

Abstract
Heme-containing chlorite dismutases (Clds) catalyze a highly unusual O–O bond forming reaction. The O–O cleaving reactions of hydrogen peroxide and peracetic acid (PAA) with the Cld from Dechloromonas aromatica (DaCld) were studied to better understand the Cl–O cleavage of the natural substrate and subsequent O–O bond formation. While reactions with H2O2 resulted in slow destruction of the heme, at acidic pH, heterolytic cleavage of the O–O bond of PAA cleanly yielded the ferryl porphyrin cation radical (Compound I). At alkaline pH, the reaction proceeds more rapidly and the first observed intermediate is a ferryl heme. Freezequench EPR confirmed that the latter has an uncoupled protein-based radical, indicating that Compound I is the first intermediate formed at all pH values and that radical migration is faster at alkaline pH. These results suggest by analogy that two-electron Cl–O bond cleavage to yield a ferryl-porphyrin cation radical is the most likely initial step in O–O bond formation from chlorite.
O–O bond breaking processes are fundamental to biological reactions involving O2 and its aqueous reduction products. O–O bond joining, formally the microscopic reverse, is much rarer. Chlorite dismutases (Clds) are heme-dependent enzymes that catalyze a biologically unusual O–O bond joining reaction, converting a single chlorite ion (ClO2−) to O2 and Cl−.1–7 This reaction is of interest as a surrogate for understanding the only other well-described O–O bond joining process in biology, catalyzed by photosystem II, as well as efforts to convert water or other reduced species to O2 using synthetic catalysts.
Clds are widespread across bacterial and archaeal species,8, 9 with only a handful known or expected to catalyze efficient chlorite decomposition, a reaction normally associated with bacterial respiration of perchlorate.10 Clds are frequently described and annotated as peroxidases, though the biochemical capabilities and biological roles of these presumably non-chlorite-reactive Clds are not entirely clear. Structurally, the Clds are part of a larger protein superfamily containing the heme-binding dye-decoloring peroxidases (DyPs) and their EfeB sequencerelatives, both of which bear striking similarities to Clds (Figure 1).7–9, 11–15 Well-characterized DyPs and EfeBs are highly active heme-dependent peroxidases, facilitating cleavage of the O–O bond of H2O2 via generation of high-valent Fe(IV)=O intermediates that serve as reactive oxidants.11–13, 16 The structural similarity between Clds and these proteins suggests that Clds, particularly those from non-perchlorate respiring bacteria and archaea, may likewise react with H2O2, organic peroxides, or peracids (ROOH), which are also much more biologically common substrates than chlorite. If such reactivity were avid, it might suggest that members of this large family of proteins serve as novel peroxidases or catalases. The reactivity of Clds with alternate substrates, however, has not previously been examined in detail.
Figure 1. DaCld monomer and active site structures (dark blue) shown overlaid with the structure of the representative dye-decoloring peroxidase from Rhodococcus jostii (RjDypB, cyan).
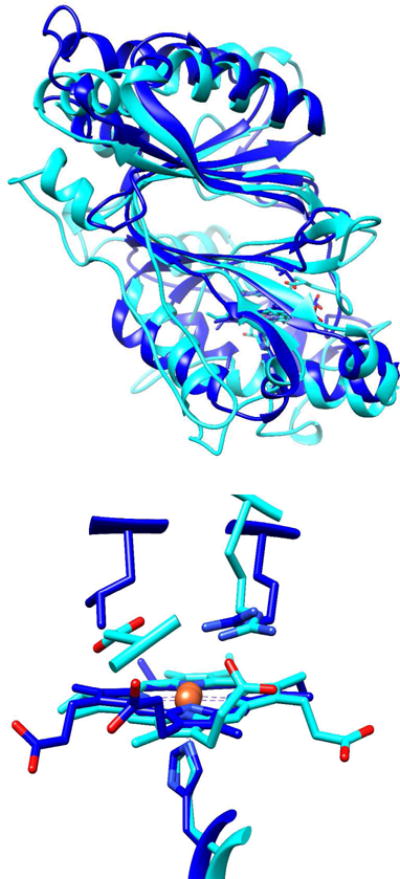
(Top) Monomers of RjDypB (PDB: 3QNR) and DaCld (PDB:3QNR) are shown as cartoons. Hemes are shown as sticks in the N-terminal domains. (Bottom) Heme-containing active sites are overlaid, showing the different orientations of the heme and catalytically important residues. RjDypB has a proximal His226 and distal Arg-Asp pair (Arg244, Asp153). DaCld likewise has a proximal His ligand to the heme (His170) and an Arg-Leu pair (Arg183, Leu185). A propionic acid side chain in DyPs is in hydrogen bonding contact with the distal Arg. This interaction is not present in chlorite dismutases, due to the heme’s flipped position in the active site.
Here, we describe steady-state, transient kinetic, and initial spectroscopic studies of the reaction of the Dechloromonas aromatica Cld (DaCld) with H2O2 and peracetic acid (PAA, CH3(CO)OOH, pKa = 8.2). These studies both reflect on the Clds’ potential biological roles as peroxide-directed catalysts and give insights into the unusual O–O bond joining reaction catalyzed by this enzyme. The use of these alternate oxidants has been especially important for achieving the latter objective because O2 generation by DaCld is extraordinarily rapid,17 preventing the direct observation of reaction intermediates via conventional stopped-flow UV/vis transient kinetic methods or allied freeze-quench techniques. We have therefore studied surrogate reactions with peroxide and peracids in order to gain indirect insight into how O2 is formed by Clds. These studies draw on the insight, originally put forward by Babcock and others, that biologically relevant O–O cleaving mechanisms are the microscopic reverse of O–O joining.18 The proposed O–O radical and nucleophilic joining mechanisms for DaCld are indeed analogous in a reverse sense to the homolytic and heterolytic mechanisms for breaking the O–O of peracid or peroxide (Scheme 1), and the high-valent heme intermediates formed in either O–O joining or scission are identical. As a consequence, we hypothesize that the identity of the intermediates in the DaCld reaction with the reduced oxygen species should report on the types of intermediates this enzyme preferentially stabilizes, and therefore the likely mechanism by which it converts chlorite to O2.
Scheme 1.

Proposed reaction mechanisms for Cl–O/O–O bond cleavage and O–O bond formation in DaCld, illustrating the relationships between the two pathways and their proposed microscopic steps. (A) Heterolytic O–O bond cleavage from the heme-PAA (OOAc−) complex (top pathway) yields Compound I and an acetate (OAc−) leaving group. Compound I is two electron equivalents above Fe(III) and therefore can catalyze two H-atom transfers from substrate (RH). Homolytic bond cleavage (bottom pathway) is an alternative, leading to Compound II and acetyl radical (OAc•). Compound II can catalyze one H-atom transfer, returning the heme to its ferric form. (B) Heterolytic bond cleavage from the analogous ferric-chlorite complex (top pathway) yields Compound I and the chlorite leaving group. Homolytic bond cleavage leads to Compound II and chlorine monoxide Recombination of the leaving group and high valent heme intermediate by a nucleophilic (top pathway) or radical (bottom pathway) process could yield a ferric-peroxyhypochlorite complex that would in turn proceed to yield chloride and dioxygen.
The results reported here support a heterolytic O–O bond cleaving mechanism for PAA over the entire range of pH for which the enzyme is stable. This suggests, by analogy, a mechanism for O2 production from chlorite involving Compound I ([Fe(IV)=O]P+•) formation followed by nucleophilic recombination of the hypochlorite leaving group (Scheme 1). Movement of the exchange coupled radical away from the porphyrin and onto the protein accelerates at higher pH, kinetically destabilizing the Compound I intermediate. Slow turnover with substrates and bleaching/degradation of the heme cofactor by H2O2 suggest that peroxidase chemistry is biologically unlikely for DaCld and closely related enzymes that lack an active site base; however, the protein is an avid peroxidase if an already-deprotonated peracid is used. These results are discussed in the context of both the likely O2-generating mechanisms and their implications for the larger superfamily.
EXPERIMENTAL PROCEDURES
Reagents
Stocks of reagent grade hydrogen peroxide (H2O2) (35% Acros) and peracetic acid (PAA) (Sigma-Aldrich) were freshly made and their concentrations determined by iodometric titration. Briefly, to 100 μL of a solution of stock was added 10 μL of 12 M HCl and ~10 μL of a solution of KI (7 mM final concentration). The resulting I2 that formed (yellow solution) was titrated by addition of 2–200 μL volumes of a 1 mM sodium thiosulfate standard (Titristar) in the presence of 2 μL of a starch indicator (Ricca Chemicals/1.0% w/v) until the blue color due to I2 disappeared. For each mole of peroxide or peracid initially present, 2 moles of sodium thiosulfate are consumed. Reductants used were guaiacol (99%, Acros), (TCI-Ace) and L-ascorbic acid (Sigma).
Protein preparation and analysis
DaCld expression and purification were carried out as previously described for both the wild-type and R183Q mutant.19 All data are referenced to the concentration of heme-bound protein subunit as determined by the pyridine hemochrome assay of Berry and Trumpower.20
Chlorite dismutase steady-state assay
Continuous oxygen production from chlorite decomposition was routinely measured using a Clark-type O2 electrode. The electrode was equilibrated at 4 °C and calibrated using the calculated O2 concentration of air-saturated water as a reference. The specific activity of the enzyme was defined as μmoles of O2 produced min−1mg−1 enzyme (4 °C, 0.1 M phosphate buffer pH, 6.8, 2 mM chlorite).
Buffers
For pH-dependent studies, the following buffers were used: 0.1 M citrate-phosphate (pH 4–8) and 0.1 M borate-phosphate or 0.2 M glycine (pH 8–10).
Steady-state kinetics
Peroxidase activity was studied by measuring the initial rates for the oxidation of guaiacol using hydrogen peroxide (H2O2) or peracetic acid (PAA). Reactions were monitored spectroscopically at 25 °C in a 1 cm pathlength quartz cuvette using a Varian Cary 50 spectrometer with temperature control from a Peltier cooler. Guaiacol oxidation was monitored at 460 nm (ε470 = 26.6 mM−1cm−1). Reactions were initiated by the addition of 1–2 μM DaCld.
Catalase activity was continuously monitored via oxygen production using a Clark-type O2 electrode. Equilibration and calibration were as described above. Reactions were carried out in a 1.5 mL chamber in which buffer alone was equilibrated for 5 min under N2 gas, bringing the starting O2 concentration to 0–50 μM. Background O2 consumption by the probe was measured prior to addition of saturating H2O2 via gas-tight Hamilton syringe. 1–2 μM DaCld was added via gas-tight syringe to initiate the reactions.
Transient kinetics
Reactions with H2O2 and PAA were monitored using a Hi-Tech SF-61DX2 stopped-flow system in single mixing mode by rapidly mixing the enzyme (~1–10 μM) with varying concentrations of PAA or H2O2. Apparent rate constants (kobs) were calculated by fitting single-exponential curves using the KinetAssyst software from Tgk Scientific. Second-order rate constants were determined under pseudo-first order conditions from the slope and intercepts of kobs versus PAA or H2O2 concentrations. Both enzyme and oxidant were dissolved in the same buffer prior to binding experiments and the pH was confirmed routinely.
Sequential-mixing stopped-flow analysis was used to measure the rates of reduction of intermediates and/or to determine how many equivalents of reductant were required to regenerate the ferric starting material (Scheme 1). The appropriate intermediate was prepared by rapidly mixing enzyme with 1 eq of PAA. The intermediate was allowed to maximize in concentration (based on kinetics obtained from single mixing experiments) before subsequent mixing with a solution containing varying concentrations of reductant (ascorbate). All measurements were carried out at 20 °C. Kinetic data were subjected to global nonlinear least squares analyses (SPECFIT program) to obtain best fit kinetic parameters and component spectra (i.e. reactant, intermediate and product spectra).
Resonance Raman spectroscopy
For RR spectroscopy, the final Cld concentrations in the PAA reaction ranged between 55 and 70 μM. Resonance Raman spectra were obtained with 406.7 nm excitation from a Kr+ laser, using the 135° backscattering geometry. The spectrometer was calibrated against Raman frequencies of toluene, dimethyl formamide, and acetone. Data were collected at ambient temperature from samples in spinning 5-mm NMR tubes. Laser power at the samples ranged from 10 to 15 mW. Spectra were collected over time in 15s acquisitions.
Freeze quench EPR
Reaction mixtures of 30–100 μM DaCld and peracetic acid (5–20-fold excess) in 0.2 M phosphate buffer at pH 5.8 or 8.0 were freeze quenched in milliseconds and seconds time regimes. Freeze quenching in the millisecond range was accomplished with a commercial rapid-mix, freeze-quench system. Room temperature reaction mixtures were sprayed into an isopentane slush at −120 °C and then packed into quartz tubes for examination by EPR spectroscopy. Reactions quenched on the seconds time scale were mixed at 4 °C, put into quartz tubes, and flash frozen in an acetone slush bath (−95 °C). Uncertainty in the quench times for these samples is estimated at ±1 s. X-band EPR spectra were recorded at 77 K using a Varian E9 spectrometer. The spectra were referenced to 2,2-diphemyl-1-picrylhydrazyl (DPPH) in a frozen toluene glass. Spin quantification was carried out with microwave power of 20 μW, where neither the DPPH standard nor the protein radical signals were saturated. Integrated intensities were determined from normalized double integrals of the derivative EPR spectra.
RESULTS
pH dependence of steady state and transient reactions
The steady state reaction of DaCld with chlorite was previously studied over a wide range of pH,21 though the transient reaction with chlorite is too fast to observe directly.22 The steady state and transient kinetics of the DaCld reactions with H2O2 and PAA were studied here at several pH values over which the enzyme is stable (pH 6–10). Though it was not possible to obtain an exact pKa, the reactions with PAA change dramatically around neutral pH (vide infra). Several results are therefore presented at pH 6 and 8, as these values illustrate the behavior of DaCld over the entire active range of acidic and alkaline pHs, respectively (Scheme 2).
Scheme 2.
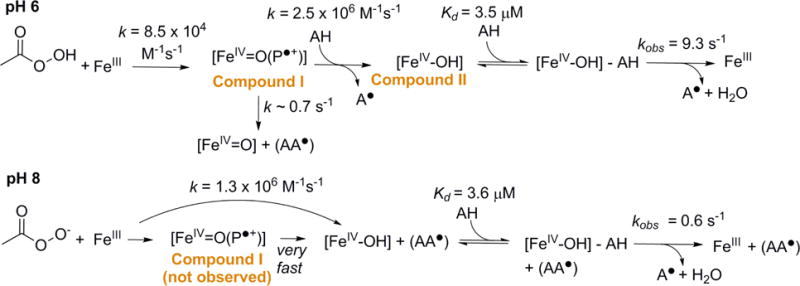
Proposed reactions between ferric DaCld and peracetic acid at pH 6 and 8. The proposed intermediates are indicated.
Catalase activity of DaCld
Rates of O2 production from DaCld/H2O2 were measured in the presence of 20 mM H2O2 from pH 4–10. In this pH range, the values obtained were not significantly above background/no enzyme controls, and well below rates measured for typical catalases.23, 24 Based on comparison of these data, DaCld does not exhibit significant catalase activity.
Steady-state kinetics of DaCld, H2O2 or peracetic acid, and guaiacol
The peroxidase reaction (Scheme 1, top) begins with peroxide activation followed by two sequential one-electron oxidations (hydrogen atom transfers) of the substrate AH:
| (3) |
| (4) |
| (5) |
Compound II is a ferryl heme, [FeIV=O], with no porphyrin or protein-based radical. The steadystate reaction of DaCld with H2O2 and the peroxidase co-substrate guaiacol (2-methoxyphenol) was studied at pH 6 and 8. At both pH values, DaCld is an inefficient catalyst for the oxidation of guaiacol by H2O2, yielding values for kcat ~103 fold and kcat/KM(H2O2) ~104–105-fold smaller than those measured for highly efficient horseradish peroxidase-C (HrP–C) at pH 7 (Table 1). The kcat/KM(H2O2) increases only slightly (~10 fold) with pH. By contrast, DaCld exhibits significantly greater peroxidase activity with PAA as the oxidizing agent, with kcat/KM(PAA) is 2–3 orders of magnitude higher than kcat/KM(H2O2).
Table 1.
Steady state kinetic constants for the reactions of DaCld and peroxidases with phenolic cosubstrates
| Enzyme/Oxidant | Co-substrate | pH | kcat (S−1) | kcat/KM(oxidant) (M−1Y−1) | Ref |
|---|---|---|---|---|---|
| DaCld/ClO2−a | None | 5.2 | 2.0(0.6)×104 | 3.2(0.4)×107 | 21 |
| 7.6 | 3.0(0.8)×103 | 6.9(0.8)×106 | |||
| DaCld/PAAb | Guaiacol | 6 | 2.8 (0.50) | 9.3 (1.8)×103 | This work |
| 8 | 0.3 (0.01) | 3.1 (0.2)×104 | This work | ||
| DaCld/H2O2b | Guaiacol | 6 | 0.2 (0.02) | 4.1 (1.5)×101 | This work |
| 8 | 0.2(0.01) | 4.8(0.9)×102 | This work | ||
| HrP-C/H2O2c | Guaiacol | 7 | 376 (30.0) | 4.5×106 | 50 |
| DyP-A/H2O2 | Pyrogallol | 4.5 | 0.31 (0.01) | 4.9(0.3)×102 | |
| DyP-A/H2O2d | ABTS | 4.5 | 68.0 (2.00) | 1.7(0.1)×104 | 16 |
| DyP-B/H2O2 | Pyrogallol | 4.5 | 2.08 (0.04) | 7.9(0.2)×104 | |
| DyP-B/H2O2d | ABTS | 4.5 | 14.1 (0.20) | 2.1 (O.l)×105 |
Buffers = 100 mM Citrate-Phosphate, 4 °C.
Guaiacol at saturating (8.35 mM) concentrations; buffers =100 mM Citrate-Phosphate, 20 °C.
KM determined from Lineweave-Burke plots; buffers = 10 mM Sodium Phosphate, 25 °C.
50 mM sodium acetate.
The kcat for the O2 producing reaction with chlorite is larger still (Table 1), possibly because this reaction depends on the formation of a single enzyme-substrate complex rather than sequential reactions with three separate substrate molecules (oxidant, HA, HA). The magnitude of kcat/KM(oxidant) is largely due to the efficiency of formation of the initial enzyme-substrate complex and appears to be greater for anionic oxidants. Chlorite is effectively deprotonated over the pH range studied (HClO2 ⇆ ClO2− + H+; pKa = 1.86)25 and kcat/KM(chlorite) is close to the limit imposed by diffusion. By contrast, the relatively high pKa of H2O2 (11.6) suggests that it is present overwhelmingly in its neutral form over the pH range studied,26 while PAA’s pKa (and kcat/KM) is intermediate between the two (CH3(CO)OOH ⇆ CH3COOO− + H+; pKa = 8.2).
Transient reaction with PAA at pH 6
Following reaction with 3–5 eq PAA, the ferric species converts isosbestically to an apparent Compound I species based on the diminished Soret band and the band energies (395 nm, shoulder at 341 nm; visible bands: 525, 550 600, 645 nm). A second-order rate constant (k = 1.9×105 M−1s−1) for its formation was determined (Figure S1).
The Compound I formed at pH 6 with fewer than 5 eq of PAA decays to a mixture of two species within 15 s, consistent with an Fe(III) high spin heme similar to the resting enzyme (ν3 1494 cm−1, ν4, 1372 cm−1) and a ferryl species (ν3, 1508 cm−1, ν4, 1378 cm−1) (Figure S2).27–34 The 77 K X-band EPR spectrum of a sample prepared in a similar manner and frozen in liquid N2/acetone has a g = 2.005 signal with a peak-to-peak line width of 18 G (Figure S3). Because this signal is readily observed at 77 K, it is concluded to arise from an organic, amino-acid-based radical (AA•) that is not exchange coupled with the ferryl: [FeIV=O] + (AA•). The isoelectronic, exchange-coupled intermediates [FeIV=O(AA•)] or [FeIV=O(P+•)], by contrast, would both be expected to exhibit very broad EPR signals at 10 K that would be undetectable at 77 K.35–37 The lack of exchange coupling with the iron suggests the Fe and AA• are separated by >10 Å. Based on current data, it is not possible to determine the location/locations of the amino acid radical, which may be inhomogeneous, or the heme oxidation state (FeIII or FeIV=O) with which the protein-based radical is associated.
Gradual decay of the [FeIV=O] to ferric Cld and/or FeIII + (AA•), which are indistinguishable by RR, is observed in the RR spectra via increasing intensity of the 1494-cm−1 ν3 and a corresponding decrease in the 1508-cm−1 ν3 intensity. These data (Figure S2) indicate that the conversion of [FeIV=O] back to resting enzyme is slow in the absence of a large excess of oxidant. Over a period of minutes, the heme-based chromophore observed for the 15 s intermediate bleaches completely, indicative of heme destruction.
The reaction of ferric DaCld with higher concentrations (>5 eq) of PAA yields Compound Imore rapidly. Its conversion to a second intermediate and subsequent heme destruction (occurring over several minutes) are more clearly observable. The second intermediate has a UV/vis spectrum consistent with either Compound II, the one-electron reduction product of Compound I, or [FeIV=O] + (AA•), which is isoelectronic with Compound I (Soret at 410 nm; α/β bands: 525, 555 nm). Both intermediates have indistinguishable UV/vis and RR spectra in CcP and similar peroxidases.38 Their Soret band extinction of ~105 M−1cm−1 reflects intact aromaticity in the porphyrin ring and contrasts with the diminished absorbance of the non-aromatic Compound I. The rate of conversion of Compound I to the second intermediate is independent of [PAA] (100–500 μM or 10–50 eq), occurring via a single exponential process with k = 0.95 s−1 (Figure S5) and maximizing at ~8.7 s after Compound I forms. [PAA] independence indicates that PAA does not act as a reducing agent toward Compound I and is consistent with a unimolecular process in which the porphyrin radical from [FeIV=O(P+•)] moves onto an amino acid to form an uncoupled radical. The 77 K EPR spectrum measured for a 15 s sample is similar in appearance to that described above for the reaction of DaCld with <5 eq of PAA. Spin quantification of the EPR signal indicates a protein-based radical in ~36% of the total enzyme.
Transient reactions of Compound I with ascorbate (pH 6)
Sequential mixing experiments with PAA and ascorbate were carried out to study the reactivity of the intermediate. The putative Compound I was generated by mixing a slight stoichiometric excess of PAA with the enzyme and aging for 2 s. It was then mixed with either 1 or 2 eq of ascorbate and the reaction to form 1-e− oxidized semidehydroascorbate followed via stopped-flow spectrophotometry. One-electron donation from ascorbate (which can in principle act as either a 1-or 2-e− donor) is the observed pathway in plant peroxidases; semidehydroascorbate is thought to dimerize over time.39, 40 Following mixing with 1 eq of ascorbate, a species consistent with Compound II, the expected 1-e− reduction product, forms (Soret band at 412 nm; α/β = 525, 555 nm). This species persists for several minutes with concurrent, gradual bleaching of the chromophore. It is important to note that, while this species is electronically consistent with assignment as Compound II, a definitive assignment will require EPR characterization of the intermediate. Reacting Compound I with 2 eq of ascorbate results in the initial formation of an apparent Compound II, maximizing after ~0.0025 s. This intermediate subsequently converts isosbestically back to the ferric form in 1.5 s (Figure 3B).
Figure 3. DaCld Compound I converts to (A) ferryl and (B) ferryl and ferric species after mixing with 1 and 2 eq ascorbate, respectively, exhibiting a complete peroxidase cycle at pH 6.
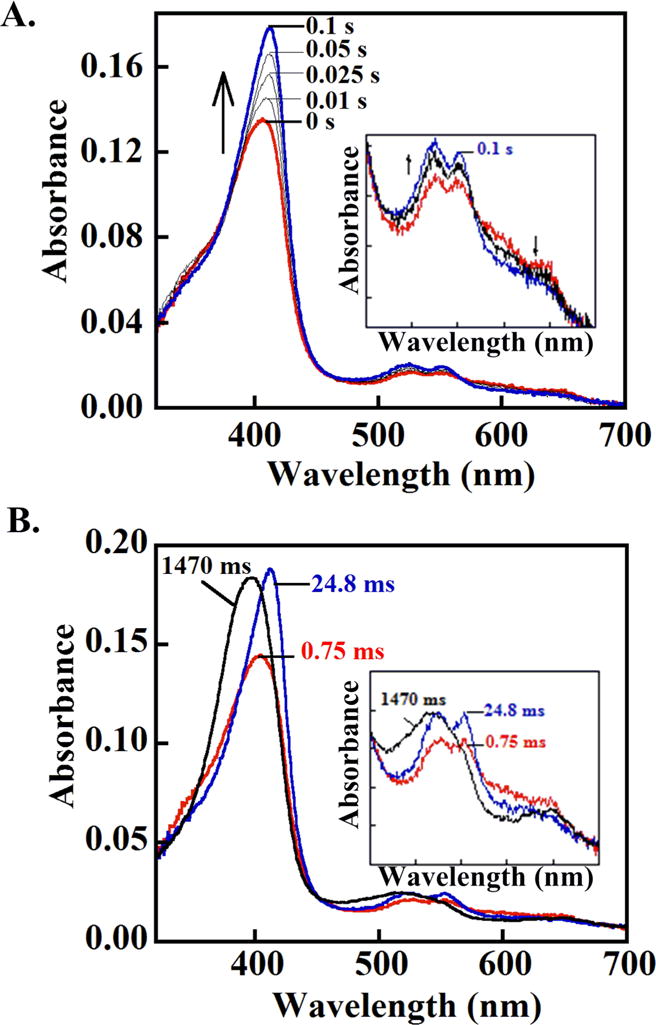
(A) DaCld (10 μM, 2.5 μM final) was mixed with a slight stoichiometric excess of peracetic acid and aged for ~2 s. The Compound I species that forms (red spectrum) was then mixed with 1 eq of ascorbate (2.5 μM final), forming the ferryl species (blue spectrum) after ~0.1 s. This species is tentatively assigned as Compound II. (B) (B) Compound I (red) was formed as in (A) and subsequently mixed with 2 eq of ascorbate (5 μM final). An intermediate with a spectrum resembling Compound II or [Fe(IV)=O] + (AA▪) forms after 2.5 ms and converts to the ferric enzyme after 1.5 s (black). Insets: visible bands (450–700 nm) shown on an expanded scale.
The reaction of Compound I with pseudo first order concentrations of ascorbate (≥ 10 eq) was examined. Rate constants for the single exponential conversion of Compound I to Compound II were fit at 415 nm, yielding a second order rate constant of 2.5×106 M−1s−1 (Figure 3S). The subsequent conversion of Compound II to the ferric species (400 nm) occurred as a single phase. The resulting kobs have a hyperbolic dependence on [ascorbate] with a maximum of 9.3 s−1 and an apparent Kd = 3.5 μM (Figure S6).
Transient reaction with PAA at pH 8
Ferric DaCld at pH 8.0 has a Soret band at 396 nm (slightly red shifted relative to pH 6) and charge-transfer bands at 510 and 645 nm (unchanged with pH).21 Following reaction with ≥3 eq of PAA, the ferric enzyme converts isosbestically to an apparent ferryl species (Soret band at 415 nm with greater absorptivity than ferric; shoulder at 350 nm; α/β bands = 525, 555 nm; Figure 2B, Table 2). The enzymatic intermediate could be [FeIV=O], the product of direct, homolytic cleavage of the PAA O–O bond (Scheme 1, top). Alternatively, it could be [FeIV=O] + (AA•), which would form via radical migration away from a preceding, undetected Compound I and which would have an identical UV/vis spectrum. Because the intermediate forms with apparent isosbestic points from the ferric species, conversion of [FeIV=O(P+•)] to [FeIV=O] + (AA•) would have to be very fast relative to the rate of [FeIV=O(P+•)] formation. Values for kobs fit at 415 nm depended linearly on [PAA] (k = 1.3×106 M−1s−1) (Figure S7). The intermediate persists for several seconds before the heme chromophore bleaches. Bleaching occurs at a faster rate at higher [PAA].
Figure 2. DaCld forms distinct intermediates at (A) acidic and (B) alkaline pH following rapid mixing with 3 eq PAA.
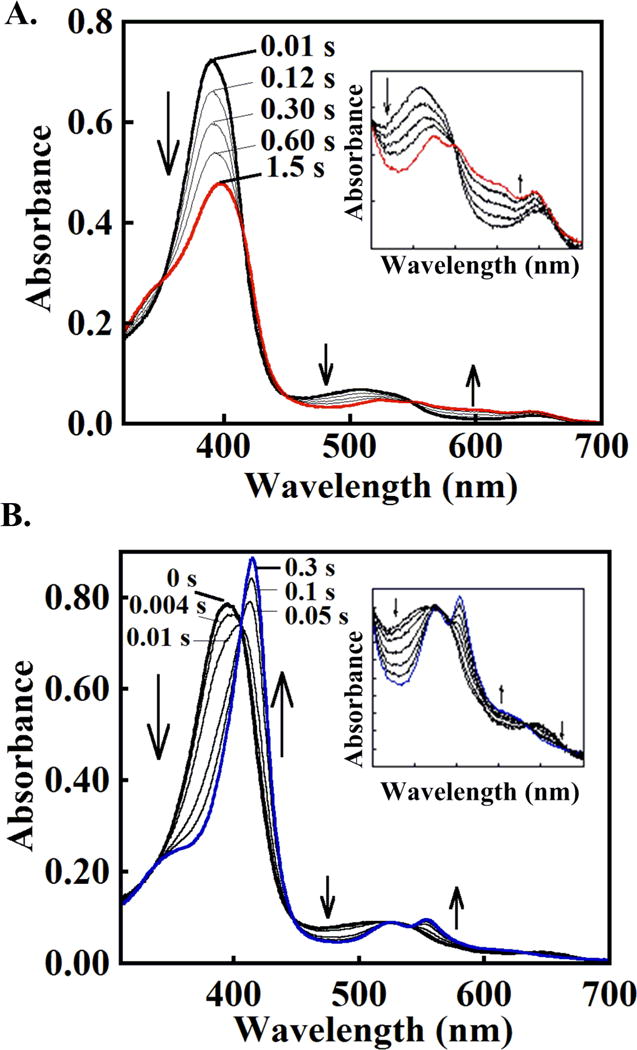
(A) Compound I forms at pH 6. ~15 μM DaCld (7.5 μM final) was mixed with 50 μM peracetic acid (25 μM final) in 0.2 M citrate-phosphate buffer (20 °C). \The initial spectrum is shown in black and the spectrum for the intermediate formed after ~1.5 seconds in red. Intervening spectra measured at the given times are gray. (B) Compound II or [Fe(IV)=O] + (AA▪) forms as the initial intermediate under similar conditions but at pH 8. The initial spectrum is shown in black and the spectrum for the ferryl intermediate that formed after ~300 ms in blue. Intervening spectra measured at the given times gray. Insets: visible bands (450 – 700 nm) shown on an expanded scale.
Table 2.
UV/visible absorbances for stable and intermediate states of DaCld and well-characterized heme peroxidases
| Species | Soretb | CT1 | β | α | CT2 | Ref |
|---|---|---|---|---|---|---|
| DaCld Ferric pH 6 | 392 (99) | 645 (2.2) | 510 | This work | ||
| DaCld Compound I | 395 | 600, 645 sh. | 525 | 550 sh. | ||
| DaCld Compound II | 412 | 525 | 555 | |||
| DaCld | 410 | None | 525 | 555 | None | |
| DaCld Ferric pH 8 | 396 (109) | 645 (3.3) | 535 sh. | 510 | ||
| DaCld Compound 0 | 408 | None | 535 | 575 sh. | None | |
| DaCld | 415 (125) | None | 525 | 555 | None | |
|
| ||||||
| Ferric HRP | 402.5 | 643 (3.23) | 530 | 580 (2.8) | 498 | 23 |
| HRP Ferrous-O2 | 417 (108) | None | 544 | 580 | None | |
| HRP Compound I | 400 | 651, 622 (6.6) | 525 | 577 (7.4) | ||
| HRP Compound II | 410 (105) | 527 | 555 | |||
|
| ||||||
| Ferric DypB | 404 | 634 | 503 | 16 | ||
| DypB Compound I | 397 | 648 | 580, 613 | |||
| Ferric DypA | 408 | 632 | 502 | |||
| DypA Compound II | 419 | 619 | 528 | 557 | ||
|
| ||||||
| Ferric EfeB | 406 | 660 | 485 | 76 | ||
| EfeB Compound I | 414 | 603 | 530 | 555 | ||
This species forms following reaction of Compound I with one eq of ascorbate.
Numbers in parentheses are extinction coefficients (mM−1cm−1). The indicated band has a shoulder = sh.
Resonance Raman data were measured for DaCld at 15 s intervals following reaction of the enzyme with ~8 eq of PAA at pH 8 (Figure 4A). The resting enzyme contains 5-coordinate high spin heme, similar to the enzyme at pH 6.0 (ν3 1494 cm−1, ν4, 1371 cm−1). Following addition of PAA, the enzyme converts to a species with a RR spectrum consistent with a ferryl (ν3 at 1508 cm−1, ν4 at 1377 cm−1).27, 28, 30, 31, 34, 41, 42 This spectrum is very similar to that measured for the [Fe(IV)=O] + (AA▪) intermediate that follows Compound I in the pH 6.0 DaCld/PAA reactions (Figure S2, Inset). The intermediate persists for 15–30 s, after which some ferric enzyme begins to be detected (intensity at 1494 cm−1 and a shift in the ν4 frequency back towards 1371 cm−1). A plot of ν4 versus time illustrates conversion of the ferryl back to the resting ferric under low [PAA], when the bleaching process is relatively slow (Figure S4).
Figure 4. Spectroscopic characterization of the pH 8 intermediate shows that it is likely a breakdown product of Compound I.
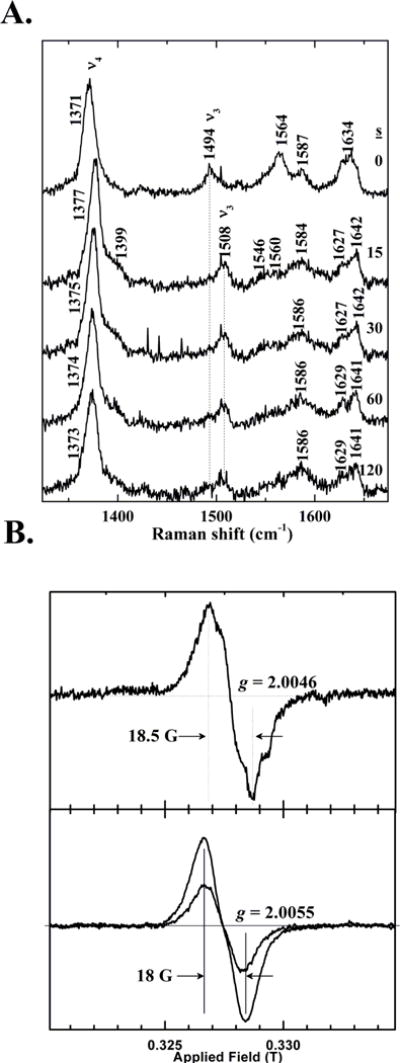
(A) High frequency resonance Raman spectroscopy confirms the presence of a ferryl intermediate in the reaction of PAA and DaCld at pH 8. Final concentrations of DaCld and peracetic acid following mixing were 58 μM and 480 mM, respectively (~8 eq PAA). Spectra were measured prior to mixing and via fifteen second acquisitions subsequently. Data were acquired at ambient temperature with 406.7 nm excitation and 12 mW power at the sample. Spectra initiated at 0, 15, 30, 60, and 120 s after mixing are plotted. (B) X-band EPR spectra of DaCld/PAA reaction mixtures measured at 77 K indicate an uncoupled [FeIV=O] + (AA•) species. Top: DaCld was reacted with 5 eq PAA acid (50.8 μM DaCld final concentration) at 22 °C. The sample was freeze-quenched into a liquid N2/isopentane slush after a 50 ms reaction time. Instrument parameters were: 200 G sweep width, 0.5 G modulation amplitude, 2.0 mW power, 100 kHz modulation frequency, and 77 K. Bottom: DaCld (22 μM, final concentration) was reacted with 9 equivalents of PAA at 22 °C. Samples were subsequently frozen in an acetone/liquid nitrogen bath at 14 s and 60 s (less intense signal). Instrument parameters were: 200 G sweep width, 4 G modulation amplitude, 2.0 mW power, 100 kHz modulation frequency, and 77 K.
The protein/PAA mixture was freeze-quenched at 77 K at varying time points following mixing and examined by EPR (Figure 4B). The poorly-resolved fine structure and the temporal dependence of the spectral amplitude, which increases over tens of milliseconds, are consistent with a protein-based radical (e.g., Tyr• or Trp•) or mixture of radicals. Comparison of the shape of the EPR spectrum at 50 ms (Figure 4B top) with ones after 15–60 seconds of reaction clearly reveals changes (Figure 4B bottom). Spin quantification of the EPR signal showed ~46% of the enzyme in the radical form at 50 ms and 38% at 15 s. The existence of radical species at 77 K in a significant fraction of the sample suggests that it is [FeIV=O] + (AA•) rather than [FeIV=O]. The time dependence of the spectral signature is consistent with changing populations of radical sites due to radical migration. Assignments of the radical sites will require multi-frequency EPR spectroscopy of WT, mutant, and isotopically labeled (Trp-and Tyr-deuterated) enzymes. These studies are ongoing and will be reported separately (A. Ivancich, personal communication).
The Soret-excited RR spectra were recorded for the same set of freeze-quenched intermediate samples at 77 K, pH 8 (data not shown). The RR data are very similar to the 22 °C spectra obtained at 15 s, showing only the ferryl species and no Fe(III). Because the EPR data indicated the presence of a substoichiometric population of radicals in the aged reactions, the freeze-quench samples must contain a mixture of [FeIV=O] + (AA•) and an EPR-silent species: [FeIV=O] or exchange coupled [FeIV=O(AA•)].
Transient reactions of the ferryl intermediate with ascorbate (pH 8)
The presumed [FeIV=O] + (AA•) species that initially forms at pH 8 was prepared and reacted with 1 eq of ascorbate in a sequential mixing experiment. The intermediate converted completely and with isosbestic points to the starting ferric enzyme (Figure 5). These data are consistent with the intermediate either being one electron equivalent above the ferric (Compound II), or with an [FeIV=O] + (AA•) species (isoelectronic with Compound I) in which only the FeIV=O reacts with ascorbate. In the latter case, the lack of reactivity between ascorbate/protein radical may be due to the radical’s inaccessibility, which might result if it migrates away from the active site to one or more locations on the protein. (See below.)
Figure 5. The Compound II or [Fe(IV)=O] + (AA▪) intermediate that forms at pH 8 does not exhibit a full peroxidase cycle, instead converting to an apparent ferric species after mixing with only 1 eq ascorbate.
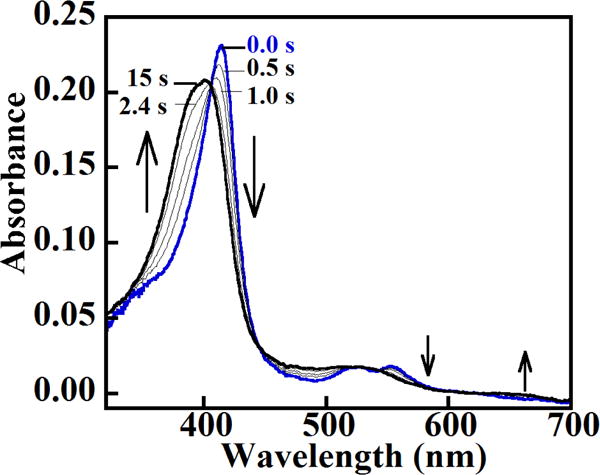
DaCld (2.5 μM final) was mixed with a slight stoichiometric excess of peracetic acid at pH 8 and aged for 1 s. The resulting ferryl species (blue spectrum) was mixed with 1 eq of ascorbate (~2.5 μM final), resulting in formation of a species with a spectrum resembling the ferric starting material (black spectrum). Intervening spectra measured at the given times are shown in gray. Insets: visible bands (450–700 nm) shown on an expanded scale.
The sequential reaction was then carried out under pseudo first-order conditions. The resulting plot of kobs versus [ascorbate] is hyperbolic with a maximum of 0.6 s−1 and apparent Kd = 3.6 μM (Figure S8). These numbers are similar to those measured for the ascorbate-mediated conversion of Compound II to the ferric at pH 6.0.
Transient reaction of DaCld with H2O2 at pH 6 and 8
Reaction of peroxidases with peroxide in the absence of reducing equivalents results in heme destruction via heme oxygenase-like pathways that have been described in several cases.17, 37, 43–50 The heme in DaCld degrades following incubation with ~500 eq of H2O2. Rapid mixing of DaCld with H2O2 (10–500 eq) results in little interpretable change in its UV/vis spectrum. Reaction with a large excess of H2O2 (2800 eq) at pH 6 converts the ferric enzyme slowly (within 2 s) to a species with a Soret band at 408 nm and visible bands at 535, 575 nm (Table 2). This spectrum is similar to that measured in peroxidases for the ferric-anion complexes Compound 0 (ferric-hydroperoxide) or Compound III (ferric-superoxide).37, 43, 50, 51 The latter intermediate forms following the reaction of ferric peroxidases or catalases with multiple equivalents of H2O2 and proceeds via more than one documented pathway.43 Kinetic traces corresponding to Compound 0/III formation (at 408 nm) fit well to single exponential curves (second-order rate constant k = 96 M−1s−1) (Figure S9). The chromophore for the Compound 0/III intermediate converts without isosbestic behavior to a verdoheme-like spectrum with a broad band at 650 nm and concomitantly bleaching Soret (k = 0.1 s−1) (Figure 6A).
Figure 6. DaCld forms a transient Compound 0 or III-like intermediate (ferric-(hydro)peroxide), rather than Compound I following reactions with excess peroxide.
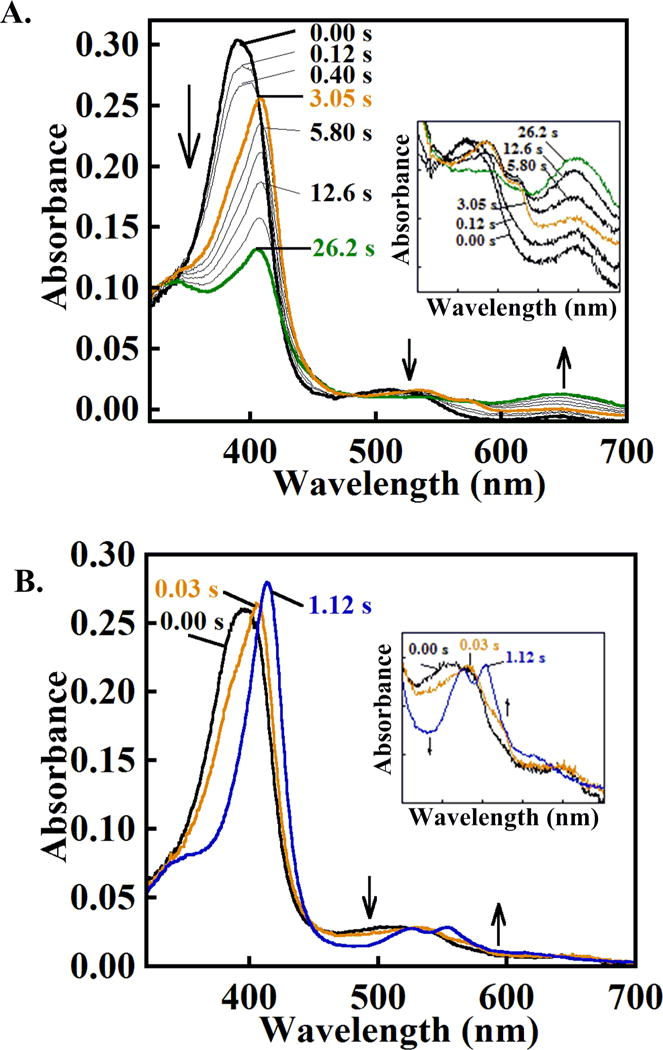
(A) DaCld was mixed with a large excess (>2800 eq) of H2O2 at pH 6: 7 μM enzyme, 3.5 μM final mixed with 20 mM H2O2,10 mM final in 0.2 M citrate-phosphate buffer at pH 6, 20 °C. The initial ferric spectrum is shown in black, the putative Compound 0 or Compound III intermediate formed after ~3 s in orange, and a spectrum suggesting verdoheme formation at 26 s in green. Intervening spectra are shown at the indicated time points in gray. The absence of isosbestic points indicates that simple conversion of one species to another is unlikely. (B) DaCld forms a transient Compound 0 or III-like intermediate spectrum after reaction with a smaller excess (>280 eq) H2O2 at pH 8. DaCld (7 μM, 3.5 μM final) was mixed with 2 mM hydrogen peroxide (1 mM final) in 0.2 M citrate-phosphate buffer at pH 8 and 20 °C. The initial spectrum is shown in black, the putative Compound 0 intermediate that formed after 0.03 seconds in orange, and the following apparent ferryl spectrum that formed after 1.1 s in blue. Intervening spectra are omitted for clarity. Top: complete spectra; bottom: visible bands shown on an expanded scale. Note the similarity in the spectra for the first intermediates, colored orange, at pH 8 and 6.
A similar Compound 0/III-like species forms more readily at pH 8 from the reaction of DaCld and an order of magnitude less (280 eq) H2O2 (1.7×104 M−1s−1, Figures 6B and S10). This species converts to an apparent ferryl (Soret maximum at 415 nm; α, β bands = 525, 555 nm) via a single exponential process. Associated kobs values depend hyperbolically on [H2O2] (maximum kobs = 7.9 s−1, Kd= 1.5 mM (Figure S10).
A hallmark of peroxidases is the rapid reaction of H2O2 with the ferric heme to form Compound I, occurring with second-order rate constants up to 107–108 M−1s−1.23, 52, 53 The second-order rate constants for the DaCld/H2O2 reaction are much smaller (1.2×102 – 1.6×104 M−1s−1, pH 6–8) and comparable to those measured for the reaction of H2O2 and ferric (met) myoglobin (102–103 M−1s−1) or for some peroxidases in which the distal His has been mutated to a hydrophobic residue (an active site configuration resembling DaCld’s).37, 54 The first detectable intermediate for the DaCld/H2O2 reaction at either pH is likewise not a peroxidase-like Compound I, but resembles Compounds 0 or III, which have very similar UV/vis spectra.37, 50, 51, 55 Accumulation of Compound 0 would suggest that deprotonation of H2O2 to form the hydroperoxy adduct, while slow relative to Compound I formation in a peroxidase, is faster than the subsequent bond cleavage steps. Compound III can form at high [H2O2] via reaction of the enzyme with multiple equivalents of H2O2. The need for high eq to form the Compound 0/III intermediate at pH 6 and its conversion to a species resembling verdoheme (though blue-shifted) suggests that the intermediate may be Compound III, which can form via Compound I, Compound II, or a [FeIV=O] + (AA•) as intermediates. At pH 8, several factors suggest the first intermediate is Compound 0: the progression from the putative Compound 0/III to a ferryl; the lack of any observed O2 release from H2O2, which would accompany conversion of Compound III to Compound II, in the steady state; the more rapid formation of this intermediate than the initial intermediate at pH 6 even at 10-fold lower H2O2; and the lack of an active site base to facilitate deprotonation of H2O2 (a factor normally limiting capture of Compound 0). Further study is needed to definitively characterize these intermediates.
DISCUSSION
The reactions of DaCld with H2O2 and PAA have been comprehensively studied in order to understand the relationships between the O–O cleaving (from H2O2, PAA) and the highly unusual O–O bond forming reactions catalyzed by several proteins in the chlorite dismutase family. The data presented here indicate that reaction with the peracetate ion proceeds rapidly with heterolytic O–O cleavage as the sole pathway, while the much less acidic H2O2 reacts sluggishly and leads to destruction of the heme. These results have important implications for the mechanism of O2 formation from chlorite and set distinct boundaries for the probable biological functions of DaCld’s diverse sequence and structural relatives (Figure 1).
PAA reacts rapidly with resting DaCld (Scheme 2) to form high-valent intermediates that differ at low and high pH (Figure 2). Under acidic conditions (pH 6), the ferric species converts neatly to the product of heterolytic bond cleavage (Compound I, [FeIV=O(P+•)]) following reaction with 3 eq of PAA. Compound I reacts rapidly and with overall second order (k = 2.6×106 M−1s−1) kinetics with ascorbate, suggesting that peroxidase reactivity is not limited by the inability of an organic substrate to dock into and react at the active site. The resulting ferryl species subsequently converts to the ferric form after reaction with a second molecule of ascorbate (Kd(apparent) = 3.5 μM; kmax = 9.3 s−1), indicating a complete peroxidase catalytic cycle (Figure 3).
The reaction between DaCld and 3 eq of PAA even at mildly alkaline pH (8) results, by contrast, in the complete conversion of the ferric starting material to what appears via UV/vis to be a ferryl species without Compound I’s porphyrin cation radical. UV/vis suggests that the heme in this species returns completely to the ferric form following reaction with 1 equivalent of ascorbate. This change can be interpreted in one of two ways. First, the reaction mechanism could undergo a pH-dependent shift from heterolytic to homolytic cleavage of the O–O bond (Scheme 1). Such a shift would be supported by structural rearrangement of the distal arginine (Arg183) side chain (Figure 1), which we have previously proposed goes from a heme-directed (closed) to a solvent-directed (open) conformation with a pKa near 6.6.21 Structural shifts in distal Arg residues have been documented in other heme systems, including peroxidases and the sensor protein FixL.56–60 A heme-oriented, positively charged Arg183 would promote heterolytic bond cleavage by polarizing the O–O bond of PAA. The closed Arg183 would likewise be optimally situated to stabilize an anionic acetate leaving group. The open orientation of the side chain predominating at alkaline pH, on the other hand, would render the already hydrophobic heme pocket even more so, stabilizing a neutral acetyl radical as the leaving group and supporting homolytic O–O cleavage. This reaction would generate Compound II as the first intermediate. Homolytic Cl–O cleavage was indeed predicted as the preferred mechanism in a recent computational study of water soluble, synthetic porphyrin models of Cld.61 A pH dependent switch from heterolytic to homolytic bond cleavage with a near-neutral pKa is consequently consistent with both the data and proposed models for small molecule and enzymatic function.
The stopped flow data can also be explained by heterolytic formation of Compound I and an acetate leaving group at all pHs. The rates of Compound I formation and of its conversion to an isoelectronic [FeIV=O] + (AA•) species would both have to accelerate with pH. The first observable intermediate under alkaline conditions would then be [FeIV=O] + (AA•) (or equivalently, [FeIV-OH] + (AA•)), which is indistinguishable via UV/vis and RR from [FeIV=O] alone. Migration of Compound I’s porphyrin cation radical onto various redox-active amino acid side chains to form (AA•) – typically oxidized tryptophans or tyrosines – occurs as part of the regular catalytic cycle in some peroxidases and as an unwanted side reaction in others.24, 35, 36 The DaCld crystal structure shows three highly conserved Trp and two Tyr residues within 5 Å of the heme (Scheme 2), any or all of which might in principle provide a conduit for radical migration.62 The observed reaction of [FeIV=O] + (AA•) with a single equivalent of ascorbate to yield a ferric species would then most likely be due to selective one-electron reduction of the ferryl iron to the ferric. This might occur if the protein radical species, which one would expect to have proceeded out of the active site, were inaccessible to the reductant.
To distinguish these two possibilities, the acidic and alkaline intermediates were examined by RR and EPR (Figure 4 and SI). RR confirmed that the entire sample converts to a ferryl species following reaction with PAA irrespective of pH, and low temperature (77 K) EPR confirmed the presence of a large population of protein-based radicals uncoupled from the ferryl heme under basic conditions. These data suggest that Compound I is indeed the initially-produced intermediate at all pH values examined here. Radical migration away from the heme to form an uncoupled amino acid radical is rapid in the absence of a peroxidase-like reducing substrate, particularly at high pH when the distal arginine is in its open form. The rapidity of this reaction is potentially responsible for the observation of some protein-based radicals even at low pH, where the initially observed Compound I intermediate may be imperfectly trapped.
If the reactions between DaCld and PAA or chlorite are analogous, cleavage of the Fe(III)O–ClO bond should be heterolytic, forming a Compound I/hypochlorite pair (Scheme 1). Nucleophilic attack of the hypochlorite upon Compound I would then yield a transient Fe(III)-OOCl− intermediate, breaking down to produce the ferric resting enzyme, O2, and Cl−. However, while PAA and chlorite are both anionic, oxygen-donating oxidants that react with ferric heme, they are different substrates, and one may ask how closely the PAA/chlorite analogy can be expected to hold. Linear free energy relationship (LFER) analyses have given insight into the kinetics and mechanisms of bond cleavage by oxidation catalysts. These analyses have shown that rate constants for RO–O(H) bond heterolysis (kRO–O(H)) depend linearly on the pKa of the leaving group (RO− + H+ ⇆ ROH).63, 64 Leaving groups that are stronger acids (and hence relatively stable anions) are associated with faster heterolytic bond scission. As leaving group pKas increase, however, the downward linear trend in kRO–O(H) values abruptly ceases and the plot becomes horizontal. In the horizontal region of the kRO–O(H) versus pKa plot, the competing homolytic pathway with its neutral radical leaving group (RO•) predominates. Heterolytic scission of the CH3(CO)O–O(H) bond of PAA results in Compound I and the relatively acidic acetate leaving group (pKa = 4.8), pointing clearly toward kinetically favorable O–O heterolysis for PAA.
The situation is more ambiguous for chlorite. The aqueous HClO/ClO + H+ pKa of 7.5 is near the heterolytic/homolytic transition for aqueous model complex reactions.65 A full LFER analysis for DaCld itself was not possible, due first to the protein’s lack of reactivity with high-pKa peroxides and peracids and second to the insolubility of other potentially interesting oxidants in water. The spectroscopy-based model suggests that the active site Arg side chain toggles between in/out conformations with a near-neutral pKa of 6.5.21 The positive charge of this side chain could stabilize the hypochlorite anion, lowering the hypochlorite pKa and enhancing the kinetic favorability of heterolytic (O)Cl–O− bond cleavage. In the out position, the neutral ClO▪ leaving group and Compound II might be favored. An unambiguous kinetic argument can therefore not be made that Cl–O cleavage, in analogy to O–O cleavage, is heterolytic across all values of pH.
The thermodynamic analogy between chlorite and PAA, however, is both closer and more strongly supportive of heterolytic bond breakage. A simple analysis of driving forces requires estimating the energetic costs of generating Compounds I or II and balancing those against the energy yielded from heterolytic or homolytic scission of the oxidant bonds. The cost of Compound I and Compound II formation in the ferric heme peroxidase HRP-C has been measured at −0.894V and −0.885V at pH 7.0, respectively (all potentials relative to the SHE).66 The DaCld heme environment is more hydrophobic than that of the peroxidases, and the more positively charged heme states should consequently be less stable.14 Consistent with this expectation, the Fe(III)|Fe(II) potential of DaCld is −0.023 V at pH 7 while the same potential for HRP–C is much more negative at –0.262V.19, 66 If the factors producing the ~+0.230 V offset in DaCld’s Fe(III)|Fe(II) potential yield a comparable shift in its intermediate|Fe(III) potentials, then these should range from +1.188–1.069 V (Compound I|Fe(III), pH 6–8) and 1.168–0.972 V (Compound II|Fe(III), pH 6–8). On the oxidant side, the standard potential (pH 0) for the two-electron PAA|acetic acid (HOOAc|HOAc) couple corresponding to heterolytic O–O cleavage was recently estimated at +1.960 V,67 which decreases to 1.636 V at pH 6.0 and 1.570 V at pH 8.0. Hence, as expected, PAA has the necessary driving force to effect two-electron oxidation of ferric DaCld to Compound I over the entire pH range investigated here, with overall reaction potentials of +0.451 V at pH 6.0 and +0.474 V at pH 8.0.
The potentials for the analogous two-electron/heterolytic couple for ClO2−|HClO (pH 6.0) or ClO2−|ClO− (pH 8.0) are 1.200 and 1.039 V, respectively. This suggests that heterolytic cleavage of the Cl–O bond by HRP–C at either pH is exergonic, with a +0.245 to 0.203 V. Consistent with this analysis, chlorite has been observed to act as a “shunt” reagent for Compound I formation in peroxidases and synthetic porphyrin complexes.68–71 However, DaCld is not as strongly oxidizing as a peroxidase. With the estimated offset in potential described above, the oxidation of ferric DaCld to Compound I by ClO2− is weakly exergonic by 0.015 V (−2.9 kJ/mol) at pH 6.0 and endergonic by 0.030 V (+5.8 kJ/mol) at pH 8.0. It is important to point out that the DaCld potentials are estimates based on potential offsets and the assumption that the ClIII|ClI potential in the catalytic site is the same as it is in aqueous solution. Hence, this analysis is qualitatively consistent with weakly driven Compound I formation from chlorite at acidic pH that becomes even more weakly driven at alkaline values.
The estimated homolytic/one-electron reduction potentials suggest more clearly that direct formation of Compound II from the DaCld/chlorite reaction is thermodynamically unlikely. The potential for the HClO2|ClO couple describing homolytic Cl–O bond cleavage has to our knowledge not been reported. Based on available free energies of formation, we estimate the standard potential of this half-cell to be +1.220 V (pH 0),26 lowering to +0.626 V at pH 6.0 and +0.389 V (−38 kJ/mol) at pH 8.0. (See supplemental information for details.) One electron oxidation of ferric DaCld by ClO2− to yield the ClO• radical and Compound II would therefore be endergonic by −0.542 V (+52 kJ/mol) at pH 6.0 and −0.582 V (+56kJ/mol) at pH 8.0. Neither the DaCld/ClO2− nor the HRP-C/ClO2− reactions are therefore likely to proceed via homolytic Cl–O cleavage at any pH investigated here. In sum, the analyses of the one-and two-electron driving forces argue for the first bond-breaking step of the catalytic cycle proceeding by heterolytic O–Cl bond cleavage to yield [FeIV=O(P+•)] (Compound I) and ClO− as a leaving group. Re-reaction of the intermediate/leaving group pair would be enforced by the sterically confined distal pocket, and the weakly basic hypochlorite (ClO− + H+ ⇆ HClO, pKa = 7.5) stabilized by the positive charge on the distal arginine.
The two possible mechanisms for O–O joining proposed here are similar to those proposed for O–O joining in the OEC reaction of photosystem II and synthetic O2-forming complexes, with either nucleophilic or radical rejoining of a sterically confined group occurring with a high valent metal-oxo complex.18,72 The complete reaction cycle is even more closely analogous to that proposed for ferric heme models and proteins that isomerize peroxynitrite to nitrate. In that case, both homolytic and heterolytic models for peroxynitrite (ON)O–O− bond scission have been proposed, leading to a Compound II/NO2▪ pair in the first case and Compound I/NO2− in the second. Steric confinement is also likely important for enforcing recombination of the metal-oxo and leaving group to produce nitrate. Interestingly, while the work reported here favors heterolytic bond cleavage followed by nucleophilic attack, recent work with myoglobin suggests Compound II/NO2▪ as the more likely reaction pathway in that case.73
Interestingly, the majority of Cld proteins likely do not react with chlorite to generate O2 and have been proposed to use peroxide as a substrate. However, the weak H2O2 reactivity observed here for DaCld suggests that peroxide reactivity is biologically unlikely for chlorite dismutases (Table 1) in spite of very close structural similarity to the very efficient dye decoloring peroxidases (DyPs) and their “EfeB” homologs. The EfeB and YfeX proteins from Rhodococcus jostii RH1 (also called DypA and DypB, respectively), for example, react avidly with H2O2.16, 74, 75 The DypA forms a ferryl (Compound II or [FeIV=O + (AA•)]) intermediate following reaction with just one equivalent of H2O2, and the DypB converts stoichiometrically to a stable Compound I (pH 7.5, 25 °C; half-life = 9 min).16, 74 Apparent Compound I intermediates have also been observed in the transient H2O2 reactions of DyPDec1 (from Thanatephorus cucumeris) and the EfeB from Escherichia coli (also known as YcdB).11, 76 In addition, characterized DyPs exhibit steady state constants with H2O2 in the range of what one would expect for efficient peroxidases (Table 1).77 Key active site differences between Dyp and Cld family proteins might be responsible for the observed differences in reactivity with H2O2 (Figure 1). First, an active site base has been viewed as a sine qua non of peroxidase chemistry, since deprotonation of H2O2 is typically necessary for making a strong complex with ferric heme.50, 78, 79 Cld family proteins have a lone cationic residue in their distal pockets that is apparently not able to act as an active site base.19 The conserved distal aspartic acid found in the DyP and EfeB members of this family, or alternatively the arginine of an Asp-Arg pair, has by contrast been proposed to serve in lieu of the conventional distal His base in these proteins.11, 74, 75 The Cld-family proteins also exhibit a 180° rotation of the heme around an axis spanning the A and C pyrrole rings relative to the proteins in the EfeB and DyP families.8 This rotation removes hydrogen bonding connections between the distal arginine and one of the propionic side chains, and appears to allow for conformational mobility of the arginine residue in the DaCld distal pocket.19
CONCLUSIONS
The reactions of DaCld with H2O2 and PAA shed light on both the likely biological roles of Clds and the mechanism by which some Clds catalyze their unusual O2 forming reaction. First, sluggish reactivity with H2O2 in the transient and steady state suggests that this and similar proteins are unlikely to act as functional peroxidases or catalases in vivo. This is not due to the inability of the reducing substrate to approach the reactive heme: rapid reaction with ascorbate is observed when peracetic acid is the oxidant, and the expected Compound I and II intermediates form. Rather, the lack of a distal base likely limits the ability of the enzyme to form the initial Compound 0 ferric-hydroperoxy complex. This structural feature sets DaCld and the whole chlorite dismutase family in strong contrast with their DyP and EfeB structural relatives, which are highly functional peroxidases that react readily with H2O2.
The reactions of DaCld with PAA further illustrate how DaCld breaks this O–O bond and suggests how, by analogy, it may be expected to form one. Heterolytic cleavage of the bound peracid O–O bond in DaCld appears to be the preferred reaction over the full range of pHs for which the protein is stable. At weakly acidic pH, Compound I is the initially observed intermediate, while at alkaline pHs (≥8), a mixture of [FeIV=O] + (AA•) and another ferryl heme species forms an order of magnitude more quickly. These observations give indirect mechanistic support for an O2 forming process via a similar microscopic reverse reaction: nucleophilic attack of a hypochlorite leaving group upon a Compound I intermediate, ultimately yielding O2.
Supplementary Material
Acknowledgments
We thank Drs. A. Ivancich and Sunil Ojha, and Garrett Moraski for helpful discussions. The members of the Broderick group at Montana State are thanked for their assistance in helping the laboratory become operational in its new location.
Abbreviations
- Compound I
[Fe(IV)=O(P+▪)] or [Fe(IV)=O(AA▪)], with the radicals exchange coupled
- Compound II
[Fe(IV)=O] (no observable protein radicals)
- [Fe(IV)=O] + (AA▪)
ferryl with an uncoupled protein-based radical
- Cld
chlorite dismutase
- DaCld
Dechloromonas aromatica Cld
- DyP
dye-decoloring peroxidase
Footnotes
Supported by the National Institutes of Health grants R01GM090260 (JLD) and GM094039 (GLR).
SUPPORTING INFORMATION
Additional supporting data and a description of the derivation of estimated redox potentials discussed here are provided as supporting information (http://pubs.acs.org).
References
- 1.Lee AQ, Streit BR, Zdilla MJ, Abu-Omar MM, DuBois JL. Mechanism of and exquisite selectivity for O–O bond formation by the heme-dependent chlorite dismutase. Proc Natl Acad Sci USA. 2008;105:15654–15659. doi: 10.1073/pnas.0804279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.vanGlinkel CG, Rikken GB, Kroon AGM, Kengen SWM. Purification and characterization of chlorite dismutase: A novel oxygen-generating enzyme. Arch Microbiol. 1996;166:321–326. doi: 10.1007/s002030050390. [DOI] [PubMed] [Google Scholar]
- 3.Stenklo K, Thorell HD, Bergius H, Aasa R, Nilsson T. Chlorite dismutase from Ideonella dechloratans. J Biol Inorg Chem. 2001;6:601–607. doi: 10.1007/s007750100237. [DOI] [PubMed] [Google Scholar]
- 4.Bender KS, O’Connor SA, Chakraborty R, Coates JD, Achenbach LA. Sequencing and transcriptional analysis of the chlorite dismutase gene of Dechloromonas agitata and its use as a metabolic probe. Appl Environ Microbiol. 2002;68:4820–4826. doi: 10.1128/AEM.68.10.4820-4826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hagedoorn PL, de Geus DC, Hagen WR. Spectroscopic characterization and ligand-binding properties of chlorite dismutase from the chlorate respiring bacterial strain GR-1. Eur J Biochem. 2002;269:4905–4911. doi: 10.1046/j.1432-1033.2002.03208.x. [DOI] [PubMed] [Google Scholar]
- 6.Mehboob F, Wolterink AFM, Vermeulen AJ, Jiang B, Hagedoorn PL, Stams AJM, Kengen SWM. Purification and characterization of a chlorite dismutase from Pseudomonas chloritidismutans. FEMS Microbiol Lett. 2009;293:115–121. doi: 10.1111/j.1574-6968.2009.01517.x. [DOI] [PubMed] [Google Scholar]
- 7.Kostan J, Sjoeblom B, Maixner F, Mlynek G, Furtmueller PG, Obinger C, Wagner M, Daims H, Djinovic-Carugo K. Structural and functional characterisation of the chlorite dismutase from the nitrite-oxidizing bacterium “Candidatus Nitrospira defluvii”: Identification of a catalytically important amino acid residue. J Struct Biol. 2010;172:331–342. doi: 10.1016/j.jsb.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Goblirsch B, Kurker RC, Streit BR, Wilmot CM, DuBois JL. Chlorite Dismutases, DyPs, and EfeB: 3 Microbial Heme Enzyme Families Comprise the CDE Structural Superfamily. J Mol Biol. 2011;408:379–398. doi: 10.1016/j.jmb.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mlynek G, Sjoeblom B, Kostan J, Fuereder S, Maixner F, Gysel K, Furtmueller PG, Obinger C, Wagner M, Daims H, Djinovic-Carugo K. Unexpected Diversity of Chlorite Dismutases: a Catalytically Efficient Dimeric Enzyme from Nitrobacter winogradsky. J Bact. 2011;193:2408–2417. doi: 10.1128/JB.01262-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coates JD, Achenbach LA. Microbial perchlorate reduction: Rocket-fuelled metabolism. Nature Rev Microbiol. 2004;2:569–580. doi: 10.1038/nrmicro926. [DOI] [PubMed] [Google Scholar]
- 11.Sugano Y, Muramatsu R, Ichiyanagi A, Sato T, Shoda M. DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family. J Biol Chem. 2007;282:36652–36658. doi: 10.1074/jbc.M706996200. [DOI] [PubMed] [Google Scholar]
- 12.Sugano Y, Matsushima Y, Tsuchiya K, Aoki H, Hirai M, Shoda M. Degradation pathway of an anthraquinone dye catalyzed by a unique peroxidase DyP from Thanatephorus cucumeris Dec 1. Biodegradation. 2009;20:433–440. doi: 10.1007/s10532-008-9234-y. [DOI] [PubMed] [Google Scholar]
- 13.Liu XH, Du Q, Wang Z, Zhu DY, Huang Y, Li N, Wei TD, Xu SJ, Gu LC. Crystal Structure and Biochemical Features of EfeB/YcdB from Escherichia coli O157 Asp(235) plays divergent roles in different enzyme-catalyzed processes. J Biol Chem. 2011;286:14922–14931. doi: 10.1074/jbc.M110.197780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goblirsch BR, Streit BR, DuBois JL, Wilmot CM. Structural features promoting dioxygen production by Dechloromonas aromatica chlorite dismutase. J Biol Inorg Chem. 2010;15:879–888. doi: 10.1007/s00775-010-0651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Geus DC, Thomassen EAJ, Hagedoorn PL, Pannu NS, van Duijn E, Abrahams JP. Crystal Structure of Chlorite Dismutase, a Detoxifying Enzyme Producing Molecular Oxygen. J Mol Biol. 2009;387:192–206. doi: 10.1016/j.jmb.2009.01.036. [DOI] [PubMed] [Google Scholar]
- 16.Roberts JN, Singh R, Grigg JC, Murphy MEP, Bugg TDH, Eltis LD. Characterization of Dye-Decolorizing Peroxidases from Rhodococcus jostii RHA1. Biochemistry. 2011;50:5108–5119. doi: 10.1021/bi200427h. [DOI] [PubMed] [Google Scholar]
- 17.Streit BR, DuBois JL. Chemical and steady state kinetic analyses of a heterologously expressed heme dependent chlorite dismutase. Biochemistry. 2008;47:5271–5280. doi: 10.1021/bi800163x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoganson CW, Pressler MA, Proshlyakov DA, Babcock GT. From water to oxygen and back again: mechanistic similarities in the enzymatic redox conversions between water and dioxygen. Biochim Biophys Acta-Bioenerg. 1998;1365:170–174. doi: 10.1016/s0005-2728(98)00057-7. [DOI] [PubMed] [Google Scholar]
- 19.Blanc B, Mayfield JA, McDonald CA, Lukat-Rodgers GS, Rodgers KR, DuBois JL. Understanding How the Distal Environment Directs Reactivity in Chlorite Dismutase: Spectroscopy and Reactivity of Arg183 Mutants. Biochemistry. 2012;51:1895–1910. doi: 10.1021/bi2017377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berry EA, Trumpower BL. Simultaneous determination of hemes a, b, and c from pyridine hemocrome spectra. Analyt Biochem. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 21.Streit BR, Blanc B, Lukat-Rodgers GS, Rodgers KR, DuBois JL. How Active-Site Protonation State Influences the Reactivity and Ligation of the Heme in Chlorite Dismutase. J Am Chem Soc. 2010;132:5711–5724. doi: 10.1021/ja9082182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dassama LMK, Yosca TH, Conner DA, Lee MH, Blanc B, Streit BR, Green MT, DuBois JL, Krebs C, Bollinger JM., Jr O-2-Evolving Chlorite Dismutase as a Tool for Studying O-2-Utilizing Enzymes. Biochemistry. 2012;51 doi: 10.1021/bi201906x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunford HB. Heme Peroxidases. Wiley-VCH; 1999. [Google Scholar]
- 24.Smulevich G, Jakopitsch C, Droghetti E, Obinger C. Probing the structure and bifunctionality of catalase-peroxidase (KatG) J Inorg Biochem. 2006;100:568–585. doi: 10.1016/j.jinorgbio.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 25.Fabian I, Gordon G. Complex-Formation Reactions Of The Chlorite Ion. Inorganic Chemistry. 1991;30:3785–3787. [Google Scholar]
- 26.Atkins P, Overton T, Rourke J, Weller M, Armstrong F, Hagerman M. Inorg Chem. 5. W. H. Freeman and Company; New York: 2009. [Google Scholar]
- 27.Proshlyakov DA, Paeng IR, Paeng KJ, Kitagawa T. Resonance Raman studies of Compound I and II of Arthromyces ramosus peroxidase: close similarities in their Raman spectra but distinct oxygen exchangeability of the Fe:O heme. Biospectroscopy. 1996;2:317–329. [Google Scholar]
- 28.Kapetanaki SM, Chouchane S, Yu S, Magliozzo RS, Schelvis JP. Resonance Raman spectroscopy of Compound II and its decay in Mycobacterium tuberculosis catalase-peroxidase KatG and its isoniazid resistant mutant S315T. J Inorg Biochem. 2005;99:1401–1406. doi: 10.1016/j.jinorgbio.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 29.Sitter AJ, Reczek CM, Terner J. Comparison of the heme structures of horseradish peroxidase compounds X and II by resonance Raman spectroscopy. J Biol Chem. 1986;261:8638–8642. [PubMed] [Google Scholar]
- 30.Chuang WJ, Heldt J, Van Wart HE. Resonance Raman spectra of bovine liver catalase compound II. Similarity of the heme environment to horseradish peroxidase compound II. J Biol Chem. 1989;264:14209–14215. [PubMed] [Google Scholar]
- 31.Chuang WJ, Van Wart HE. Resonance Raman spectra of horseradish peroxidase and bovine liver catalase compound I species. Evidence for predominant 2A2u pi-cation radical ground state configurations. J Biol Chem. 1992;267:13293–13301. [PubMed] [Google Scholar]
- 32.Hashimoto S, Tatsuno Y, Kitagawa T. Resonance Raman evidence for oxygen exchange between the FeIV = O heme and bulk water during enzymic catalysis of horseradish peroxidase and its relation with the heme-linked ionization. Proc Natl Acad Sci U S A. 1986;83:2417–2421. doi: 10.1073/pnas.83.8.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terner J, Palaniappan V, Gold A, Weiss R, Fitzgerald MM, Sullivan AM, Hosten CM. Resonance Raman spectroscopy of oxoiron(IV) porphyrin pi-cation radical and oxoiron(IV) hemes in peroxidase intermediates. J Inorg Biochem. 2006;100:480–501. doi: 10.1016/j.jinorgbio.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Paeng KJ, Kincaid JR. The resonance Raman spectrum of horseradish peroxidase Compound I. J Am Chem Soc. 1988;110:7913–7915. [Google Scholar]
- 35.Singh R, Switala J, Loewen PC, Ivancich A. Two [Fe(IV)=O Trp*] intermediates in M. tuberculosis catalase-peroxidase discriminated by multifrequency (9–285 GHz) EPR spectroscopy: reactivity toward isoniazid. J Am Chem Soc. 2007;129:15954–15963. doi: 10.1021/ja075108u. [DOI] [PubMed] [Google Scholar]
- 36.Ivancich A, Dorlet P, Goodin DB, Un S. Multifrequency high-field EPR study of the tryptophanyl and tyrosyl radical intermediates in wild-type and the W191G mutant of cytochrome c peroxidase. J Am Chem Soc. 2001;123:5050–5058. doi: 10.1021/ja0036514. [DOI] [PubMed] [Google Scholar]
- 37.Rodríguez-López JN, Lowe DJ, Hernández-Ruiz J, Hiner AN, García-Cánovas F, Thorneley RN. Mechanism of reaction of hydrogen peroxide with horseradish peroxidase: identification of intermediates in the catalytic cycle. J Am Chem Soc. 2001;123:11838–11847. doi: 10.1021/ja011853+. [DOI] [PubMed] [Google Scholar]
- 38.Pond AE, Bruce GS, English AM, Sono M, Dawson JH. Spectroscopic study of the Compound ES and the oxoferryl Compound II states of cytochrome c peroxidase: comparison with the Compound II of horseradish peroxidase. Inorg Chem Acta. 1998;275–276:250–255. [Google Scholar]
- 39.Raven EL. Understanding functional diversity and substrate specificity in haem peroxidases: what can we learn from ascorbate peroxidase? Nat Prod Rep. 2003;20:367–381. doi: 10.1039/b210426c. [DOI] [PubMed] [Google Scholar]
- 40.Raven EL, Lad L, Sharp KH, Mewies M, Moody PC. Defining substrate specificity and catalytic mechanism in ascorbate peroxidase. Biochem Soc Symp. 2004:27–38. doi: 10.1042/bss0710027. [DOI] [PubMed] [Google Scholar]
- 41.Sitter. Resonance Raman spectroscopic evidence for heme iron hydroxide ligation in peroxidase alkaline forms. J Biol Chem. 1988;263:13032. [PubMed] [Google Scholar]
- 42.Hashimoto S, Teraoka J, Inubushi T, Yonetani T, Kitagawa T. Resonance Raman study on cytochrome c peroxidase and its intermediate. Presence of the Fe(IV) = O bond in compound ES and heme-linked ionization. J Biol Chem. 1986;261:11110–11118. [PubMed] [Google Scholar]
- 43.Vlasits J, Jakopitsch C, Bernroitner M, Zamocky M, Furtmüller PG, Obinger C. Mechanisms of catalase activity of heme peroxidases. Arch Biochem Biophys. 2010;500:74–81. doi: 10.1016/j.abb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 44.Arnao MB, Acosta M, del Rio JA, Garcia-Canovas F. Inactivation of peroxidase by hydrogen peroxide and its protection by a reductant agent. Biochim Biophys Acta. 1990;1038:85–89. doi: 10.1016/0167-4838(90)90014-7. [DOI] [PubMed] [Google Scholar]
- 45.Hiner ANP, Rodriguez-Lopez JN, Arnao MB, Raven EL, Garcia-Canovas F, Acosta M. Kinetic study of the inactivation of ascorbate peroxidase by hydrogen peroxide. Biochem J. 2000;348:321–328. [PMC free article] [PubMed] [Google Scholar]
- 46.Arnao MB, Acosta M, Delrio JA, Varon R, Garciacanovas F. A Kinetic-Study On The Suicide Inactivation Of Peroxidase By Hydrogen-Peroxide. Biochim Biophys Acta. 1990;1041:43–47. doi: 10.1016/0167-4838(90)90120-5. [DOI] [PubMed] [Google Scholar]
- 47.Arnao MB, HernandezRuiz J, Varon R, GarciaCanovas F, Acosta M. The inactivation of horseradish peroxidase by m-chloroperoxybenzoic acid, a xenobiotic hydroperoxide. J Molec Cat A. 1995;104:179–191. [Google Scholar]
- 48.Rodriguez Lopez JN, Hernandez Ruiz J, Garcia Canovas F, Thorneley RNF, Acosta M, Arnao MB. The inactivation and catalytic pathways of horseradish peroxidase with m-chloroperoxybenzoic acid - A spectrophotometric and transient kinetic study. J Biol Chem. 1997;272:5469–5476. doi: 10.1074/jbc.272.9.5469. [DOI] [PubMed] [Google Scholar]
- 49.Hernandez-Ruiz J, Arnao MB, Hiner ANP, Garcia-Canovas F, Acosta M. Catalase-like activity of horseradish peroxidase: relationship to enzyme inactivation by H2O2. J Biol Chem. 2001;354:107–114. doi: 10.1042/0264-6021:3540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.RodriguezLopez JN, Smith AT, Thorneley RNF. Role of Arginine 38 in horseradish peroxidase a critical, residue for substrate binding and catalysis. J Biol Chem. 1996;271:4023–4030. doi: 10.1074/jbc.271.8.4023. [DOI] [PubMed] [Google Scholar]
- 51.Loew GH, Harris DL. Role of the heme active site and protein environment in structure, spectra, and function of the cytochrome p450s. Chem Rev. 2000;100:407–419. doi: 10.1021/cr980389x. [DOI] [PubMed] [Google Scholar]
- 52.Erman JE, Vitello LB, Miller MA, Shaw A, Brown KA, Kraut J. Histidine-52 is a Critical Residue for Rapid Formation of Cytochrome-C Peroxidase Compound-i. Biochemistry. 1993;32:9798–9806. doi: 10.1021/bi00088a035. [DOI] [PubMed] [Google Scholar]
- 53.Loo S, Erman JE. A kinetic study of the reaction between cytochrome c peroxidase and hydrogen peroxide. Dependence on pH and ionic strength. Biochemistry. 1975;14:3467–3470. doi: 10.1021/bi00686a027. [DOI] [PubMed] [Google Scholar]
- 54.Egawa T, Shimada H, Ishimura Y. Formation of compound I in the reaction of native myoglobins with hydrogen peroxide. J Biol Chem. 2000;275:34858–34866. doi: 10.1074/jbc.M004026200. [DOI] [PubMed] [Google Scholar]
- 55.Harris DL, Loew GH. Investigation of the proton-assisted pathway to formation of the catalytically active, ferryl species of P450s by molecular dynamics studies of P450eryF. J Am Chem Soc. 1996;118:6377–6387. doi: 10.1021/ja954101m. [DOI] [PubMed] [Google Scholar]
- 56.Edwards SL, Poulos TL. Ligand binding and structural perturbations in cytochrome c peroxidase. A crystallographic study. J Biol Chem. 1990;265:2588–2595. [PubMed] [Google Scholar]
- 57.Smulevich G, Feis A, Howes BD. Fifteen years of Raman spectroscopy of engineered heme containing peroxidases: what have we learned? Acc Chem Res. 2005;38:433–440. doi: 10.1021/ar020112q. [DOI] [PubMed] [Google Scholar]
- 58.Gong W, Hao B, Chan MK. New mechanistic insights from structural studies of the oxygen-sensing domain of Bradyrhizobium japonicum FixL. Biochemistry. 2000;39:3955–3962. doi: 10.1021/bi992346w. [DOI] [PubMed] [Google Scholar]
- 59.Balland V, Bouzhir-Sima L, Kiger L, Marden MC, Vos MH, Liebl U, Mattioli TA. Role of arginine 220 in the oxygen sensor FixL from Bradyrhizobium japonicum. J Biol Chem. 2005;280:15279–15288. doi: 10.1074/jbc.M413928200. [DOI] [PubMed] [Google Scholar]
- 60.Jasaitis A, Hola K, Bouzhir-Sima L, Lambry JC, Balland V, Vos MH, Liebl U. Role of distal arginine in early sensing intermediates in the heme domain of the oxygen sensor FixL. Biochemistry. 2006;45:6018–6026. doi: 10.1021/bi060012i. [DOI] [PubMed] [Google Scholar]
- 61.Keith JM, Abu-Omar MM, Hall MB. Computational investigation of the concerted dismutation of chlorite ion by water-soluble iron porphyrins. Inorg Chem. 2011;50:7928–7930. doi: 10.1021/ic2009732. [DOI] [PubMed] [Google Scholar]
- 62.Blanc B, Rodgers KR, Lukat-Rodgers GS, DuBois JL. Understanding the roles of strictly conserved tryptophan residues in O2 producing chlorite dismutases. Dalton Trans. 2013;42:3156–3169. doi: 10.1039/c2dt32312e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Balasubramanian PN, Bruice TC. Oxygen transfer involving nonheme iron: the influence of leaving group ability on the rate constant for oxygen transfer to (EDTA)Fe(III) from peroxycarboxylic acids and hydroperoxides. Proc Natl Acad Sci U S A. 1987;84:1734–1738. doi: 10.1073/pnas.84.7.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ortiz-Maldonado M, Gatti D, Ballou DP, Massey V. Structure-function correlations of the reaction of reduced nicotinamide analogues with p-hydroxybenzoate hydroxylase substituted with a series of 8-substituted flavins. Biochemistry. 1999;38:16636–16647. doi: 10.1021/bi991603u. [DOI] [PubMed] [Google Scholar]
- 65.Bruice TC, Zipplies MF, Lee WA. The pH dependence of the mechanism of reaction of hydrogen peroxide with a nonaggregating, non-mu-oxo dimerforming iron (III) porphyrin in water. Proc Natl Acad Sci U S A. 1986;83:4646–4649. doi: 10.1073/pnas.83.13.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Battistuzzi G, Bellei M, Bortolotti CA, Sola M. Redox properties of heme peroxidases. Arch Biochem Biophys. 2010;500:21–36. doi: 10.1016/j.abb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 67.Awad MI, Denggerile A, Ohsaka T. Electroreduction of Peroxyacetic Acid at Gold Electrode in Aqueous Media. J Electrochem Soc. 2004;151:E358–E363. [Google Scholar]
- 68.Hewson WD, Hager LP. Mechanism Of The Chlorination Reaction Catalyzed By Horseradish-Peroxidase With Chlorite. J Biol Chem. 1979;254:3175–3181. [PubMed] [Google Scholar]
- 69.Slaughter LM, Collman JP, Eberspacher TA, Brauman JI. Radical autoxidation and autogenous O-2 evolution in manganese-porphyrin catalyzed alkane oxidations with chlorite. Inorg Chem. 2004;43:5198–5204. doi: 10.1021/ic049922j. [DOI] [PubMed] [Google Scholar]
- 70.Shahangian S, Hager LP. The Reaction Of Chloroperoxidase With Chlorite And Chlorine Dioxide. J Biol Chem. 1981;256:6034–6040. [PubMed] [Google Scholar]
- 71.Jakopitsch C, Spalteholz H, Fürtmuller PG, Arnhold J, Obinger C. Mechanism of reaction of horseradish peroxidase with chlorite and chlorine dioxide. J Inorg Biochem. 2008;102:293–302. doi: 10.1016/j.jinorgbio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 72.Betley TA, Wu Q, Van Voorhis T, Nocera DG. Electronic Design Criteria for O–O Bond Formation via Metal-Oxo Complexes. Inorg Chem. 2008;47:1849–1861. doi: 10.1021/ic701972n. [DOI] [PubMed] [Google Scholar]
- 73.Lee JB, Hunt JA, Groves JT. Mechanisms of iron porphyrin reactions with peroxynitrite. J Am Chem Soc. 1998;120:7493–7501. [Google Scholar]
- 74.Singh R, Grigg JC, Armstrong Z, Murphy MEP, Eltis LD. Distal Heme Pocket Residues of B-type Dye-decolorizing Peroxidase: Arginine but not aspartate is essential for peroxidase activity. J Biol Chem. 2012;287:10623–10630. doi: 10.1074/jbc.M111.332171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yoshida T, Tsuge H, Konno H, Hisabori T, Sugano Y. The catalytic mechanism of dye-decolorizing peroxidase DyP may require the swinging movement of an aspartic acid residue. FEBS J. 2011;278:2387–2394. doi: 10.1111/j.1742-4658.2011.08161.x. [DOI] [PubMed] [Google Scholar]
- 76.Sturm A, Schierhorn A, Lindenstrauss U, Lilie H, Brüser T. YcdB from Escherichia coli Reveals a Novel Class of Tat-dependently Translocated Hemoproteins. J Biol Chem. 2006;281:13972–13978. doi: 10.1074/jbc.M511891200. [DOI] [PubMed] [Google Scholar]
- 77.Kim SJ, Shoda M. Purification and characterization of a novel peroxidase from Geotrichum candidum Dec 1 involved in decolorization of dyes. Applied and Environmental Microbiology. 1999;65:1029–1035. doi: 10.1128/aem.65.3.1029-1035.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Erman JE, Vitello LB, Miller MA, Kraut J. Active-Site Mutations in Cytochrome-C Peroxidase - a Critical Role for Histidine-52 in the Rate of Formation of Compound-I. J Am Chem Soc. 1992;114:6592–6593. [Google Scholar]
- 79.Vitello LB, Erman JE, Miller MA, Mauro JM, Kraut J. Effect of Asp-235-]Asn Substitution on the Absorption-Spectrum and Hydrogen-Peroxide Reactivity of Cytochrome-C Peroxidase. Biochemistry. 1992;31:11524–11535. doi: 10.1021/bi00161a034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.