

Abstract
The protocols described in this unit were developed to monitor membrane traffic in cultured cell monolayers that display hepatic polarity. In general, the assays are designed to visualize and/or quantitate membrane trafficking by monitoring the fates of antibodies bound to specific membrane proteins. We first describe how to infect cells with recombinant adenovirus, the preferred method for introducing exogenous genes into hepatic cells. We next provide a morphological assay to monitor basolateral to apical transcytosis. In a supporting protocol, we describe how to visualize apical recycling and/or retention. In an additional supporting protocol, we provide a semi-quantitative method to measure the relative extents of apical delivery. Finally, we describe quantitative assays to measure basolateral internalization and recycling. The methods presented in this unit provide a relatively simple, yet powerful approach to examining hepatic membrane traffic.
Keywords: hepatocytes, apical, basolateral, transcytosis, membrane, traffic
INTRODUCTION
Polarized Hepatocytes
Epithelial cells are vital for the success of multi-cellular organisms. They line the body’s surface and the lumen of all organs that are in direct contact with the environment, providing a selective barrier between the external and internal worlds. They accomplish this task by cementing themselves together with the formation of intercellular junctions that restrict distinct activities to specific plasma membrane domains (either apical or basolateral). The functional asymmetry (or polarity) is mirrored by the asymmetrical distribution of membrane proteins. Each domain is characterized by distinct subsets of plasma membrane proteins; few proteins have been identified that distribute equally between the two domains.
Hepatocytes are the major epithelial cell of the liver. Arranged in cords, they abut adjacent hepatocytes (lateral domain) and face at least two blood sinusoids (basal domain; Fig. 15.17.1). An anastomizing network of grooves between adjacent cells forms the bile canaliculi (apical domain). Because of this complex architecture, hepatocytes do not have a single basolateral-to-apical axis as do the simple bipolar epithelial cells lining kidney tubules or the intestinal lumen. Furthermore, the hepatic apical domain is unique among all other polarized cells both in composition and in its specialized exocrine functions, which include biliary transport of bile acids and products of detoxification, the release of phospholipids and cholesterol, and delivery of polymeric immunoglobulin A (pIgA; part of our mucosal immune system). The basolateral domain (basal + lateral) communicates with the circulation. It is responsible for the transport of glucose, bile acids, and amino acids into the blood. The majority of plasma proteins and lipoproteins are synthesized in hepatocytes and secreted at the basolateral domain. Moreover, macromolecules, such as insulin and pIgA, are internalized from the circulation at this domain. Because these domain-specific functions depend on the maintenance of the polarized phenotype, it is of fundamental importance to understand the mechanisms responsible for the establishment and maintenance of hepatic polarity. Answers to these fundamental questions come, in part, from understanding protein traffic in polarized epithelial cells.
Figure 15.17.1.
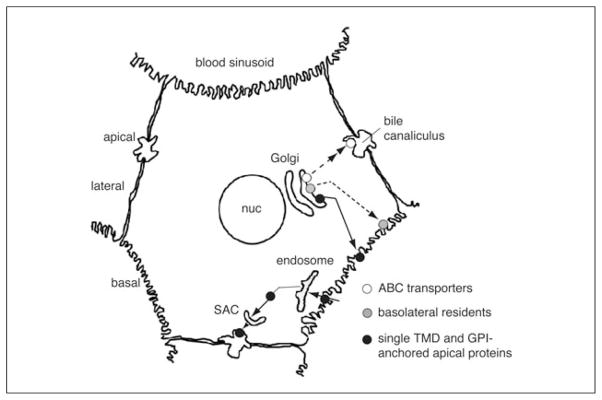
Protein traffic in polarized hepatocytes. Protein traffic patterns of resident hepatic plasma membrane proteins are shown. Newly synthesized basolateral proteins are transported directly from the TGN to the basolateral membrane. In contrast, newly synthesized single trans-membrane domain (TMD) and GPI-anchored proteins take the indirect route where they are transported from the TGN to the basolateral membrane, selectively internalized, and transcytosed to the apical membrane. En route to the apical surface, they traverse at least two known compartments: the early endosome and the subapical compartment (SAC). Newly synthesized multi-spanning apical transporters are directly transported from the TGN to the apical membrane. Although the transporters do not traverse the SAC en route to the apical surface, they may traverse an as yet unidentified compartment (not shown).
The Model Systems
The assays described in this unit are designed to monitor membrane traffic in cultured cell monolayers that display hepatic polarity (Fig. 15.17.2). Traditionally, intact animals or isolated perfused livers have been used to examine hepatic membrane traffic. Although these studies provided a wealth of information, there are disadvantages to using such systems for mechanistic studies. Not only is there considerable physiologic variation among animals, it is also difficult to quickly alter experimental parameters (e.g., addition or withdrawal of inhibitors, changes in temperature), which is often required for such studies. To overcome these experimental barriers, many researchers have turned to in vitro systems. The challenge here has been to maintain cells in culture that retain their hepatocyte-specific characteristics and domain polarity, i.e., maintain specific canalicular and sinusoidal plasma membrane domains.
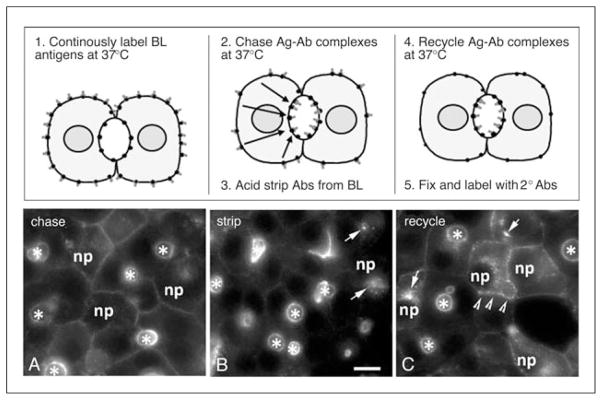
Figure 15.17.2.

Visualizing basolateral to apical transcytosis in polarized hepatic cells using an antibody-based, morphological assay. In the top panels, a schematic for the assay is provided and the basic steps of the procedure are listed. Cells are first cooled to 4°C to stop membrane traffic and incubated with specific primary antibodies. After removing the unbound antibodies, cells are warmed to 37°C for various times, fixed, and permeabilized and the locations of the primary antibodies are detected with fluorescently labeled secondary antibodies. Bottom panels, WIF-B cells were surface labeled with specific antibodies to APN for 20 min at 4°C (A). After removing excess antibodies, the antigen-antibody complexes were chased for 45 (B) or 90 min (C) at 37°C. Asterisks mark the bile-canalicular spaces. Arrows in B point to the sub-apical compartment. Abbreviations: BL, basolateral; Ag, antigen; Ab, antibody; 2°, secondary. Bar: 10 μm.
Although isolated hepatocytes have been a popular model system for the past 30 years or more, they may not be the most suitable system. Within hours of culture, they lose their liver-specific phenotype, including the loss of cell surface polarity. Similarly, isolated hepatocyte couplets (two cells adjoined via a bile canaliculus) are only useful for short-term studies (Boyer, 1997). However, two cultured hepatic cell lines are used widely for studies of polarized membrane traffic: the human hepatoma cell line, HepG2, and the rat-human hybrid, WIF-B cells. Although studies in HepG2 cells have contributed to our understanding of mechanisms regulating membrane traffic (Zegers and Hoekstra, 1998; van Ijzendoorn and Hoekstra, 1999; Roelofsen et al., 2000; De Marco et al., 2002), the extent of polarity in these cells is variable, with as little as 10% of the population exhibiting a polarized phenotype. Thus, examination of polarized traffic using biochemical approaches is not possible. In contrast, 70% to 90% of mature WIF-B cells in culture exhibit phase-lucent structures that are functionally and compositionally analogous to the bile canaliculi, and domain-specific distributions of proteins and lipids are maintained (Ihrke et al., 1993; Shanks et al., 1994).
The Assays
In general, the assays described in this unit visualize/measure membrane trafficking by monitoring the fates of antibodies bound to specific membrane proteins. Because tight junctions restrict access to the apical surface, only basolateral antigens are labeled. The assays are straightforward (see Figs. 15.17.2-3). In general, cells are first cooled to 4°C to stop membrane traffic and incubated with specific primary antibodies. After removing the unbound antibodies, cells are warmed to 37°C for various times to allow trafficking of antigen-antibody complexes. Alternatively, antibodies are continuously internalized from the basolateral surface at 37°C before washing and subsequent chase. For morphological studies, cells are fixed, permeabilized, and the locations of the primary antibodies detected with fluorescently labeled secondary antibodies. Alternatively, primary antibodies can be directly labeled with a fluorophore and followed in living cells in real-time. For biochemical assays, biotinylated antibodies are used, and after the desired time of chase, cells are lysed and populations measured on streptavidin-coated 96-well plates.
Figure 15.17.3.
Visualizing apical retention/recycling in polarized hepatic cells using an antibody-based, morphological assay. In the top panels, a schematic for the assay is provided and the basic steps of the procedure are listed. Cells are incubated with specific primary antibodies to an apical membrane protein either at 4°C (see Basic Protocol 1) or continuously at 37°C for up to one hour to increase labeling intensity. Cells are washed to remove unbound antibodies and incubated at 37°C for another hour to concentrate the antibody-antigen complexes at the apical surface. Residual basolaterally bound antibodies are stripped with isoglycine, and the remaining antibodies allowed to traffic for an additional hour or more to allow recycling. Bottom panels, WIF-B cells were continuously labeled at the basolateral surface with specific antibodies to APN for 1 hr at 37°C, washed, and the antibody-antigen complexes chased to the apical membrane for an additional hour. The image in (A) shows both polarized and nonpolarized WIF-B cells after antibody labeling and chase. (B) APN antibodies remaining at the basolateral membrane were stripped with isoglycine for 2 min at 37°C, such that only the APN-antibody complexes present at the apical membrane in polarized cells or in the intracellular “apical compartment” in nonpolarized cells (marked with arrows) remain. Cells in (C) were incubated an additional hour at 37°C after stripping. Only apical proteins in the nonpolarized cells recycled to the plasma membrane (marked with arrowheads). Asterisks mark bile canalicular spaces; selected nonpolarized cells are indicted with np. Abbreviations: BL, basolateral; Ag, antigen; Ab, antibody; 2°, secondary. Bar: 10 μm.
In Basic Protocol 1, we describe how to infect cells with recombinant adenovirus, the preferred method for introducing exogenous genes or knocking down gene expression in hepatic cells. In Basic Protocol 2, we provide a morphological assay to monitor basolateral to apical transcytosis, and in Support Protocol 2, we describe an assay to visualize apical recycling and/or retention. In Support Protocol 1, we provide a semi-quantitative method to measure the relative extents of apical delivery. In Basic Protocol 3, we describe quantitative assays to measure basolateral internalization and recycling. The biochemical and morphological methods presented in this unit provide a relatively simple yet powerful approach to examining hepatic membrane traffic.
BASIC PROTOCOL 1: RECOMBINANT ADENOVIRUS INFECTION
Because mature hepatic cells in culture are terminally differentiated, they are recalcitrant to conventional transfection methods (e.g., lipophilic reagents) that require active cellular division. We have found that recombinant adenovirus is the most effective method for exogenous gene expression. Infection occurs regardless of mitotic activity, has a natural tropism for liver, and is efficient (>90% for most viruses), allowing for biochemical analysis of overexpressing cells. In addition, protein expression is robust, mostly uniform, and importantly, retention of polarity is excellent. Although less widely used, recombinant lentiviruses can also be used to express exogenous genes in hepatic cells. Both viruses can also be used to knockdown gene expression when the first 300 base pairs of the gene of interest are inserted in the anti-sense orientation. In addition, shRNA adenoviruses are emerging as a means for knocking down expression of specific proteins.
There are many commercial sources of adenoviruses expressing a host of genes. For example, Eton, Imgenex, and SignaGene have large repositories of premade recombinant adenoviruses. If the adenovirus for a gene of interest is not commercially available, many commercial sources provide custom adenovirus production services (e.g., Cell BioLabs, Viraquest, Rockland, and SignaGen, among many others). Recombinant adenoviruses can easily be made in-house using a variety of adenovirus vector systems. For example, PAdEAsy (Agilent), RAPAd (Cell BioLAbs), and the Gateway pAd/CMV/V5-Dest (Invitrogen) systems are widely used. More recently, shRNA adenovirus production services and vector systems are also commercially available (e.g., SignaGen). For an example of how adenoviruses are generated, see previously published protocols (He, 2004).
HepG2 cells are commercially available (ATCC# HB-8065), but the WIF-B cells are not. They can be provided by the corresponding author of this unit, as well as from Drs. A. Hubbard (Johns Hopkins School of Medicine) and D. Cassio (INSERM), the co-discoverers of the cell line. Furthermore, researchers that routinely use WIF-B cells in their research are asked to distribute them when asked. Rat liver hepatocytes can be easily isolated using a two-step collagenase perfusion method and seeded onto collagen-coated coverslips (Shenvi et al., 2008). Because hepatocyte couplets are especially adherent to glass, they can be enriched from primary hepatocyte preparations by seeding onto glass coverslips directly; alternatively, they can be enriched by centrifugal elutriation (Boyer, 1997). Isolated hepatocytes and couplets should be used within 24 to 48 hr after harvesting. In general, cells that are to be transfected are grown on 22 × 22–mm glass coverslips. Both WIF-B and HepG2 cells are cultured using standard methods (Sandell and Sakai, 2011; also see UNIT 1.1) in a humidified 7% CO2 incubator at 37°C. HepG2 cells are grown in Dulbecco’s modified Eagle medium and WIF-B cells in F12 (Coon’s or Khaighn’s modification) containing 4% to 5% fetal bovine serum. WIF-B cultures additionally contain 10 μM hypoxanthine, 40 nM aminopterin, and 1.6 μM thymidine. Cells are seeded 1.0–1.3 × 104 cells/cm2 and grown for 8 to 12 days (WIF-B) or 3 to 5 days (HepG2) until they have reached maximum density and polarity.
NOTE: To use recombinant adenovirus or lentivirus, you must have proper institutional approval to handle Biosafety Level 2 (BSL-2) materials and the approved procedures must be used.
NOTE: WIF-B cells (derived from the WIF12-1 clone) carry the complete set of rat chromosomes and 7–11 human chromosomes (Ihrke et al., 1993; Shanks et al., 1994). Although the full complement of rat genes is likely expressed, some human genes may be activated (Griffo et al., 1993) such that both may need to be targeted in knockdown experiments.
NOTE: All steps are performed in laminar flow hood using sterile technique (Sandell and Sakai, 2011).
NOTE: All solutions (except virus stocks) are prewarmed to 37°C.
Materials
Purified adenovirus or lentivirus (see protocol Introduction)
Ice
Serum-free medium (e.g., Coon’s or Khaighn’s modified F12 medium; Sigma-Aldrich)
Cells grown on 22 × 22–mm glass coverslips in 35-mm dishes or 6-well dishes
Complete medium [e.g., Coon’s or Khaighn’s modified F12 medium (Sigma) supplemented with fetal bovine serum (Gemini Bio-Products)]
Ice bucket
Analog vortex mixer
1.5-ml microcentrifuge tubes
Vacuum/aspirator
-
Thaw adenovirus stock aliquots on ice and place ice bucket in the laminar flow hood.
Some adenoviruses are labile when thawed. We recommend that the adenovirus stocks not be outside of the −80°C freezer for long, and that the entire infection procedure take only 10 to 15 min. -
Immediately before diluting, vortex the virus stock using a medium-speed setting to disperse clumped and settled particles. In a 1.5-ml microcentrifuge tube, dilute the virus to the desired concentration in 1.0 ml serum-free medium.
The optimal viral concentration for >90% expression in WIF-B cells is 2.5 × 1010 virus particles/ml. However, expression efficiencies can vary among viruses and among cell types. A few dilutions may need to be tried to get optimal infection and expression.Vortex again immediately before applying diluted virus to coverslips. Retrieve the cells grown on coverslips from 37°C incubator and rinse with 2 to 3 ml serum-free medium.
-
Aspirate the serum-free medium from the cells using a vacuum/aspirator and add 1.0 ml of the virus prepared in step 2.
If the virus stock is precious, 100 μl of the diluted virus can be placed in a small “puddle” directly on the coverslip. To do this, coverslips are pushed into the middle of the well, and the medium around them aspirated completely. If the coverslip is touching the side of the well or too much media remains, the surface tension of the “puddle” will break and the coverslip will dry out. Return the dishes to the 37°C incubator and infect for 30 to 60 min.
-
Add 2 to 3 ml complete medium to each dish (without removing virus) and place the dishes back into the 37°C incubator.
In general, 16- to 24-hr incubations are sufficient for robust protein expression. If using recombinant anti-sense or shRNA adenovirus to knockdown protein expression, longer incubations (e.g., 48 to 72 hr) are needed.
BASIC PROTOCOL 2: BASOLATERAL TO APICAL TRANSCYTOSIS ASSAY
Different epithelial cells use different routes to transport newly synthesized apical proteins to their correct destination. In simple epithelial cells, such as MDCK cells, proteins take the “direct” route; they are delivered from the TGN directly to the apical surface. In hepatocytes, single transmembrane domain and glycophosphatidylinositol-anchored apical residents take the “indirect route” (Bartles et al., 1987; Bartles and Hubbard, 1988; Schell et al., 1992). They are first transported from the TGN to the basolateral plasma membrane where they are selectively retrieved by endocytosis and transcytosed to the apical surface (Fig. 15.17.1). Other epithelial cells, e.g., Caco-2 cells, use a combination of both pathways. Thus, hepatocytes represent one extreme in the possibilities, and provide a unique format to study transcytosis.
NOTE: Although the assays described in this unit were designed for use in WIF-B cells, they may be optimized for other hepatic cell types. For example, the morphological assays can likely be performed in HepG2 cells and isolated hepatocyte couplets. In addition, if the yield of couplets is high, the biochemical assays may be used to measure basolateral internalization and recycling. A paraformaldehyde/methanol fixation method is described below, which is well-suited to preserve the unique hepatic domain architecture, but many other fixation procedures can be used (e.g., Asai, 2008; also see UNIT 4.3).
Materials
Cells grown on 22 × 22–mm glass coverslips in 35-mm dishes or 6-well dishes
Complete medium [e.g., Coon’s or Khaighn’s modified F12 medium (Sigma) supplemented with fetal bovine serum (Gemini Bio-Products)]
Ice
Primary antibodies specific to antigen(s) of choice (e.g., anti-CD59, AbCam; anti-dipeptidyl peptidase, Life Span Biosciences; or anti-scavenger receptor B1, Novus Biologicals)
1 × PBS (see recipe)
4% paraformaldehyde/PBS (see recipe)
Methanol (production grade)
1% bovine serum albumin/PBS (see recipe)
Fluorophore-conjugated secondary antibodies (e.g., Alexa 488 or 568-conjugated antibodies; Invitrogen)
0.2% bovine serum albumin/PBS (see recipe)
Mounting medium (e.g., Fluoromount from Electron Microscopy Sciences or ProLongGold from Invitrogen)
1.5-ml microcentrifuge tubes
6-well dishes
Ice bucket
Vacuum/aspirator
Stainless steel forceps, super-fine tip
Humidified chamber
Glass slides
Epifluorescence microscope equipped with 60× or 100× objectives
Digital camera (e.g., Coolsnap HQ2 digital camera from Photometrics) and image capture software (e.g., IPLabs from Biovision)
Surface label
NOTE: All steps are done on ice, in the cold room with prechilled solutions.
-
1
Place the cells grown on coverslips into new 6-well dishes.
-
2
Add 2 to 3 ml prechilled complete medium to each well and place the dishes on ice in an ice bucket in a cold room (4°C) and cool down for 5 min to stop membrane movement.
-
3
Aspirate all liquid using a vacuum/aspirator and push the coverslips into the middle of the well with forceps.
If the coverslip is touching the side of the well, the surface tension of the “puddle” will break and the coverslip will dry out. -
4
Add 100 μl of the primary antibodies diluted to the desired concentration in complete medium to each coverslip and incubate for 15 to 30 min at 4°C on ice.
In general, a good starting dilution for most antibodies is 1:50 (see Critical Parameters).If longer labeling times are chosen, we suggest using carbonate-free medium buffered with 10 mM HEPES to avoid changes in pH. -
5
Briefly rinse each coverslip with complete medium to remove most of the excess antibodies.
-
6
Wash each coverslip by incubating on ice with 2 ml complete medium three times for 2 min each. For the 0 min time point, or if you are only interested in detecting surface-associated antigens, proceed to the cell fixation steps after washing.
Transcytosis assay
NOTE: Steps are performed at 37°C with prewarmed solutions.
-
7
Replace cold, complete medium with 2 to 3 ml fresh complete medium prewarmed to 37°C.
-
8
Return the cells to the 37°C incubator for the desired time of chase. For many antigens, apical delivery is detected after 1 hr. For complete apical delivery, up to 3 hr may be required.
Alternatively, continuous antibody uptake may be used to label the structures of the entire transcytotic pathway. For these experiments, coverslips are placed in 6-well dishes and 100 μl of the primary antibodies diluted to the desired concentration are added to each coverslip. The dishes are carefully returned to the 37°C incubator for the desired times. The coverslips are then rinsed and washed three times, each time for 2 min with complete medium and then fixed and stained.
Perform cell fixation and permeabilization
NOTE: All steps are done on ice with prechilled solutions.
NOTE: This method allows for the superior preservation of domain architecture; however, other standard cell fixation/permeabilization methods may be used.
-
9
Briefly rinse each coverslip once with PBS.
-
10
Add 1.0 ml ice-cold 4% PFA/PBS to each coverslip for 1 min and gently mix after each addition to ensure the coverslip is fully immersed.
-
11
Add 1.0 ml prechilled methanol to each coverslip and mix gently.
-
12
Add an additional 2.0 ml methanol to each coverslip.
-
13
Aspirate the liquid and add another 2.0 ml methanol, aspirate again, and quickly add another 2.0 ml methanol. Let cells sit on ice for 10 min.
If many coverslips are being processed at once, most of the methanol can be decanted directly into the sink or proper waste container to speed up aspiration. -
14
Aspirate the methanol and quickly replace with 2 ml PBS.
-
15
Discard the PBS and briefly rinse the coverslips a second time with PBS, then rehydrate the cells by incubating three times for 5 min with 2 ml PBS at room temperature.
-
16
Continue with immunofluorescence staining.
You can also store the fixed cells in 0.02% azide/PBS at 4°C (most structures and antigens are stable now).
Perform immunofluorescence staining
-
17
Aspirate the PBS and block the cells (nonspecific binding sites) by adding 1 to 2 ml of 1% BSA/PBS for at least 15 min at room temperature.
-
18
Place the coverslips in a humidified chamber.
The humidified chamber can be made using a large 15-cm plastic petri dish or other container with moistened filter paper in the bottom upon which Parafilm is placed. As you place the coverslip into the chamber, remove excess liquid by placing the edge of the coverslip gently against a tissue wipe. This will wick the fluid off. -
19
Add 100 μl secondary antibodies diluted to the desired concentration in 1.0% BSA/PBS to each coverslip and incubate 30 min at room temperature.
Be sure not to let the coverslip dry out as you are adding the antibody solutions. -
20
Place the coverslips back into the 6-well dish and wash two times for 5 min each in 0.2% BSA/PBS.
-
21
Wash the coverslips once for 5 min in PBS.
-
22
Remove the excess liquid and mount the coverslip(s) onto glass slides with a drop (~30 μl) of mounting medium.
-
23
Allow the mounting medium to harden for 60 min to overnight at room temperature before viewing under the microscope (Fluoromount-G can also harden overnight at 4°C; complete curing of ProLong Antifade may take 2 days, but cells can be viewed earlier).
Micrographs are taken using a cooled CCD camera and imaging analysis software.
SUPPORT PROTOCOL 1: SEMI-QUANTITATIVE METHOD TO MEASURE EXTENT OF APICAL DELIVERY
Because the apical membrane in polarized hepatic cells is not experimentally accessible, direct measurement of apical delivery cannot be done. However, we have developed a semi-quantitative fluorescence-based assay that measures the relative extent of transcytotic delivery to the apical membrane.
Additional Materials (also see Basic Protocol 2)
Slides prepared using Basic Protocol 2
Image analysis software (e.g., ImageJ, National Institutes of Health)
Additional reagents and equipment for principles of imaging an analysis (Asai, 2008)
Visualize and digitize random epifluorescent fields from each slide (for principles of imaging and analysis, see Asai, 2008).
-
From micrographs, measure the average pixel intensities of selected regions of interest (ROI) placed at the apical or basolateral membrane of the same polarized cell.
The Measure ROI tool of the ImageJ software can be used for these measurements. -
Obtain the average background pixel intensity by placing an ROI at a nonlabeled region of the cell.
In general, the ROI areas are 30 to 50 pixels2 and multiple ROIs are collected in the same cell to verify that representative intensities are measured. Subtract the average background pixel intensity from each apical and basolateral value.
-
Determine a ratio of apical to basolateral average fluorescence intensity.
An increased ratio indicates increased apical delivery.
SUPPORT PROTOCOL 2: MORPHOLOGY-BASED RECYCLING/APICAL RETENTION ASSAY
For these assays, cells that have been antibody-labeled and processed up to step 8 of Basic Protocol 2 are further processed to visualize recycling and/or apical retention. In general, the remaining surface-associated antibodies are stripped with a low pH buffer allowing the researcher to monitor the fate of internalized antibody-antigen complexes after apical delivery (see Fig. 15.17.3).
Additional Materials (also see Basic Protocol 2)
Cells on coverslips (see step 8 from Basic Protocol 2)
Isoglycine, pH 2.5 (see recipe)
Rinse the cells briefly three times, each time with prewarmed PBS.
Aspirate the PBS. Add 0.5 ml isoglycine prewarmed to 37°C to each coverslip and incubate at room temperature for 2 min while shaking gently in your hands. Start the timer after adding isoglycine to the first coverslip.
Immediately decant the isoglycine (just dump into the sink) and add 2 ml prewarmed PBS to each coverslip in the same order that the isoglycine was added.
Briefly rinse the coverslips three times, each time with prewarmed PBS, then twice with prewarmed complete medium.
-
Aspirate the complete medium. Add prewarmed complete medium and place the cells into a 37°C incubator. Let the cells recover for at least 10 min before adding any pharmacological agents. Recycle for desired times, usually 1 to 2 hr.
We recommend time points of 0, 2, 5, 15, 30, and 60 min for these assays. In most cases, recycling will reach saturation after 1 hr. Proteins with slower recycling rates may require 2-hr incubations. Briefly rinse the coverslips three times, each time with PBS. Aspirate the PBS and fix and stain as described in Basic Protocol 2 (see steps 10 to 23).
-
Measure relative apical retention as described in Support Protocol 1.
If antigen-antibody complexes recycle into intracellular structures, the procedure can be modified by collecting average pixel intensities for background, for the apical surface, and for the intracellular structures. After background subtraction, a ratio of the apical versus intracellular fluorescence intensities can be determined.
BASIC PROTOCOL 3: QUANTITATIVE BASOLATERAL INTERNALIZATION AND RECYCLING ASSAYS
For these assays, purified, biotinylated antibodies are used to specifically label proteins at the basolateral surface in live cells. After the desired time of continuous labeling at 37°C, the remaining cell surface-associated antibodies are eluted with a low pH buffer. Cells are then lysed, and aliquots of the eluate and lysate are incubated in streptavidin-coated 96-well plates. The captured antibodies are detected with HRP-conjugated secondary antibodies, followed by colorimetric detection of the substrate.
Materials
0.5 M Tris base
Cells grown on 22 × 22–mm glass coverslips in 35-mm or 6-well dishes
Primary antibodies specific to antigen(s) of choice
Complete medium (with serum)
Ice
1× PBS (see recipe), ice-cold
Isoglycine, pH 2.5 (see recipe)
Low pH lysis buffer, pH 2.5 (see recipe)
High pH lysis buffer, pH 7.75 (see recipe)
0.4% Trypan blue stain (Invitrogen)
Wash buffer (see recipe)
Biotinylated affinity-purified antibodies or total IgG purified from serum or ascites
Neutralized isoglycine (see recipe)
HRP-conjugated secondary antibodies (e.g., anti-rabbit or mouse IgG peroxidase conjugates from Sigma-Aldrich)
HRP Substrate Detection kit (Bio-Rad Laboratories)
Stainless steel forceps, super-fine tip Kimwipes
37°C incubator
1.5-ml microcentrifuge tubes
Cell scraper
35-mm dishes
Light microscope equipped with a low-powered objective
Reacti-Bind Neutravidin-Coated Polystyrene Plates (Thermo Fisher Scientific)
Humidified chamber (see Basic Protocol 2)
Microplate reader
Internalization
-
1
Prelabel 1.5-ml microcentrifuge tubes containing 60 μl Tris base. Set the tubes aside for use in step 8.
-
2
With forceps, place the coverslips in the middle of the wells in a new 6-well dish. Aspirate any excess medium transferred, or wick off any excess medium first by placing edge against a Kimwipe.
-
3
Gently add 100 μl antibodies diluted to the desired concentration in complete medium to each coverslip.
-
4
Carefully place the dish into a 37°C incubator and continuously label the cells for the desired time points.
We recommend time points of 0, 2, 5, 15, 30, and 60 min for these assays. Proteins with slower internalization rates may require 2-hr labeling incubations.Do these time points in duplicate. In addition, it is easiest to add antibodies so that all time points expire at the same time. To avoid cooling of the cells to room temperature when adding antibody for a new time point, dishes may be placed on a Styrofoam pad while outside the hood. -
5
After continuous labeling, place the 6-well dishes on ice and wash the cells three times, each time for 2 min with ice-cold complete medium.
-
6
Rinse the cells twice, each time with ice-cold PBS.
-
7
Elute surface-bound antibodies from the cells with 300 μl isoglycine at room temperature for 5 min.
-
8
Recover the eluates with a pipet and place them into prelabeled 1.5-ml microcentrifuge tubes that each contain 60 μl Tris base (this neutralizes the eluted antibodies; see step 1).
-
9
Rinse the cells three times, each time with PBS. Place the 6-well dish on ice and lyse with 300 μl low pH lysis buffer for 30 min.
-
10
Scrape the cells from the coverslip using a cell scraper and place the extract in prelabeled microcentrifuge tubes containing 60 μl of Tris base.
-
11
Correct the elution samples (step 8) for detergent by adding 40 μl of 10× high pH lysis buffer.
Final volumes: Elutions (surface-associated) = 400 μl
Lysates (internalized population) = 360 μl.
Recycle
For these assays, cells that have been continuously labeled with biotinylated antibodies are eluted as above (steps 1 to 8) and re-incubated at 37°C for the desired times. Cells are eluted again (representing the recycled population) and then lysed. The eluates and lysates are processed as for the internalization assays.
-
12
Prewarm isoglycine, complete medium, and PBS to 37°C.
-
13
Prelabel 1.5-ml microcentrifuge tubes containing 60 μl Tris base and set aside for use in steps 16 and 20.
-
14
Repeat steps 1 to 6. Continuously label all cells for 1 hr.
-
15
Elute cells with 300 μl isoglycine for 2 min at room temperature. Set the timer after addition of isoglycine to the first coverslip.
-
16
Recover the eluates from at least a pair of coverslips and place into prelabeled microcentrifuge tubes containing 60 μl of Tris base (see step 13) to neutralize the solution.
This sample is referred to as total surface bound. -
17
Immediately discard the isoglycine from the remaining coverslips (just dump it in the sink) and rinse two to three times with 2 ml PBS. Add 2 ml PBS to the coverslips in the same order that the isoglycine was added.
-
18
Rinse the cells two to three times with complete medium at 37°C and allow recycling for the desired times at 37°C.
We recommend time points of 0, 2, 5, 15, 30, and 60 min for these assays. Proteins with slower recycling rates may require 2-hr incubations. -
19
Quickly rinse the coverslips with prewarmed PBS and elute with 300 μl isoglycine for 5 min at room temperature.
-
20
Recover the eluates and place into prelabeled microcentrifuge tubes containing 60 μl of Tris base (see step 13).
-
21
Lyse the cells as described in steps 9 and 10.
-
22
Correct the elution samples from step 16 and 20 for detergent concentration by adding 40 μl of 10× high pH lysis buffer.
Final volumes: Total surface bound = 400 μl
Eluted samples (recycled population) = 400 μl
Lysates (the intracellular population) = 360 μl.
Perform trypan blue exclusion to assess cell viability
During the first stripping step in the recycling assay, a significant percentage of the cells will not survive. Thus, the calculated values must be corrected for the percent of surviving cells. Cell viability can be measured using trypan blue exclusion on one or two coverslips processed in parallel. In general, the numbers of cells surviving are similar after 0 to 60 min of recycling, but it is recommended to assay cells after 60 min of recycling.
-
23
Place the coverslip into a 35-mm dish.
-
24
Briefly rinse the cells with PBS at room temperature.
-
25
Add 1 ml PBS and 1.0 ml 0.4% trypan blue dissolved in PBS to the dish.
-
26
Incubate for 3 min at room temperature.
-
27
Count the number of total cells and number of blue cells on a tissue culture microscope using a low-powered objective. Count at least ten random fields.
-
28
Calculate percent cells surviving by dividing the number of blue cells by the total number of cells.
Quantitative measurements
-
29
Wash the wells of the neutravidin-coated 96-well plate three times, each time with 100 μl wash buffer.
-
30
Serially dilute the purified biotinylated antibodies in neutralized isoglycine to produce a standard curve. Start at the top of the plate and add 200 μl antibody solution (well A1; see sample plate setup). The remaining seven wells below should contain 100 μl neutralized isoglycine. Remove 100 μl of the antibody solution and place into the next well (B1). Pipet up and down several times to mix and remove 100 μl from that well and place into the next well. Continue down the plate until well G1; remove 100 μl from this well and discard. Each well should have only 100 μl. Include a blank (H1, 100 μl of neutralized isoglycine) to account for background color development.
The starting dilution (for A1) should be ~1:10,000, but it depends on the antibody concentration of the stock solution and the extent of biotinylation. -
31
For each sample, add 100 μl of the undiluted sample to one well. The well below should contain 100 μl neutralized isoglycine. In addition, add 100 μl of the undiluted sample to this well and pipet up and down several times to mix. Remove 100 μl from this well and discard.
-
32
Incubate 2 hr at room temperature in a humidified chamber.
-
33
Wash as in step 29.
-
34
Add 100 μl HRP-conjugated secondary antibodies diluted 1:1000 in wash buffer to each well.
-
35
Incubate 30 min at room temperature.
-
36
Wash as in step 29.
-
37
Proceed with colorimetric assay as described by the manufacturer. For the Bio-Rad kit, prepare the solution (9 parts Solution A with 1 part Solution B) right before use.
The solutions should be at room temperature. -
38
Allow color development at room temperature; this usually takes about 1 hr. Read the plate at OD405 using a microplate reader.
See Table 15.17.1 for a sample plate setup for an experiment with six time points (0 to 60 min).
Table 15.17.1.
Sample Plate Setup for an Experiment with Six Time Points (0 to 60 min)
| Ab dilution | Eluted samplesa | Lysed samplesa | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| A | 1:10,000 | 0′ undil | 5′ undil | 30′ undil | 0′ undil | 5′ undil | 30′ undil |
| B | 1:20,000 | 1:2 | 1:2 | 1:2 | 1:2 | 1:2 | 1:2 |
| C | 1:40,000 | 0′ undil | 5′ undil | 30′ undil | 0′ undil | 5′ undil | 30′ undil |
| D | 1:80,000 | 1:2 | 1:2 | 1:2 | 1:2 | 1:2 | 1:2 |
| E | 1:160,000 | 2′ undil | 15′ undil | 60′ undil | 2′ undil | 15′ undil | 60′ undil |
| F | 1:320,000 | 1:2 | 1:2 | 1:2 | 1:2 | 1:2 | 1:2 |
| G | 1:640,000 | 2′ undil | 15′ undil | 60′ undil | 2′ undil | 15′ undil | 60′ undil |
| H | Blank | 1:2 | 1:2 | 1:2 | 1:2 | 1:2 | 1:2 |
Undil, undiluted.
Perform calculations
-
39
Subtract the blank value from each point (the blanks have been incubated with secondary antibodies and processed with the detection kit).
-
40
Generate a standard curve from the antibody serial dilution plotting ng protein on the x axis and OD405 readings on the y axis.
-
41
Determine the ng protein in each sample.
-
42
Calculate the final protein amounts by multiplying the undiluted eluted samples by 4 and the undiluted lysates by 3.6. The 1:2 dilutions should be multiplied by 8 and 7.6, respectively. Determine the average values for the two dilutions. Finally, average these values for the duplicate coverslips for each time point.
-
43
Calculate the percent internalized or recycled. Correct the recycled values for percent cells surviving (see steps 23 to 28).
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Bovine serum albumin/PBS, 0.2%
For 50 ml:
10 ml 1% BSA/PBS (see recipe)
40 ml ddH2O
Make fresh and leave at room temperature
Bovine serum albumin/PBS, 1%
For 50 ml:
0.5 g bovine serum albumin (BSA)
50 ml 1× phosphate-buffered saline (PBS; see recipe)
-
Make fresh and leave at room temperature
Gently layer BSA on top of PBS and let dissolve slowly. Mixing will cause the BSA to clump.
High pH lysis buffer, 10×
For 100 ml:
5.84 g octylglucoside (200 mM final; if octylglucoside is included in the low pH lysis buffer)
1% Triton X-100
1.5 g glycine (200 mM final)
0.87 g NaCl (150 mM final)
Adjust pH to 7.75 using 10 N NaOH
Store up to 6 months at 4°C
Isoglycine
For 100 ml:
3.0 g glycine (400 mM final)
8.7 g NaCl (150 mM final)
Adjust pH to 2.5 using concentrated HCl
Store up to 6 months at 4°C
Low pH lysis buffer
For 100 ml:
584 mg octylglucoside (20 mM final), optional
0.1% Triton X-100
1.5 g glycine (200 mM final)
0.87 g NaCl (150 mM final)
Adjust pH to 2.5 using concentrated HCl
Store up to 6 months at 4°C
Neutralized isoglycine
For 100 ml:
75 ml isoglycine
15 ml 0.5 M Tris base
10 ml of 10× high pH lysis buffer (see recipe)
Store up to 6 months at 4°C
4% Paraformaldehyde/PBS
For 10 ml:
2.5 ml of 16% PFA (Electron Microscopy Sciences)
1 ml of 10× PBS (see recipe)
6.5 ml ddH2O
Prepare fresh and store on ice
PBS, 1×
For 1 liter:
100 ml of 10× PBS (see recipe)
900 ml ddH2O
Store up to 6 months at 4°C
PBS, 10×
For 1 liter:
2.0 g KCl (2.6 mM final)
2.0 g KH2PO4 (1.5 mM final)
80.0 g NaCl (136 mM final)
12.2 g Na2HPO4 (8.5 mM final)
Adjust pH to ~7.4 using 10 N NaOH
Store up to 1 year at room temperature
Wash buffer
For 100 ml:
0.1 g BSA (0.1% final)
0.05% Tween-20
0.87 g NaCl (150 mM final)
0.61 g Tris base (50 mM final)
Adjust pH to 7.2
Store up to 6 months at 4°C
COMMENTARY
Background Information
Exogenous protein expression
Insights into the mechanisms regulating polarized membrane traffic often require the judicious use of exogenously expressed proteins. For example, expression of mutant regulatory proteins is used to assess their effects on certain trafficking steps. Generally, stable or transient transfections allow for exogenous protein expression in a variety of polarized epithelial cell types. However, hepatic cells are recalcitrant to conventional transfection methods. Instead, recombinant adenovirus is the method of choice to deliver genes to polarized WIF-B cells and hepatocytes in vivo (Bastaki et al., 2002). Because adenovirus has a natural tropism for liver, its infection efficiency is high (>90% for most viruses). Although expression is transient (~4 days), it is robust and reasonably uniform. Importantly, retention of hepatic polarity is excellent in infected cells. Of course, there are caveats to the use of a recombinant protein approach. Since overexpression can lead to protein mislocalization, it is important to know the levels and locations of endogenous proteins and those of the exogenously expressed protein to fully interpret results. However, overexpression can be exploited when the right question is asked. For example, saturation of the apical secretory pathway with protein overexpression in MDCK cells revealed that mechanisms with finite capacities exist (Marmorstein et al., 2000).
Antibody-based assays
Although we are still unable to measure endocytosis or recycling from the apical surface (see below), we have made excellent progress in developing methods to monitor internalization and recycling from the basolateral membrane and transcytotic delivery to the apical surface (Tuma et al., 2002; Nyasae et al., 2003). In general, these assays rely on the use of antibodies that recognize external epitopes of proteins present (at least transiently) at the basolateral surface. Because tight junctions restrict access to the apical surface, only proteins at the basolateral surface are labeled.
The validity of the antibody trafficking method depends on the assumption that the antibodies are serving as faithful reporters of the membrane proteins to which they are bound, i.e., that the trafficking behavior is not an antibody-induced phenomenon. To test this assumption, several control experiments were originally performed (Ihrke et al., 1998). These controls included: the use of Fab fragments (to exclude the possibility for antigen cross-linking by divalent IgG); the use of antibodies to track proteins with different intracellular fates (to ensure those fates were observed); and measurements to determine whether antibodies were degraded or released into the medium (to determine stability of the trafficked antigen-antibody complexes). Furthermore, labeled antigens were found to reach their steady-state distributions at rates similar to those observed in hepatocytes in vivo, as measured by quantitative biochemical techniques (Bartles et al., 1987; Schell et al., 1992; Maurice et al., 1994; Ihrke et al., 1998). In addition, the quantitative assays for measuring basolateral internalization and recycling are very sensitive; <0.2 ng of bound antibody can be detected. Importantly, the kinetics measured for transferrin receptor using these assays were consistent with those previously reported from other systems, further validating this antibody-based approach (Tuma et al., 2002).
Critical Parameters
The architectural complexity of the liver has limited the ability to examine bile canalicular events. Unlike the lumenal domains of kidney or intestinal epithelia, the hepatocyte lumenal surfaces are branching networks between adjacent cells that are not easily accessible to experimental manipulation. Although HepG2 and WIF-B cells have emerged as in vitro polarized hepatic cell models, the canalicular domain is also sequestered from the external milieu (Fig. 15.17.2) and is not amenable to the types of experimentation performed in simpler, columnar epithelial cells. Although we are able to visualize apical protein dynamics in WIF-B and HepG2 cells, the assays to measure them directly are still lacking. Nonetheless, the assays described in this unit allow for the examination of many important membrane traffic steps.
The success of the assays described in this unit hinges upon finding appropriate antibodies for antigen labeling. First, because live cells are surface labeled in all of these assays, it is important to choose antibodies only to extracellular epitopes of the membrane protein to be examined. This also means that only proteins that span the membrane and have external epitopes can be examined using these assays. When possible, we suggest using monoclonal antibodies. Here, all antibodies share the same affinity for the antigen (unlike polyclonal antibodies), and there is less background with these preparations. Because antibodies (whether monoclonal or polyclonal) vary greatly in their antigen affinities, and because the abundance of membrane proteins also varies, each antibody selected must be independently optimized for use. We suggest systematically testing increasing antibody dilutions across a wide range of concentrations. For ascites or whole serum, we recommend dilutions from 1:25 all the way to 1:1000. If using purified antibodies, we recommend a range from 0.05 to 10 μg/ml. Although these ranges seem extensive, we routinely use antibodies at dilutions from both ends of this spectrum. Similarly, conditions for isoglycine stripping must be determined for each antibody used. There are a couple of parameters that can be altered to optimize efficient antibody stripping. We recommend slight increases in stripping time at room temperature or a reduced stripping time (1 to 2 min) at 37°C. In addition, we have found that decreasing the antibody concentrations during cell labeling can lead to better stripping. Nonetheless, we have found that some antibodies cannot be used in assays that involve antibody stripping. They have such high affinity for their antigens that the conditions required for stripping drastically diminishes or inhibits cell viability. Other antibodies may have too low affinities to be used in any uptake assay because they dissociate from their antigen during the experiment (see below).
Another critical parameter to consider when doing these experiments is monitoring tight junction integrity. If tight junctions are leaky, antibodies may surface-label apical antigens, thereby confounding interpretation of the trafficking results. We routinely check for tight junction integrity by processing a 0 min chase sample for immunostaining of the surface-labeled primary antibodies. If the tight junctions are intact, the bound antibodies should only be detected at the basolateral surface (see Fig. 15.17.2). Finally, we also suggest that morphological analysis be performed alongside the quantitative assays. It is important to determine whether the antibody-antigen complexes are trafficking to the predicted location (e.g., the apical surface). Furthermore, significant loss of labeling intensity over time suggests that, due to low affinity for the antigen, unbound/dissociated antibody is transported to lysosomes and degraded. If this and/or aberrant patterns are observed, the antibody selected is likely not appropriate for use in these assays.
Anticipated Results
The results of a typical basolateral to apical transcytosis assay (Basic Protocol 2) are shown in Figure 15.17.2. At 0 min chase, only basolateral staining of the newly synthesized apical resident protein en route to the apical surface is observed. However, after 45 min, apical labeling is observed along with intracellular labeling of sub-apical structures. These structures represent the previously described sub-apical compartment (Ihrke et al., 1998). By 90 min, nearly all of the antigen-antibody complexes are detected at the apical surface, indicating successful delivery. Although a 90 min chase is typically long enough to detect apical labeling, it should be noted that different proteins are delivered to the apical surface with different kinetics. For example, antibodies to aminopeptidase N (APN) and polymeric IgA receptor show maximal apical labeling relatively quickly, after only 1 to 2 hr. However, 5′ nucleotidase-antibody complexes take ~3 hr to reach steady state–like distributions (Ihrke et al., 1998).
In Figure 15.17.3, the results of a typical apical recycling/retention assay are shown. First, the antibody-antigen complexes are concentrated at the apical surface by incubating surface-labeled cells for 1 hr. The excess antibody is washed away and the cells are chased an additional hour at 37°C (Fig. 15.17.3A). Any residual antibodies at the basolateral surface are stripped with isoglycine (Fig. 15.17.3B). It is important to note that 10% to 30% of mature WIF-B cells are nonpolarized. In these nonpolarized cells, apical proteins travel to the so-called “apical compartment” (marked with arrows in Fig. 15.17.3B), juxtanuclear structures that contain only other apical proteins (Tuma et al., 2002). Cells were incubated another hour at 37°C to allow recycling of antigen-antibody complexes. In general, only the apical proteins in nonpolarized WIF-B cells recycle from the intracellular “apical compartment” to the plasma membrane (marked with arrowheads), whereas in polarized cells, they remain at the apical surface (Fig. 15.17.3C). We have found that apical protein recycling to the basolateral plasma membrane is observed in latrunculin-treated cells, suggesting that apical retention is actin-dependent (Tuma et al., 2002).
Time Considerations
In general, the actual adenovirus infection (Basic Protocol 1) takes only 10 to 15 min, but an overnight incubation (16 to 24 hr) is needed for robust protein expression. Sufficient knockdown of protein expression often requires longer incubation times after infection (48 to 72 hr). The transcytosis assay (Basic Protocol 2) takes ~1 to 3 hr. The incubation time for complete apical delivery will vary due to the kinetics of the protein of interest. While a minimum of 45 min chase may be sufficient for some proteins, others may require chase times up to 3 hr for complete delivery. It should be noted that the medium and dishes must be prechilled for surface labeling and prewarmed for chase times. The recycling/apical retention assay (Support Protocol 2) requires an additional ~2 to 3 hr depending on the length of recycling. In addition, allot 3 hr for immunostaining. In general, these assays can be completed in one full day. The image capture and analysis can take from a few hours to half a day. Quantitative recycling and internalization (Basic Protocol 3) requires ~4 hr, depending on the length of time needed for efficient recycling and internalization of the protein of interest. Allot an additional ~4 hr for quantitative measurements and calculations.
Literature Cited
- Asai DJ. Immunofluorescence microscopy. Curr Protoc Essential Lab Tech. 2008:9.2.1–2.21. [Google Scholar]
- Bartles JR, Hubbard AL. Plasma membrane protein sorting in epithelial cells: Do secretory pathways hold the key? Trends Biochem Sci. 1988;13:181–184. doi: 10.1016/0968-0004(88)90147-8. [DOI] [PubMed] [Google Scholar]
- Bartles JR, Feracci HM, Stieger B, Hubbard AL. Biogenesis of the rat hepatocyte plasma membrane in vivo: comparison of the pathways taken by apical and basolateral proteins using subcellular fractionation. J Cell Biol. 1987;105:1241–1251. doi: 10.1083/jcb.105.3.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastaki M, Braiterman LT, Johns DC, Chen YH, Hubbard AL. Absence of direct delivery for single transmembrane apical proteins or their “Secretory” forms in polarized hepatic cells. Mol Biol Cell. 2002;13:225–237. doi: 10.1091/mbc.01-07-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL. Isolated hepatocyte couplets and bile duct units-novel preparations for the in vitro study of bile secretory function. Cell Biol Toxicol. 1997;13:289–300. doi: 10.1023/a:1007435408208. [DOI] [PubMed] [Google Scholar]
- De Marco MC, Martin-Belmonte F, Kremer L, Albar JP, Correas I, Vaerman JP, Marazuela M, Byrne JA, Alonso MA. MAL2, a novel raft protein of the MAL family, is an essential component of the machinery for transcytosis in hepatoma HepG2 cells. J Cell Biol. 2002;159:37–44. doi: 10.1083/jcb.200206033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffo G, Hamon-Benais C, Angrand PO, Fox M, West L, Lecoq O, Povey S, Cassio D, Weiss M. HNF4 and HNF1 as well as a panel of hepatic functions are extinguished and reexpressed in parallel in chromosomally reduced rat hepatoma-human fibroblast hybrids. J Cell Biol. 1993;121:887–898. doi: 10.1083/jcb.121.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC. Adenoviral vectors. Curr Protoc Hum Genet. 2004;40:12.4.1–12.4.25. doi: 10.1002/0471142905.hg1204s40. [DOI] [PubMed] [Google Scholar]
- Ihrke G, Neufeld EB, Meads T, Shanks MR, Cassio D, Laurent M, Schroer TA, Pagano RE, Hubbard AL. WIF-B cells: An in vitro model for studies of hepatocyte polarity. J Cell Biol. 1993;123:1761–1775. doi: 10.1083/jcb.123.6.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrke G, Martin GV, Shanks MR, Schrader M, Schroer TA, Hubbard AL. Apical plasma membrane proteins and endolyn-78 travel through a subapical compartment in polarized WIF-B hepatocytes. J Cell Biol. 1998;141:115–133. doi: 10.1083/jcb.141.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Csaky KG, Baffi J, Lam L, Rahaal F, Rodriguez-Boulan E. Saturation of, and competition for entry into, the apical secretory pathway. Proc Natl Acad Sci USA. 2000;97:3248–3453. doi: 10.1073/pnas.070049497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice M, Schell MJ, Lardeux B, Hubbard AL. Biosynthesis and intracellular transport of a bile canalicular plasma membrane protein: Studies in vivo and in the perfused rat liver. Hepatology. 1994;19:648–655. doi: 10.1002/hep.1840190316. [DOI] [PubMed] [Google Scholar]
- Nyasae LK, Hubbard AL, Tuma PL. Transcytotic efflux from early endosomes is dependent on cholesterol and glycosphingolipids in polarized hepatic cells. Mol Biol Cell. 2003;14:2689–2705. doi: 10.1091/mbc.E02-12-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofsen H, Wolters H, Van Luyn MJ, Miura N, Kuipers F, Vonk RJ. Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology. 2000;119:782–793. doi: 10.1053/gast.2000.17834. [DOI] [PubMed] [Google Scholar]
- Sandell L, Sakai D. Mammalian cell culture. Curr Protoc Essential Lab Tech. 2011:4.3.1–4.3.32. [Google Scholar]
- Schell MJ, Maurice M, Stieger B, Hubbard AL. 5′ nucleotidase is sorted to the apical domain of hepatocytes via an indirect route. J Cell Biol. 1992;119:1173–1182. doi: 10.1083/jcb.119.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks MS, Cassio D, Lecoq O, Hubbard AH. An improved rat hepatoma hybrid cell line. Generation and comparison with its hepatoma relatives and hepatocytes in vivo. J Cell Sci. 1994;107:813–825. doi: 10.1242/jcs.107.4.813. [DOI] [PubMed] [Google Scholar]
- Shenvi SV, Dixon BM, Shay KP, Hagen TM. A rat primary hepatocyte culture model for aging studies. Curr Protoc Toxicol. 2008;37:14.7.1–14.7.10. doi: 10.1002/0471140856.tx1407s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma P, Nyasae L, Hubbard A. Non-polarized cells selectively sort apical proteins from cell surface to a novel compartment, but lack apical retention mechanisms. Mol Biol Cell. 2002;13:3400–3415. doi: 10.1091/mbc.02-04-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ijzendoorn SC, Hoekstra D. Polarized sphingolipid transport from the subapical compartment: Evidence for distinct sphingolipid domains. Mol Biol Cell. 1999;10:3449–3461. doi: 10.1091/mbc.10.10.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegers MMP, Hoekstra D. Mechanisms and functional features of polarized membrane traffic in epithelial and hepatic cells. Biochem J. 1998;336:257–269. doi: 10.1042/bj3360257. [DOI] [PMC free article] [PubMed] [Google Scholar]