insoluble material scraped into 500 μl of SDS/PAGE sample buffer. For phosphoprotein analysis by SDS/PAGE immunoblot, cells were lysed by scraping into 500 μlof4°C SDS/PAGE sample buffer and immediately boiled for 5 min.
-free HBSS with 25 mM mannose and 0.5 mM phloridzin for 20 min at 37°C. The buffer was then exchanged isoosmotically for media with 25 mM glucose at 37°C. Drugs were included in both buffers. For analysis of detergent-soluble and -insoluble fractions, cells were lysed in 500 μl (per 10 cm2) of csk buffer [50 mM 2-(N-morpholino)ethanesulfonic acid, pH 7.4/3 mM EGTA/5 mM MgCl2/0.5% Triton X-100/2 mM Na2P2O7/1 mM Na3VO4/0.5 μM calyculin A/1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride/0.8 μM aprotinin/0.5 mM bestatin/15 μM E-64/20 μM leupeptin/10 μM pepstatin A), as described (9), and monolayers extracted for 2 min at room temperature with gentle rocking. The csk buffer, representing the detergent-soluble fraction, was removed and residual detergent-insoluble material scraped into 500 μl of SDS/PAGE sample buffer. For phosphoprotein analysis by SDS/PAGE immunoblot, cells were lysed by scraping into 500 μlof4°C SDS/PAGE sample buffer and immediately boiled for 5 min.
After SDS/PAGE and transfer to poly(vinylidene difluoride), replicate membranes were immunoblotted by using antibodies to diphosphorylated p38 MAP kinase (Cell Signaling Technology, Beverly, MA), total p38 MAP kinase (Santa Cruz Biotechnology), total ezrin (3C12, Sigma), epitope-tagged ezrin (P5D4, Sigma), or actin (AC15, Sigma). Affinity-purified polyclonal antisera to threonine 567-phosphorylated ezrin was prepared as described (13). Antibodies were detected with affinity-purified goat anti-mouse or -rabbit IgG peroxidase conjugated antibodies (Cell Signaling Technology) and chemiluminescence on BioMax MR film (Eastman Kodak). Signal intensity, corrected for background, was determined by densitometry by using imagej 1.29 (National Institutes of Health, Bethesda).
Fluorescence Microscopy and Image Analysis. Monolayers were fixed in 1% paraformaldehyde in PBS for 15 min as described (14). They were then permeabilized with 0.1% Triton X-100 for detection of surface and intracellular antigen. Permeabilization was omitted for detection of surface antigens, as described (12, 14). NHE3 was detected by incubation with affinity-purified rabbit polyclonal antisera (15) raised in animals immunized with peptides corresponding to the NHE3 intracellular C-terminal tail (residues 666–813) and an extracellular loop (residues 301–332). No staining of nonpermeabilized monolayers was detected by using antisera from animals immunized with the C-terminal NHE3 tail alone. Threonine 567 phosphorylated ezrin was detected only after permeabilization with affinity-purified rabbit polyclonal antisera (13). Both primary antisera were then labeled with Alexa-594 conjugated goat anti-rabbit antibodies (Molecular Probes). In some cases, F-actin was labeled with Alexa-488 conjugated phalloidin, with permeabilization only after surface NHE3 labeling was complete. Stained monolayers were mounted in Slowfade (Molecular Probes) and images collected via a ×100 PLAN APO objective using a Leica DMLB epifluorescence microscope equipped with an 88000 filter set (Chroma Technology, Brattleboro, VT) and Roper Coolsnap HQ camera (Roper Scientific, Duluth, GA) controlled by metamorph 6.1 (Universal Imaging, Downingtown, PA). Representative xy-fields, viewing the monolayer en face from the apical aspect, were chosen for quantitative analysis based on staining quality and cell distribution, as assessed using a nuclear stain (Hoechst 33258) by an observer blinded to the experimental conditions. The apical membrane was located based on brush border F-actin distribution. Based on the z-depth of the widefield images, the entire brush border thickness was imaged by this approach. Selective imaging of surface NHE3 was accomplished by immunolabeling of nonpermeabilized preparations, as described (12, 14). Postacquisition analyses of mean pixel intensity were performed by using metamorph. In all image acquisition and analysis, identically timed exposures within a single experiment were used for quantitative comparisons. For data presentation, postacquisition processing of all images within a single figure was identical.
Statistical Analysis. All experiments were performed with duplicate or greater samples in each individual experiment. Results are expressed as mean ± standard deviation. Student's t test was used to compare the data of different conditions.
Results
Ezrin Activation Follows Initiation of Na+-Glucose Cotransport. We have previously shown that initiation of SGLT1-dependent Na+-glucose cotransport leads to activation of NHE3 via a p38 MAP kinase-dependent signaling pathway (1). NHE3 activity is known to be regulated by the NHE regulatory factors NHERF-1 and -2 (E3KARP). In turn, NHERF-1 and -2 bind to ezrin, potentially linking the NHE3–NHERF complex to F-actin. Thus, we hypothesized that NHE3 activation after initiation of Na+-glucose cotransport might be regulated by ezrin. Activation of ezrin is generally accomplished by release of intramolecular ezrin interactions, allowing ezrin to link target molecules, such as NHERF, to F-actin. To evaluate ezrin activation after initiation of Na+-glucose cotransport we assessed ezrin association with the detergent-insoluble cytoskeletal fraction. An increase in cytoskeletal-associated (detergent-insoluble) ezrin was first detectable 60 sec after initiation of Na+-glucose cotransport and reached 135 ± 8% of control within 180 sec (Fig. 1A, P < 0.02). Notably, a transient decrease in cytoskeletal-associated ezrin was reproducibly detected within 30 sec after the initiation of Na+-glucose cotransport.
Fig. 1.

Ezrin is activated after initiation of Na+-glucose cotransport. (A) Ezrin associates with the cytoskeleton after Na+-glucose cotransport. Cytoskeletal (detergent-insoluble) and cytosolic (detergent-soluble) ezrin fractions were separated at indicated times after initiation of Na+-glucose cotransport. Immunoblot for ezrin showed progressive increases in cytoskeletal ezrin that were accompanied by corresponding decreases in cytosolic ezrin. Densitometric analysis of triplicate samples from this experiment is shown. Results are typical of at least four independent experiments, each with triplicate samples. (B) Ezrin is phosphorylated threonine 567 after Na+-glucose cotransport. Monolayers were lysed at indicated times after initiation of Na+-glucose cotransport and immunoblotted for threonine 567 phospho-ezrin and total ezrin. Densitometric analysis of triplicate samples from this experiment is shown. Results are typical of at least seven independent experiments, each with triplicate samples. (C and D) Ezrin phosphorylation at threonine 567 after Na+-glucose cotransport is most prominent at the apical membrane and perijunctional cytoskeleton. Monolayers were fixed before (C) and 240 sec after (D) initiation of Na+-glucose cotransport and stained for threonine 567 phosphorylated ezrin. The representative images shown were taken at a z-plane just subapical to the microvilli at identical exposures. A marked enhancement in ezrin phosphorylation is evident and, when assessed quantitatively, represents a mean 140 ± 5% increase in phosphorylated ezrin pixel intensity 240 sec after initiation of Na+-glucose cotransport (P < 0.001, n = 20).
Ezrin unfolding and subsequent association with the cytoskeleton are specifically triggered by phosphorylation at threonine 567. Thus, we assessed ezrin phosphorylation using phosphospecific antisera that recognizes ezrin phosphorylated at that site. Threonine 567 phosphorylation increased to 215 ± 19% of control 240 sec after initiation of Na+-glucose cotransport (Fig. 1B, P < 0.02). Similar to ezrin association with the cytoskeleton, a transient decrease in ezrin phosphorylation was reproducibly seen within the first 30 sec after initiation of Na+-glucose cotransport.
Ezrin phosphorylation was also assessed morphologically in cultured monolayers after initiation of Na+-glucose cotransport. Both total and phosphorylated ezrin were predominantly detected in the apical cytoplasm. Phosphorylated ezrin in particular was concentrated at cell boundaries, where it colocalized with the perijunctional actomyosin ring, regardless of Na+-glucose cotransport activity. Ezrin phosphorylation within the apical cytoplasm and particularly within the perijunctional actomyosin ring was markedly increased by initiation of Na+-glucose cotransport (Fig. 1 C and D). Quantitative analysis of the intensity of the phosphorylated ezrin signal showed that it increased to 140 ± 5% of control 240 sec after initiation of Na+-glucose cotransport (P < 0.001). Thus, ezrin phosphorylation was increased by Na+-glucose cotransport beneath the brush border, the site of NHE3 activation.
Expression of Mutant Ezrins Blocks NHE3-Dependent Cytoplasmic Alkalinization. To further evaluate the role of ezrin in NHE3 activation by Na+-glucose cotransport, we stably transfected Caco-2 cells with three ezrin constructs: a full length wild-type ezrin, an ezrin with a tyrosine to phenylalanine point mutation at position 353 (F353Y), and a truncated construct that includes only the N-terminal 309 residues (Nter) of ezrin. Each of the expressed ezrin constructs included a C-terminal vesicular stomatitis virus G epitope tag and has been described (6, 16). The Nter ezrin construct disrupts ezrin function more completely than F353Y and has been characterized in multiple cell types. In epithelial cells, Nter ezrin impairs morphogenic and motogenic responses, whereas expression of a similar truncated ezrin in T cells blocks the polarization that occurs after conjugation with antigen-presenting cells (13, 17). Expression in transfected cells was confirmed by immunoblot and immunofluorescence by using an antibody against the vesicular stomatitis virus G epitope tag. We also verified that expression of wild-type and dominant negative ezrins did not impair SGLT1 expression and function. Apical SGLT1-mediated glucose uptake was comparable in the transfected clones and the parent cell line (data not shown). Finally, to determine whether expression of dominant negative ezrin constructs disrupted basal NHE3 function, we evaluated NHE3-dependent recovery after cytoplasmic acidification. Cells in nominally 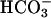 -free media were acidified by NH4Cl pulse and pHi recovery assessed in the presence of 50 μM HOE694 (18, 19), which inhibits NHE1 and NHE2 but not NHE3. Rates of pH recovery were comparable in cells expressing wild-type and mutant ezrin constructs (Fig. 5, which is published as supporting information on the PNAS web site), showing that basal NHE3 function was not globally disrupted by expression of dominant negative ezrins. Thus, SGLT1-dependent glucose uptake and basal NHE3 function were both intact in cell lines stably transfected with wild-type and dominant negative ezrin constructs.
-free media were acidified by NH4Cl pulse and pHi recovery assessed in the presence of 50 μM HOE694 (18, 19), which inhibits NHE1 and NHE2 but not NHE3. Rates of pH recovery were comparable in cells expressing wild-type and mutant ezrin constructs (Fig. 5, which is published as supporting information on the PNAS web site), showing that basal NHE3 function was not globally disrupted by expression of dominant negative ezrins. Thus, SGLT1-dependent glucose uptake and basal NHE3 function were both intact in cell lines stably transfected with wild-type and dominant negative ezrin constructs.
We have previously shown that Na+-glucose cotransport triggers cytoplasmic alkalinization that requires NHE3 (1). Because ezrin can regulate NHE3 function, we studied the role of ezrin in this NHE3-dependent cytoplasmic alkalinization using Caco-2 cells expressing the transfected ezrin constructs. In cells expressing transfected wild-type ezrin, cytoplasmic alkalinization after initiation of Na+-glucose cotransport was identical to that of the control parental cell line (Fig. 2A). Although cytoplasmic alkalinization was triggered by Na+-glucose cotransport in Y353F ezrin expressing cells, the magnitude of alkalinization in three independent clones was reduced by 36 ± 8% (Fig. 2 A, P < 0.01). In contrast, three independent clones expressing Nter ezrin all failed to respond to initiation of Na+-glucose cotransport with cytoplasmic alkalinization following initiation of Na+-glucose cotransport (Fig. 2 A). These data indicate that ezrin is critically involved in activation of NHE3 by Na+-glucose cotransport. Because basal NHE3 function is intact in these Nter ezrin-expressing cells, this represents a failure to increase NHE3 activity after initiation of Na+-glucose cotransport rather than a global loss of NHE3 function.
Fig. 2.

Dominant negative ezrin constructs inhibit NHE3-dependent cytoplasmic alkalinization after Na+-glucose cotransport. (A) pHi increases rapidly after initiation of Na+-glucose cotransport in Caco-2 cells expressing wild-type (wt) ezrin (circles). Although pHi also increased in Caco-2 cells expressing F353Y ezrin (squares), the magnitude of the response was always less than that seen in the parental line or in cells expressing wild-type ezrin. In contrast, Caco-2 cells expressing Nter ezrin (triangles) failed to increase pHi after initiation of Na+-glucose cotransport. Results are typical of more than 10 independent experiments with three separate clones expressing dominant negative ezrin constructs. (B) Inhibition of NHE1 and NHE2 with 50 μM HOE694 (triangles) has no significant effect on the pHi response to Na+-glucose cotransport in cells expressing wild-type ezrin. In contrast, inhibition of NHE3 with 0.1 μM S3226 (squares) markedly reduced cytoplasmic alkalinization, confirming the role of NHE3 in this process. Results are typical of more than five independent experiments. (C) Inhibition of NHE1 and NHE2 with 50 μM HOE694 (triangles) has no significant effect on the pHi response to Na+-glucose cotransport in cells expressing F353Y ezrin. In contrast, inhibition of NHE3 with 0.1 μM S3226 (squares) markedly inhibited cytoplasmic alkalinization, confirming the role of NHE3 in this process. Results are typical of more than five independent experiments.
Consistent with the reported IC50 for NHE3 of 0.02 μM, 0.1 μM S3226, a dose that inhibits NHE3 partially but does not inhibit NHE1 or NHE2 (20), inhibited cytoplasmic alkalinization after initiation of Na+-glucose cotransport by 41 ± 8% in cells not transfected with exogenous ezrins (1). Increasing S3226 to 1 μM, a dose that inhibits NHE3 more completely but may partially inhibit NHE1 (20), increased this inhibition to 66 ± 1% (1), but further increases in S3226 dose did not inhibit alkalinization further. The residual S3226-resistant alkalinization response was not due to other Na+-H+ exchangers, because 1 μM S3226 and 50 μM HOE694 (to inhibit NHE1 and NHE2) in combination inhibited alkalinization by 77 ± 6%, only 11% more than 1 μM S3226 without HOE694 (Fig. 6, which is published as supporting information on the PNAS web site). Thus, although NHE3 activation is responsible for the bulk of cytoplasmic alkalinization after initiation of Na+-glucose cotransport, up to 11% (77-66%) of alkalinization may be due to NHE1 or NHE2 and up to 23% (100-77%) may be due to other processes that are likely not Na+-H+ exchangers, because they cannot be inhibited by HOE694 or S3226. Although presently not identified, a role for 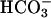 transporters can also likely be excluded, because these studies were done in nominally
transporters can also likely be excluded, because these studies were done in nominally 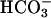 -free media.
-free media.
At 0.1 μM, S3226 prevented alkalinization by 40 ± 6% in cells transfected with wild-type ezrin (Fig. 2B) and by 60 ± 5% in cells expressing F353Y ezrin (Fig. 2C). Inhibition of NHE1 and NHE2 with 50 μM HOE694 did not significantly inhibit cytoplasmic alkalinization after Na+-glucose cotransport in cells expressing wild-type or F353Y ezrin constructs (Fig. 2 B and C), and treatment with 1 μM S3226 and 50 μM HOE694 in combination inhibited alkalinization in cells transfected with wild-type or F353Y ezrin to similar extents. Inhibition of p38 MAP kinase prevented cytoplasmic alkalinization in cells transfected with either wild-type or F353Y ezrin (data not shown). Thus, like the parental cells, the alkalinization observed in wild-type and F353Y ezrin-expressing cells requires p38 MAP kinase-dependent NHE3 activation.
Translocation of NHE3 to the Apical Membrane Is Associated with Cytoplasmic Alkalinization and Is Inhibited by Dominant Negative Ezrin. The data above show that NHE3 activation is necessary for cytoplasmic alkalinization after Na+-glucose cotransport but do not reveal the mechanism of NHE3 activation. We considered NHE3 phosphorylation as a potential regulatory mechanism (21), but, although documented, the role of phosphorylation in NHE3 regulation remains to be established. We also considered trafficking of NHE3 between intracellular vesicles and the apical membrane, which has been described as a mechanism of acute NHE3 up-regulation (22, 23). To explore this possibility, we measured surface NHE3 before and after initiation of Na+-glucose cotransport. Surface expression of NHE3 increased markedly within 30 seconds of Na+-glucose cotransport and then remained elevated (Fig. 3). Total NHE3 content, as assessed by immunofluorescent staining of permeabilized monolayers, did not change over the same interval. Thus, the observed increase in surface NHE3 expression is consistent with recruitment of NHE3 to the apical membrane from intracellular pools and correlates temporally with onset of cytoplasmic alkalinization after initiation of Na+-glucose cotransport.
Fig. 3.

Na+-glucose cotransport activates ezrin-dependent translocation of NHE3 to the apical membrane. (A) Initiation of Na+-glucose cotransport resulted in a marked increase in NHE3 detectable at the apical surface of fixed nonpermeabilized Caco-2 monolayers expressing wild-type (closed circles) but not Nter (open circles) ezrin. The data shown were obtained from quantitative analysis of images like those in B–E. The representative images shown (B–E) were taken at the z-plane of the apical membrane at identical exposures. This immunofluorescent method of quantifying surface NHE3 was method was validated based on comparison with cell surface biotinylation studies, performed as described (34), in which similar data were obtained (Fig. 7, which is published as supporting information on the PNAS web site). (B and C) NHE3 is detected only at the apical membrane of Caco-2 monolayers expressing wild-type ezrin before initiation of Na+-glucose cotransport (B). By 240 sec after initiation of Na+-glucose cotransport (C), surface NHE3 labeling increased by 102 ± 6% (P < 0.001, n = 9). (D and E) NHE3 is detected at the apical membrane of Caco-2 monolayers expressing Nter ezrin before initiation of Na+-glucose cotransport (D). By 240 sec after initiation of Na+-glucose cotransport (E), surface NHE3 labeling increased by 15 ± 8% (P > 0.05, n = 9).
Because Nter ezrin expression prevented cytoplasmic alkalinization after initiation of Na+-glucose cotransport, we considered the hypothesis that the mechanism of Nter ezrin action was inhibition of NHE3 recruitment to the apical membrane. In contrast to the NHE3 recruitment apparent in cells transfected with wild-type ezrin, surface NHE3 expression did not increase after initiation of Na+-glucose cotransport in Nter ezrin-transfected cells (Fig. 3). Thus, Nter ezrin prevents NHE3 activation by blocking NHE3 recruitment to the apical membrane after initiation of Na+-glucose cotransport.
Ezrin Activation Is p38 MAP Kinase-Dependent. The data show that Na+-glucose cotransport results in ezrin activation, NHE3 translocation, and NHE3-dependent cytoplasmic alkalinization. Disruption of ezrin signaling prevents both NHE3 translocation and NHE3-dependent cytoplasmic alkalinization. Previous analyses of signaling between NHE3 and SGLT1 have shown that p38 MAP kinase is required for NHE3 activation to occur (1). Thus, we asked whether activation of p38 MAP kinase and ezrin phosphorylation occurred in parallel. Diphosphorylation of p38 MAP kinase correlates with kinase activation in general, and we have shown that this correlation is also true under the conditions studied here (1). As shown in Fig. 4A, p38 MAP kinase diphosphorylation occurred with kinetics that paralleled ezrin phosphorylation. We considered three possibilities: (i) p38 MAP kinase and ezrin phosphorylation are independent events, both activated by Na+-glucose cotransport; (ii) p38 MAP kinase is activated by ezrin; and (iii) ezrin is activated by p38 MAP kinase. Because the first hypothesis can be inferred only after disproving the second and third hypotheses, we began by asking whether p38 MAP kinase activation required ezrin function. This was assessed by using cell lines expressing transfected wild-type or Nter ezrin dominant negative ezrin. p38 MAP kinase phosphorylation occurred normally after initiation of Na+-glucose cotransport in both wild-type (Fig. 4A) and Nter (Fig. 4B, P < 0.01) ezrin-expressing cell lines. This result shows that the portion of the signaling pathway that links Na+-glucose cotransport to p38 MAP kinase activation is intact in Nter ezrin-expressing cells and does not explain the failure of NHE3-dependent cytoplasmic alkalinization in these cells. Thus, this result excludes the possibility that p38 MAP kinase is activated by ezrin.
Fig. 4.

Ezrin activation is prevented by p38 MAP kinase inhibition. (A) p38 activation occurs in parallel with ezrin activation. Monolayers expressing wild-type ezrin were lysed at indicated times after initiation of Na+-glucose cotransport and immunoblotted for diphosphorylated p38 MAP kinase, total p38 MAP kinase, threonine 567 phosphorylated ezrin, and total ezrin. Densitometric analysis of triplicate samples from a single experiment are shown. p38 MAP kinase and ezrin were phosphorylated with similar kinetics. Results are typical of more than seven independent experiments, each with triplicate samples. (B) Dominant negative ezrin expression does not prevent p38 MAP kinase activation. Monolayers expressing Nter dominant negative ezrin kinase were lysed at indicated times after initiation of Na+-glucose cotransport and immunoblotted for diphosphorylated p38 MAP kinase and total p38 MAP kinase. p38 MAP kinase phosphorylation was similar to that following initiation of Na+-glucose cotransport in cells expressing wild-type or F353Y ezrin. Densitometric analysis of triplicate samples from this representative experiment are shown. Results are typical of three or more independent experiments, each with triplicate samples. (C) Inhibition of p38 MAP kinase prevents ezrin activation. Monolayers pretreated with 10 μM PD169316, to inhibit p38 MAP kinase, were lysed at indicated times after initiation of Na+-glucose cotransport and immunoblotted for threonine 567 phosphorylated ezrin and total ezrin. Increases in ezrin phosphorylation were inhibited completely. Densitometric analysis of triplicate samples from this experiment is shown. Results were similar by using the related p38 MAP kinase inhibitor SB202190, whereas the structurally similar inactive compound SB202474 had no effect on ezrin phosphorylation. Results are typical of more than four independent experiments, each with triplicate samples.
To determine whether ezrin phosphorylation depended on p38 MAP kinase activation, ezrin phosphorylation was assessed when p38 MAP kinase activity was blocked. Inhibition of p38 MAP kinase prevented 84 ± 16% of ezrin phosphorylation after initiation of Na+-glucose cotransport (Fig. 4C, P < 0.01). Thus, ezrin phosphorylation after Na+-glucose cotransport depends on p38 MAP kinase activation. These data establish a signaling cascade whereby Na+-glucose cotransport leads to p38 MAP kinase activation and, in turn, ezrin and NHE3 activation. The data show that each event requires the previous event to occur and verify that inhibition of distal events, e.g., ezrin activation by expression of Nter ezrin, does not prevent proximal events, e.g., Na+-glucose cotransport and p38 MAP kinase activation.
Discussion
Intestinal absorption of Na+ and glucose are essential for maintenance of fluid and electrolyte balance. We have previously shown that Na+ absorption via the apical Na+-H+ exchanger NHE3 is activated by SGLT1-mediated Na+-glucose cotransport (1). This NHE3 activation is associated with mild cytoplasmic alkalinization of ≈0.1 pH units, reflecting increases in H+ efflux and Na+ influx. In nonpolarized cells, this enhancement of NHE3-mediated Na+ influx in response to SGLT1-mediated Na+ (and glucose) influx would seem counterproductive. However, in polarized absorptive cells, it may indicate that, in addition to transporting glucose across the apical membrane, SGLT1 is functioning as a sensor that signals the presence of nutrients in the intestinal lumen, as has been suggested for SGLT3 in muscle and cholinergic neurons (24). Thus, activation of NHE3, the major route of intestinal Na+ absorption (25) participates in an SGLT1-triggered transition to active nutrient and ion absorption. In this way, SGLT1 may be activating a cascade of events that causes a functional phenotypic shift in the absorptive enterocyte.
Although we have shown that SGLT1-dependent regulation of NHE3 requires p38 MAP kinase activation, the mechanisms by which p38 MAP kinase triggers NHE3 regulation have not been identified. One potential intermediate is the cytoskeletal linker protein ezrin (6). Ezrin is an important modulator of cortical actin structure (26, 27), receptor and transporter function (3, 28, 29), and cell survival (16, 30) that interacts with NHE3 and some other transporters via NHERF1/EBP50 and NHERF2/E3KARP (3, 28, 29). In the present study, we aimed to define the role of ezrin in SGLT1 and p38 MAP kinase regulation of NHE3.
We first asked whether ezrin was activated after initiation of Na+-glucose cotransport. Ezrin activation is known to result in increased association with F-actin. The data show that ezrin association with the cytoskeleton increases after initiation of Na+-glucose cotransport. Moreover, the kinetics of this increased cytoskeletal association match the kinetics of NHE3 activation. In most cases, ezrin associates with F-actin after phosphorylation at threonine 567. This phosphorylation releases intramolecular interactions between the amino and carboxy halves of ezrin, allowing the C terminus to associate with the cytoskeleton. We found that, like cytoskeletal association, ezrin phosphorylation at threonine 567 increased after initiation of Na+-glucose cotransport. Immunofluorescent analysis of threonine 567 phosphorylated ezrin showed that the increase occurred in ezrin localized beneath the brush border, where surface NHE3 is localized. We also detected a transient decrease in ezrin phosphorylation at early time points that was not mirrored by decreases in p38 MAP kinase phosphorylation or NHE3 activity. The significance of this exception is unknown, but this phenomenon has also been reported in the earliest stages of ezrin-dependent T cell activation and has been postulated to represent transient ezrin release to permit cytoskeletal reorganization (17).
We asked whether stable expression of dominant negative ezrin constructs could prevent NHE3 activation. Expression of Nter ezrin completely blocked NHE3 activation after initiation of Na+-glucose cotransport. In contrast, expression of F353Y ezrin effected a partial inhibition of NHE3 activation. Both SGLT1-dependent Na+-glucose cotransport and basal NHE3 function were intact in the transfected cell lines. Thus, the effects of dominant negative ezrin expression were due to defects in NHE3 up-regulation but not basal NHE3 function.
The best-characterized mechanism of NHE3 up-regulation is recruitment to the plasma membrane (22, 23). The data show that this is also the mechanism by which Na+-glucose cotransport activates NHE3. Moreover, this recruitment does not occur in Nter ezrin-transfected cells, demonstrating that NHE3 recruitment to the apical membrane requires ezrin activation. Although the specific role of ezrin in this process is not yet defined, it is notable that ezrin has been reported to associate with annexin II, a putative regulator of endocytic traffic, in cholesterol-rich membranes (31), where NHE3 is also found (32).
Activation of both ezrin and p38 MAP kinase occurred in parallel. Thus, we considered three possibilities: (i) that p38 MAP kinase and ezrin phosphorylation are independent events activated by Na+-glucose cotransport; (ii) that p38 MAP kinase is activated by ezrin; and (iii) that ezrin is activated by p38 MAP kinase. The data show that, whereas p38 MAP kinase is phosphorylated normally in cells expressing Nter ezrin, ezrin phosphorylation did not increase in cells treated with p38 inhibitors. Thus, ezrin is activated by a pathway that requires p38 MAP kinase.
Although the data do show an essential role for p38 MAP kinase in ezrin activation after Na+-glucose cotransport, they do not allow discrimination between direct phosphorylation of ezrin by p38 MAP kinase and p38 MAP kinase-dependent activation of down-stream kinase(s) that phosphorylate ezrin. The sequence surrounding threonine 567 lacks proline residues that are a hallmark of MAP kinase target motifs. Thus, p38 MAP kinase may not be the kinase directly responsible for intracellular ezrin phosphorylation. Previous studies have suggested that activation of the ezrin family member moesin can be mediated by rho kinase (33). However, Na+-glucose cotransport-dependent NHE3 activation is not affected by inhibition of rho kinase (1). Additionally, inhibition of rho kinase did not inhibit ezrin phosphorylation at T567 (J.R.T., H.S., and Y. W., unpublished data). Thus, it is unlikely that rho is involved in this p38 MAP kinase-dependent pathway of ezrin activation.
In summary, these data show that ezrin activation is a critical intermediate in recruitment and up-regulation of NHE3 after Na+-glucose cotransport. This requires phosphorylation of ezrin at threonine 567 and increased association with the cytoskeleton. The data also show that p38 MAP kinase activation is necessary for ezrin phosphorylation. Thus, in addition to defining a role for ezrin in this signaling pathway, this is a demonstration of a p38 MAP kinase-dependent mechanism of ezrin activation.
Supplementary Material
Acknowledgments
We gratefully acknowledge the expert technical assistance of Eric D. Black in the early phases of this work and helpful discussions with Drs. Monique Arpin, Mark Donowitz, and Gail Hecht. Wild-type, F353Y, and Nter ezrin constructs were generously provided by Dr. Monique Arpin (Institut Curie, Paris). This work was supported by National Institutes of Health Grants R01DK61931 (to J.R.T.), R01AI050098 (to J.K.B.), and R01DK038510 (to E.B.C.); the University of Chicago Digestive Disease Center (P30DK42086); and the University of Chicago Cancer Center (P30 CA14599).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MAP, mitogen-activated protein; pHi, cytoplasmic pH; Nter ezrin, N-terminal 309 residues of ezrin.
References
- 1.Turner, J. R. & Black, E. D. (2001) Am. J. Physiol. 281, C1533-C1541. [DOI] [PubMed] [Google Scholar]
- 2.Thwaites, D. T., Kennedy, D. J., Raldua, D., Anderson, C. M., Mendoza, M. E., Bladen, C. L. & Simmons, N. L. (2002) Gastroenterology 122, 1322-1333. [DOI] [PubMed] [Google Scholar]
- 3.Yun, C. H., Lamprecht, G., Forster, D. V. & Sidor, A. (1998) J. Biol. Chem. 273, 25856-25863. [DOI] [PubMed] [Google Scholar]
- 4.Yun, C. H., Oh, S., Zizak, M., Steplock, D., Tsao, S., Tse, C. M., Weinman, E. J. & Donowitz, M. (1997) Proc. Natl. Acad. Sci. USA 94, 3010-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamprecht, G., Weinman, E. J. & Yun, C. H. (1998) J. Biol. Chem. 273, 29972-29978. [DOI] [PubMed] [Google Scholar]
- 6.Algrain, M., Turunen, O., Vaheri, A., Louvard, D. & Arpin, M. (1993) J. Cell Biol. 120, 129-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gary, R. & Bretscher, A. (1995) Mol. Biol. Cell 6, 1061-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura, F., Amieva, M. R. & Furthmayr, H. (1995) J. Biol. Chem. 270, 31377-31385. [DOI] [PubMed] [Google Scholar]
- 9.Gautreau, A., Louvard, D. & Arpin, M. (2000) J. Cell Biol. 150, 193-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crepaldi, T., Gautreau, A., Comoglio, P. M., Louvard, D. & Arpin, M. (1997) J. Cell Biol. 138, 423-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner, J. R., Rill, B. K., Carlson, S. L., Carnes, D., Kerner, R., Mrsny, R. J. & Madara, J. L. (1997) Am. J. Physiol. 273, C1378-C1385. [DOI] [PubMed] [Google Scholar]
- 12.Turner, J. R., Lencer, W. I., Carlson, S. & Madara, J. L. (1996) J. Biol. Chem. 271, 7738-7744. [DOI] [PubMed] [Google Scholar]
- 13.Allenspach, E. J., Cullinan, P., Tong, J., Tang, Q., Tesciuba, A. G., Cannon, J. L., Takahashi, S. M., Morgan, R., Burkhardt, J. K. & Sperling, A. I. (2001) Immunity 15, 739-750. [DOI] [PubMed] [Google Scholar]
- 14.Zolotarevsky, Y., Hecht, G., Koutsouris, A., Gonzalez, D. E., Quan, C., Tom, J., Mrsny, R. J. & Turner, J. R. (2002) Gastroenterology 123, 163-172. [DOI] [PubMed] [Google Scholar]
- 15.Musch, M. W., Bookstein, C., Xie, Y., Sellin, J. H. & Chang, E. B. (2001) Am. J. Physiol. 280, G687-G693. [DOI] [PubMed] [Google Scholar]
- 16.Gautreau, A., Poullet, P., Louvard, D. & Arpin, M. (1999) Proc. Natl. Acad. Sci. USA 96, 7300-7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delon, J., Kaibuchi, K. & Germain, R. N. (2001) Immunity 15, 691-701. [DOI] [PubMed] [Google Scholar]
- 18.Counillon, L., Scholz, W., Lang, H. J. & Pouyssegur, J. (1993) Mol. Pharmacol. 44, 1041-1045. [PubMed] [Google Scholar]
- 19.Schmid, A., Scholz, W., Lang, H. J. & Popp, R. (1992) Biochem. Biophys. Res. Commun. 184, 112-117. [DOI] [PubMed] [Google Scholar]
- 20.Schwark, J. R., Jansen, H. W., Lang, H. J., Krick, W., Burckhardt, G. & Hropot, M. (1998) Pflügers Arch. 436, 797-800. [DOI] [PubMed] [Google Scholar]
- 21.Zizak, M., Lamprecht, G., Steplock, D., Tariq, N., Shenolikar, S., Donowitz, M., Yun, C. H. & Weinman, E. J. (1999) J. Biol. Chem. 274, 24753-2478. [DOI] [PubMed] [Google Scholar]
- 22.Ambuhl, P. M., Yang, X., Peng, Y., Preisig, P. A., Moe, O. W. & Alpern, R. J. (1999) J. Clin. Invest. 103, 429-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janecki, A. J., Janecki, M., Akhter, S. & Donowitz, M. (2000) J. Biol. Chem. 275, 8133-8142. [DOI] [PubMed] [Google Scholar]
- 24.Diez-Sampedro, A., Hirayama, B. A., Osswald, C., Gorboulev, V., Baumgarten, K., Volk, C., Wright, E. M. & Koepsell, H. (2003) Proc. Natl. Acad. Sci. USA 100, 11753-11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultheis, P. J., Clarke, L. L., Meneton, P., Miller, M. L., Soleimani, M., Gawenis, L. R., Riddle, T. M., Duffy, J. J., Doetschman, T., Wang, T., et al. (1998) Nat. Genet. 19, 282-285. [DOI] [PubMed] [Google Scholar]
- 26.Takeuchi, K., Sato, N., Kasahara, H., Funayama, N., Nagafuchi, A., Yonemura, S. & Tsukita, S. (1994) J. Cell Biol. 125, 1371-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsukita, S. & Yonemura, S. (1999) J. Biol. Chem. 274, 34507-34510. [DOI] [PubMed] [Google Scholar]
- 28.Sun, F., Hug, M. J., Lewarchik, C. M., Yun, C. H., Bradbury, N. A. & Frizzell, R. A. (2000) J. Biol. Chem. 275, 29539-29546. [DOI] [PubMed] [Google Scholar]
- 29.Cao, T. T., Deacon, H. W., Reczek, D., Bretscher, A. & von Zastrow, M. (1999) Nature 401, 286-290. [DOI] [PubMed] [Google Scholar]
- 30.Krieg, J. & Hunter, T. (1992) J. Biol. Chem. 267, 19258-19265. [PubMed] [Google Scholar]
- 31.Harder, T., Kellner, R., Parton, R. G. & Gruenberg, J. (1997) Mol. Biol. Cell 8, 533-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li, X., Galli, T., Leu, S., Wade, J. B., Weinman, E. J., Leung, G., Cheong, A., Louvard, D. & Donowitz, M. (2001) J. Physiol. 537, 537-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeon, S., Kim, S., Park, J. B., Suh, P. G., Kim, Y. S., Bae, C. D. & Park, J. (2002) J. Biol. Chem. 277, 16576-16584. [DOI] [PubMed] [Google Scholar]
- 34.Lencer, W. I., Moe, S., Rufo, P. A. & Madara, J. L. (1995) Proc. Natl. Acad. Sci. USA 92, 10094-10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}